

Abstract
Background
Previous studies suggest that transforming growth factor- beta (TGF-∃) provokes cardiac hypertrophy and myocardial fibrosis; however, it is unclear whether the deleterious effects of TGF-∃ signaling are conveyed through SMAD-dependent or SMAD-independent signaling pathways.
Methods and Results
To determine the contribution of SMAD dependent signaling to cardiac remodeling, we performed transaortic constriction (TAC) in SMAD3 null (SMAD3−/−) and littermate control mice (age 10–12 weeks). Cumulative survival 20 days post-TAC was significantly less in the SMAD3 −/− mice when compared to littermate controls (43.6% vs 90.9%, p<0.01). TAC resulted in a significant increase in cardiac hypertrophy in the SMAD3 −/− mice, denoted by an increase in the heart-weight-to-tibial length ratio and increased myocyte cross-sectional area. Loss of SMAD3 signaling also resulted in a significant 60% decrease in myocardial fibrosis (p < 0.05). A microRNA microarray showed that 55 microRNAs were differentially expressed in littermate and SMAD3−/− mice, and that 10 of these microRNAs were predicted to bind to genes that regulate the extracellular matrix. Of these 10 candidate microRNAs, both miR-25 and miR-29a were sufficient to decrease collagen gene expression when transfected into isolated cardiac fibroblasts in vitro.
Conclusions
The results suggest that SMAD3 signaling plays dual roles in the heart: one beneficial role by delimiting hypertrophic growth, and the other deleterious by modulating myocardial fibrosis, possibly through a pathway that entails accumulation of microRNAs that decrease collagen gene expression.
Keywords: Fibrosis, microRNA, pressure overload, hypertropy
INTRODUCTION
Members of the transforming growth factor-beta (TGF-∃) superfamily are multifunctional cytokines that play an important role in cell growth, differentiation and tissue repair in a variety of organs, including the heart.1,2 The TGF-∃ superfamily can be broadly divided into two major groups: the TGF-∃/activin subfamily and the bone morphogenic protein (BMP)/growth and differentiation (GDF) subfamily. TGF-∃ family members exert their effects by binding to heterodimeric cell surface receptors, consisting of membrane bound type 1 serine/threonine receptor kinases (Activin like kinases [ALK] 1–7) and membrane bound type II serine/threonine receptor kinases (T∃RII, ActRII, ActRIIB, BMPRII and AMHR).3 After a ligand/type I/type II receptor complex is formed, the constitutively activated type II receptor transphosphorylates and activates the type I receptor, which in turn phosphorylates downstream cytoplasmic mediators belonging to the SMAD family (for mammalian homologue of Sma proteins in Caenorhabditis Elegans and mothers against decapentaplegic (MAD) proteins in Drosophila). 3,4) Members of the SMAD family of proteins fall into three structurally and functionally distinct groups: receptor SMADs (R-SMADs [SMAD 1,2,3,5,8]), co-mediator Co-SMADs (SMAD 4), and the inhibitory SMADs (I-SMADs [SMAD 6,7]). All three functional classes of SMADs and the majority of SMAD isoforms have been detected in the cardiovascular system.2 TGF-∃ family members can also signal in SMAD independent manner, through activation of TGF-∃ induced kinase-1 (TAK1), mitogen activated protein kinases (MAPK), Rho-kinases and Akt.1,3,5
Prior studies suggest that the TGF-∃/activin and BMP/GDF family members exert distinct effects in the heart. That is, whereas TGF-∃ family members predominately mediate deleterious effects in the adult heart, including cardiac hypertrophy and myocardial fibrosis,6–11 BMP family members are essential for heart development and promote differentiation of cardiac myocytes. This diversity of effects is thought to arise, at least in part, secondary to the variety of signaling pathways that are activated by members of the transforming growth factor-beta (TGF-∃) superfamily. For example, TGF-∃, which binds to a complex of T∃RII and ALK5, results in the activation of SMAD 2 and 3, whereas BMP binds to BMPR-II and ALK2, 3, or 6 and activates SMADs 1, 5, or 8. What remains unclear, however, is whether the deleterious effects of TGF-∃ signaling are conveyed through SMAD-dependent2 or SMAD-independent signaling pathways. 12 Accordingly, to determine the contribution of SMAD3 dependent signaling to cardiac remodeling, we subjected SMAD3 deficient mice to transaortic constriction. Here we show that loss of SMAD3 dependent signaling results in increased pressure overload induced myocardial fibrosis that is accompanied by the differential expression of two micro-RNAs (miR-25 and miR-29a) in vivo, that are sufficient to decrease collagen gene expression in isolated cardiac fibroblasts in vitro, thus providing a potentially novel mechanism for SMAD3-mediated fibrosis. We also show that loss of SMAD3 mediated signaling results in increased mortality and increased concentric cardiac hypertrophy, suggesting that SMAD3 mediated signaling is cardioprotective following hemodynamic pressure overloading.
METHODS
Generation of SMAD3 Deficient Mice
The SMAD3 deficient mice used herein were generated by targeted disruption of exon 8 of the SMAD3 gene, and were the generous gift of Dr. Anita Roberts. The cardiac phenotype of these mice has been reported elsewhere.13 Heterozygous mice (SMAD3+/−) on a C57BL/6J background were backcrossed in order to generate mice that were null for SMAD3 (SMAD3−/−). Littermates with both SMAD3 alleles intact (SMAD3+/+) were used as the appropriate controls. All mice were housed under standard environmental conditions and were fed commercial chow and tap water ad libitum. All studies conformed with the principles of the National Institutes of Health "Guide for the Care and Use of Laboratory Animals,” and were approved by the Baylor College of Medicine Animal Care and Use Committee.
Effect of Pressure Overload on Survival in SMAD3 −/− Mice
Transverse aortic constriction
Hemodynamic pressure overloading was achieved by transverse aortic constriction (TAC) in 10- to 12 week old male and female SMAD3−/− and littermate control mice, exactly as described.14 To ensure that the mice were subjected to a significant hemodynamic pressure overload, we used a a 20-MHZ pulsed Doppler probe coupled to a fast-Fourier transform spectrum analyzer (model SP-25A, Medasonics, Newark, CA) to record flow velocity in the right and left carotid arteries day 24–48 hours after TAC. For the studies reported herein, we only used mice that had Doppler flow velocity ratios > than 5:1. Previously, we have shown that this degree of hemodynamic pressure overload leads to significant increase in cardiac hypertrophy, without a significant increase in morbidity and mortality.14 Sham operated SMAD3−/− and littermates were used as the appropriate controls. Mice were sacrificed at day 10 and day 20 day post-TAC, and the hearts the hearts perfusion fixed in a formaldehyde-zinc buffer (Z-fix, Anatech Ltd., Battlecreek, MI) for histological analysis. At the time of terminal sacrifice the mice were observed for signs of volume overload (pleural effusions and/or pericardial effusions). Wet and dry lung weights were measured at baseline and 20 days post banding, in order to determine the wet-to-dry lung ratio for each mouse. Dry lung weights were measured after dessicating the lungs in a convection oven at 55°C for 3 hours. We also attempted to measure ambulatory heart rate monitoring in the littermate control and SMAD3 −/− mice. However, because of their relatively smaller body weights, the SMAD3 −/− mice failed to thrive and/or died after implantation of the telemetry devices (TA10EA-F20, Datasciences, Minneapolis). As a result the Baylor Animal Use and Care Committee requested that we discontinue implantation of the telemetry devices in the SMAD3 null mice.
Effect of Pressure Overload on Cardiac Structure in SMAD3 −/− Mice
Cardiac Hypertrophy
Cardiac hypertrophy was assessed by determining the heart-weight-to-tibial length ratio heart-weight-to-body-weight ratios in littermate control and SMAD−/− mice at baseline and 10 and 20 days post-TAC.15 Cardiac myocyte cross-sectional area was determined at identical time points using lectin staining, exactly as described.16 A minimum of 100 myocytes from each heart were examined at mid-myocardial levels under 400× magnification.
Two-dimensionally targeted M-mode echocardiography was used to measure LV structure following hemodynamic overloading, as described.14 Images were obtained at baseline and day 10 and 20 in the banded and sham-operated animals. Anesthesia was induced using 1% isoflurane, and was maintained using 0.5% isoflurane. Throughout the 2-D echocardiographic studies, care was taken to maintain the heart rate > 400 beats per minute by adjusting the level of anesthesia. All 2-D and M-mode echocardiographic measurements were performed by an investigator who was blinded to the experimental that group being studied.
Myocardial Fibrosis
Perfusion fixed hearts from SMAD3−/− and littermate control mice obtained at baseline and day 10 and day 20 post-TAC or sham operation were embedded in paraffin, and stained using the picrosirius red technique, as described.17 To calculate the extent of myocardial fibrosis, the LV was divided into eight quadrants, and the percent area of extracellular picrosirius red staining was computed from random images obtained in each quadrant. Large epicardial arteries and/or veins were excluded, as well as any cutting and/or compression artifacts. At least 16–24 measurements were made for each heart from each group being studied.
Effect of Pressure Overload on Myocardial Inflammation in SMAD3−/− Mice
To determine whether hemodynamic pressure overloading resulted in increased myocardial inflammation in the SMAD3−/− mice, hearts were perfusion fixed at baseline and on day 10 and 20 post-TAC, as described above. Transverse myocardial sections were stained with hematoxylin and eosin and observed by light microscopy at 200× magnification for the degree of cellular infiltration. Three to four axial cross-sections from each heart at the mid-papillary level were identified and each section was divided into quadrants and examined by an independent investigator in a blinded manner. The degree of myocardial inflammation was scored semi-quantitatively in the following manner: 0 = no infiltrate; 1+ = infiltrates involving <25% myocardium; 2+ = infiltrates involving 25% to 50%; 3+ infiltrates involving 50% to 75% of the myocardium; and 4+ = infiltrates involving 75% to 100% of the myocardium, as described by Kanda et al.18
Role of MicroRNAs in Myocardial Fibrosis
Analysis of microRNA Expression in SMAD3 −/− mice
To determine whether microRNAs that target genes that regulate the extracellular matrix were expressed differentially in the littermate control and SMAD3 −/− mice post-TAC, we performed a microRNA microarray. Total RNA was extracted from SMAD3 −/− and littermate control mouse hearts 10 days after sham operation or TAC, using TRIzol reagent (Invitrogen, Carlsbad, CA). The manufacturer’s protocol was modified for the enrichment of small RNAs by overnight precipitation using excess volume of isopropyl alcoholisopropyl alcohol (1.5x TRIzol volume), at −80°C. 100 :g of RNA were sent to L.C. sciences for microRNA microarray.19 RNA from sham operated littemate control and SMAD3 −/− mice and littemate control and SMAD3 −/− mice exposed to TAC was labeled with Cy3 and Cy5 fluorescent dyes, and each pair of samples was hybridized to a single chip that held 380 mature microRNA probes that are listed in Sanger miRBase Release 9.2 (http://www.sanger.ac.uk/Software/Rfam/mirna/) along with control probes used for standardization and quality control. Signal intensities from the array output were normalized after background subtraction using preset control probe intensities. MicroRNAs that were differentially expressed (p<0.01 by ANOVA) were identified and candidate microRNAs that were predicted to control fibrosis-related genes (see results) were selected for further experiments.
Validation of microRNA targets
To determine whether the microRNAs identified in the microarray affected collagen gene expression, we transfected select microRNAs (see Results) into cardiac fibroblasts, and then examined collagen I and collagen III gene expression using quantitative RT-PCR. Briefly, mouse cardiac fibroblasts were prepared as previously described from hearts of 9- to 12-wk-old C57BL/6 mice. Cells from passages 3 to 5 were cultured in DMEM containing 10% FBS but without antibiotics. Twenty-four hours after plating, the cells were treated with synthetic microRNAs (20–50 pM) that were designed to mimic endogenous precursor microRNAs (Pre-miRs, Ambion Inc, Foster City, CA). Transfection was carried out according to manufacturer’s protocol in serum free media using lipofectamine 2000 (Invitrogen, Inc.). To confirm transfection of microRNAs into the cardiac fibroblasts, we used a Cy3- labeled pre-miR negative control (Cat#AM17120, Ambion, Inc.), a non-targeting microRNA tagged at 5’ end with a Cy3- fluorophor. To determine the transfection efficiency, we examined the expression levels of the Cy3-tagged microRNAs by immunofloresence in fibroblast cultures at 6 hours, 24 hours, 48 hours and 72 hours after transfection. In preliminary control experiments we determined that a concentration of 30 pM of oligonucleotide and 2 :l lipofectamine per ml of culture medium resulted in minimum cell toxicity and maximum transfection efficiency (98% efficiency). After optimization, transfection was carried out using pre-miR precursors of selected microRNAs or unlabeled pre-miR microRNA precursor negative control (Cat#AM17110, Ambion Inc.).
Collagen I (COL1A1 and COL1A2) and collagen III (COL3A1) gene expression were determined using real-time PCR. Briefly, total RNA was isolated from cardiac fibroblasts 48 hours after transfection, using TRIzol reagent (Invitrogen, Carlsbad CA). One :g of RNA was converted to cDNA using a high-capacity cDNA reverse transcription kit with random hexamers (Applied Biosytems, Foster City, CA). The primers for COL1A1, COL1A2 and COL3A1, and their corresponding FAM labeled Taqman MGB probes (Taqman gene expression assays, Applied Biosystems) were used to perform real-time PCR of the cDNA samples on an ABI 7300 real-time PCR system (Applied Biosystems). 18S ribosomal RNA was used as a RNA loading control and expression levels of target genes in samples were normalized to a control sample using the comparative Ct ())Ct) method as described before .20
Statistical Analysis
Each value is expressed as mean ∀ SEM. Survival of SMAD3 and littermate mice were compared using the Kaplan-Meier method. Two-way ANOVA was used to test for mean differences in wet-dry lung weight ratios, cardiac hypertrophy, heart weight, body weight, heart weight to tibial length ratios, heart-weight to body-weight ratios, left ventricular wall thickness, radius to height ratio, left ventricular internal diastolic dimension (LVIDD), fractional shortening (FS), fibrillar collagen content, myocardial inflammatory scores and Mac-2 positive cell counts in the SMAD3 and littermate mice at baseline, and 10 and 20 days post aortic banding. Doppler flow velocity ratios between right and left carotid arteries were compared in the SMAD3 and littermate mice using t-test. Where appropriate, post-hoc ANOVA testing (PLSD test) was used to assess mean differences between groups at a given time point. Significant differences were said to exist at a value of p<0.05.
RESULTS
Effect of Pressure Overload on Survival in SMAD3 −/− Mice
Figure 1A depicts the Kaplan-Meier analysis for the littermate control mice and SMAD3−/− mice post-TAC. As shown, the cumulative survival at 20 days post-TAC was significantly less in the SMAD3−/− mice when compared to littermate controls (43.6% vs 90.9%, p<0.01). Importantly, there were no deaths in the littermate control or SMAD3 −/− mice that underwent sham surgery. Inspection of the Kaplan Meier curves showed that the deaths in the SMAD3 −/− mice began several days after surgery and continued throughout the 20-day time of observation. In order to determine the potential mechanism(s) for the increased mortality in the SMAD3 −/− mice post-TAC, we examined the mice for pleural and/or pericardial effusions at the time of terminal sacrifice or following sudden death. This analysis disclosed that there was no evidence of pericardial or pleural effusions in the SMAD3 −/− mice. We also examined the wet-to-dry ratio of the lungs in the littermate control mice and the SMAD3 −/− mice at baseline and at 20 days post-TAC. Although there was a small but statistically significant (p < 0.05) increase in the wet-to-dry ratio in both groups of mice post-TAC (Figure 1B), there was no significant increase in the wet-to-dry ratio of the lungs in the SMAD3 −/− mice and when compared to the littermate control mice post-TAC (p > 0.05). Taken together, these observations suggest that heart failure was unlikely to be the proximate cause of the increase in mortality in the pressure overloaded SMAD3 −/− mice. As noted above, it was not possible to implant telemetry devices in the SMAD3 −/− mice; therefore, we could not determine whether the increase in mortality in the SMAD3 −/− mice was secondary to brady- or tachyarrhythmias.
Figure 1.
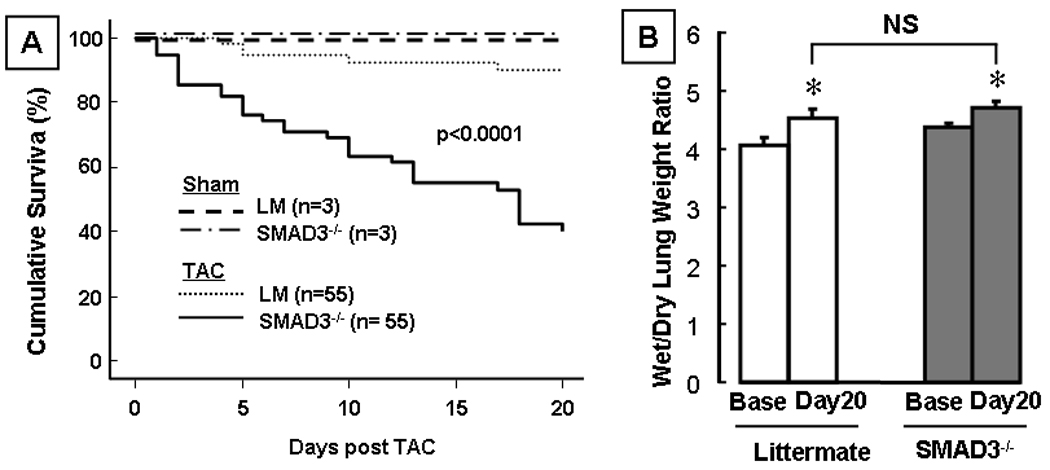
Effects of transaortic constriction (TAC) on survival and pulmonary congestion. Panel A: Kaplan Meier curves depicting survival in littermate (LM) and SMAD3 −/− mice after transverse aortic constriction (TAC) and sham operation. (n=116 mice). Panel B: Wet-to-dry lung ratio (n = 3–6 mice/group) in littermate control and SMAD3 −/− mice at baseline (Base) and 20 days (20d) post-TAC. (* p<0.05 compared to same group at baseline; NS = non-significant).
Effect of Pressure Overload on Cardiac Structure in SMAD3 −/− Mice
Cardiac Hypertrophy
Morphologic examination of transverse sections of the heart at the mid-papillary level at baseline, 10 and 20 days post-TAC revealed that both littermate control and SMAD3 −/− mice developed concentric hypertrophy after banding. However, the salient finding shown by Figure 2 is that the degree of cardiac hypertrophy was significantly greater in the SMAD3 −/− mice at day 10 and 20 post-TAC when compared to littermate control mice. Figure 2B shows that although the heart-weight- to-tibial length (HW/TL) ratio were similar in the SMAD3 deficient mice at baseline, the heart-weight- to-tibial length (HW/TL) ratio was significantly greater in the SMAD3 −/− mice by day 10 (p < 0.05) and day 20 (p < 0.05) post-TAC. Similar results were obtained when heart-weight to body-weight ratios were determined (Table 1). Two-D echocardiographic analysis (Figure 2C) of SMAD3 −/− and littermate control mice showed that LV wall thickness was increased significantly in the SMAD3 −/− mice at day 10 and day 20 post-TAC (p<0.05) and the r/h ratio was decreased significantly (p < 0.05) in the SMAD3 −/− mice at day 20 post-TAC, consistent with a greater degree of concentric hypertrophy in the SMAD3 null mice (Figure 2C, 2D). There were no significant differences in the LV end-diastolic dimension nor fractional shortening in SMAD3 −/− mice relative to littermate control mice post-TAC (Table 2).
Figure 2.
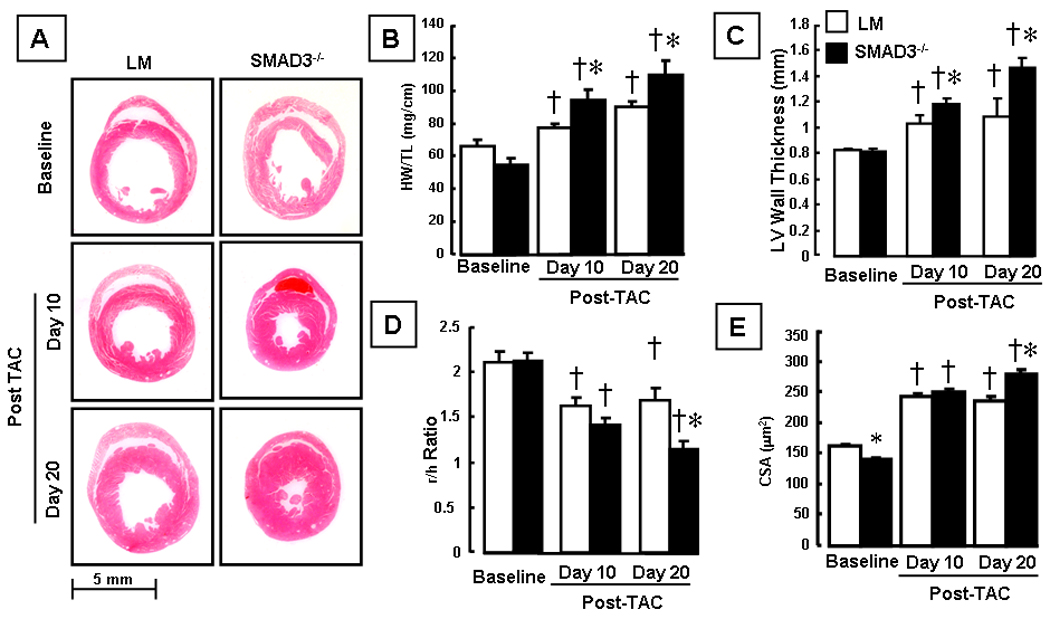
Effect of hemodynamic pressure overload on cardiac structure. Panel A: representative hematoxylin and eosin stained myocardial sections of littermate (LM) and SMAD3 −/− mice at baseline and 10 days and 20 days post-TAC. Panel B: Heart weight to tibial length ratio (HW/TL) at baseline, 10 days and 20 days post TAC in LM and SMAD3 −/− mice (n= 9 to 16 mice/group). Panel C: 2-D echocardiographic determined LV wall thickness at baseline, 10 days and 20 days post TAC in LM and SMAD3 −/− mice (n = 3–13 mice/group/time). Panel D: 2-D echocardiographic determined LV radius divided by LV wall thickness (r/h ratio) at baseline, 10 days and 20 days post TAC in LM and SMAD3 −/− mice (n = 3 mice/group/time). Panel C: Myocyte cross-sectional area (CSA, :m2) at baseline, 10 and 20 days post-TAC in LM and SMAD3 −/− mice (n = 100 myocytes/group/time). († = p < 0.01 vs baseline; * = p < 0.01 vs same group at a given time point).
Table 1.
Morphometric Data from Littermate Control Mice and SMAD3−/− Mice
| Parameter | Baseline | 10 days post-TAC | 20 days post-TAC | |||
|---|---|---|---|---|---|---|
| Littermate | SMAD3−/− | Littermate | SMAD3−/− | Littermate | SMAD3−/− | |
| HW | 124.8 ± 4.5 | 105.5 ± 7.3 | 165.3 ± 12.5* | 175.2 ± 13.2* | 169.6 ± 5.7* | 198.2 ± 13.3*,** |
| BW | 28.86 ± 0.9 | 23.1 ± 1.36** | 27.07 ± 0.80 | 21.84 ± 1.00# | 27.7 ± 1.31 | 21.1 ± 1.31** |
| HM/BW | 4.35 ± 0.1 | 4.56 ± 0.16 | 6.05 ± 0.33* | 8.10 ± 0.63*# | 6.29 ± 0.34* | 9.51 ± 0.51*# |
Morphologic examination of littermate control and SMAD3 −/− mice (n=10–20 mice group/time) was performed at baseline and 10 and 20 days post-TAC. (Key: BW = body weight, HW = heart weight, TAC = transaortic constriction;
p<0.001 vs baseline;
p<0.05 vs littermate control;
p < 0.001 vs littermate control).
Table 2.
Echocardiographic Data from Littermate Control Mice and SMAD3−/− Mice
| Parameter | Baseline | 10 days post-TAC | 20 days post-TAC | |||
|---|---|---|---|---|---|---|
| Littermate | SMAD3−/− | Littermate | SMAD3−/− | Littermate | SMAD3−/− | |
| LVEDD | 3.4 ± 0.14 | 3.4 ± 0.11 | 3.3 ± 0.12 | 3.3 ± 0.15 | 3.5 ± 0.14 | 3.3 ± 0.12 |
| FS (%) | 41.0 ± 3.4 | 38.9 ± 2.2 | 34.5 ± 3.5 | 40.3 ± 2.0 | 33.2 ± 2.0 | 34.1 ± 6.0 |
2-D echocardiographic analysis of littermate control and SMAD3 −/− mice (n=3–8 mice group/time) was performed at baseline and 10 and 20 days post-TAC. There was no significant different in the LVEDD and the % FS between the groups of mice. (Key: FS = fractional shortening, LVEDD = left ventricular end-diastolic dimension, TAC = transaortic constriction). (All p values are non-significant; n=3 or more mice/group/time point)
To determine whether the increase in cardiac hypertrophy in the SMAD deficient mice was secondary to an increase in cardiac myocyte cell size, we also examined myocyte cross-sectional area using lectin staining. As shown in Figure 2E, the myocyte cross-sectional area at baseline was significantly lower in SMAD3 −/− mice compared to littermates, consistent with the lower HW/TL ratios at baseline. However, the cross-sectional area of the myocytes in the SMAD3 −/− mice was significantly (p<0.01) greater 20 days post-TAC compared when compared to littermate controls. Taken together, these results suggest that SMAD3 signaling negatively modulates cardiac hypertrophic growth following hemodynamic pressure overloading.
Myocardial Fibrosis
To determine whether SMAD3 contributed significantly to the increase in myocardial fibrosis that is observed following hemodynamic pressure overloading, we assessed collagen content in SMAD3 −/− mice at 10 and 20 days post-TAC using the picrosirius red technique. Figure 3A depicts representative pircosirius red stained myocardial sections of littermate control and SMAD3 null mice at baseline and days 10 and 20 post-TAC, whereas Figure 3B summarizes the results of group data. As shown in Figure 3B the percent area of the LV that stained positively with picrosirius red increased in both the littermate control and SMAD3 null mice at day 10 and day 20 post-TAC, consistent with the known effects of pressure overload on myocardial fibrosis. Whereas there was a significant overall increase in myocardial fibrosis in the littermate control mice (p < 0.05 by ANOVA), the increase in fibrosis was not significant statistically in the SMAD3 null mice (p > 0.05 by ANOVA). Importantly, the extent of picrosirius red staining was significantly less in the SMAD null mice compared to littermate control mice at days 10 and 20 post-TAC.
Figure 3.
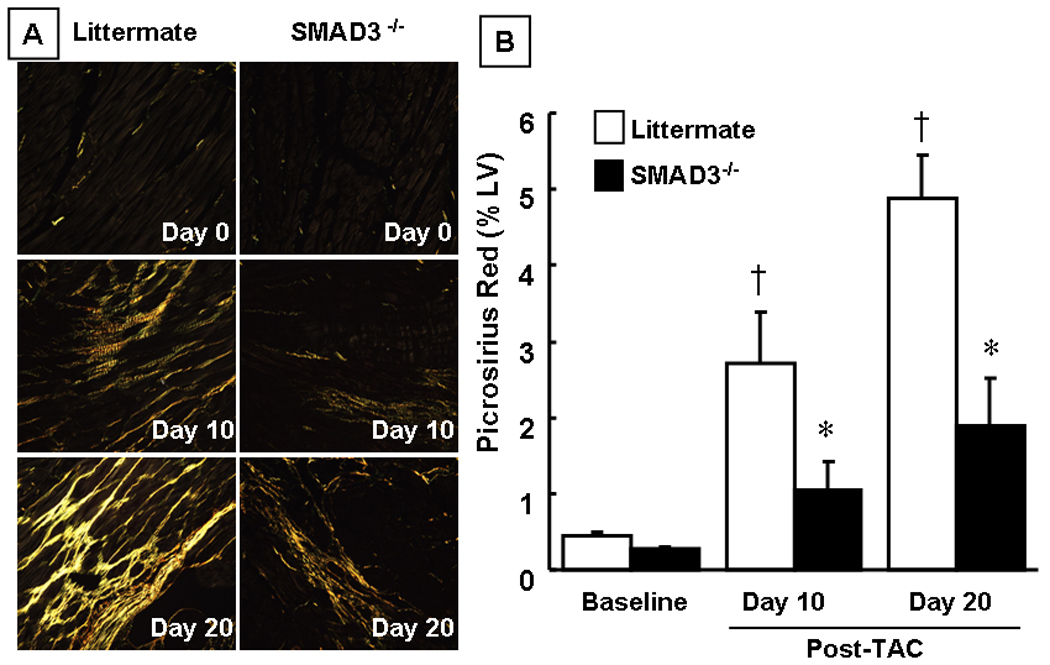
Effect of hemodynamic pressure overload on myocardial fibrosis. Panel A: representative picrosirius red stained images of littermate (LM) and SMAD3 −/− mice at baseline and 10 days and 20 days post-TAC depicting myocardial collagen (yellow) under polarized light. Panel B: Group data of the percent of LV myocardial fibrosis at baseline, 10 and 20 days post-TAC (n = 3 – 10 mice/group/time) († = p < 0.01 vs baseline; * = p < 0.05 vs same group at a given time point).
Effect of Pressure Overload on Myocardial Inflammation in SMAD3 −/− Mice
Insofar as TGF-∃ suppresses inflammation, we asked whether the cardiac hypertrophic phenotype and/or decreased survival in the SMAD3 −/− mice was secondary to increased myocardial inflammation. Figure 4A shows representative hematoxylin and eosin stained sections at baseline and at day 10 post-TAC, and Figure 4 B summarizes the results of group data. Although there was a significant increase in the inflammatory infiltrate in both the littermate control and the SMAD3 deficient mice at 10 and 20 days post-TAC when compared to baseline (p=0.02 by ANOVA), there was no significant difference in the inflammatory infiltrate score between the littermate controls and SMAD3 −/− mice at either day 10 (p=0.51) or day 20 (p=0.64) post-TAC.
Figure 4.
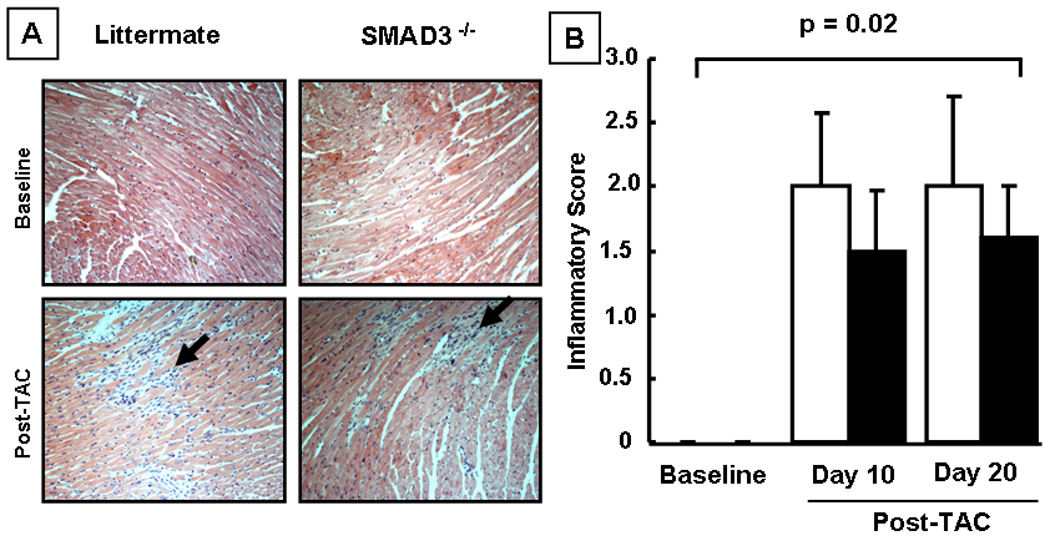
Effect of hemodynamic pressure overload on myocardial inflammation. Panel A: representative hematoxylin and eosin stained myocardial sections of littermate (LM) and SMAD3 −/− mice at baseline and 10 days post-TAC (arrows depict inflammatory cells). Panel B: group data of the semi-quantitative inflammatory scores at baseline, 10 and 20 days post TAC (n = 5 – 10 mice/group/time). NS = non-significant
Role of MicroRNAs in Myocardial Fibrosis
Analysis of microRNA Expression in SMAD3 −/− mice
Recent studies have uncovered a potentially important role for a family of tiny regulatory RNAs, known as microRNAs (miRNAs or miRs), in the control of cardiac remodeling and myocardial fibrosis (reviewed in 21). Insofar as recent studies suggest that SMAD dependent signaling governs the processing of mature microRNAs,22 we performed a microRNA microarray analysis in sham operated and banded (day 10) littermate control and SMAD3 −/− mice. Changes in the expression levels of microRNAs in SMAD3 −/− banded, SMAD3 −/− sham operated, littermate banded and littermate sham operated mice were considered to be statistically significant at the p < 0.01 level. As shown by the heat map in Figure 5A, there were 55 microRNAs that were differentially expressed in banded littermate control and SMAD3−/− mice compared to sham operated littermate control and SMAD3−/− control mice (p<0.05 by ANOVA). Specifically, 19 microRNAs were up-regulated and 18 microRNAs were down-regulated in the banded littermate and SMAD3−/− mice relative to sham-operated littermate control and SMAD3−/− mice; 10 microRNAs were down-regulated in banded littermate control mice and up-regulated in banded SMAD3−/− mice and 8 microRNAs were up-regulated in banded littermate controls but down-regulated in banded SMAD3−/− mice relative to sham operated littermate control or SMAD3−/− mice.
Figure 5.
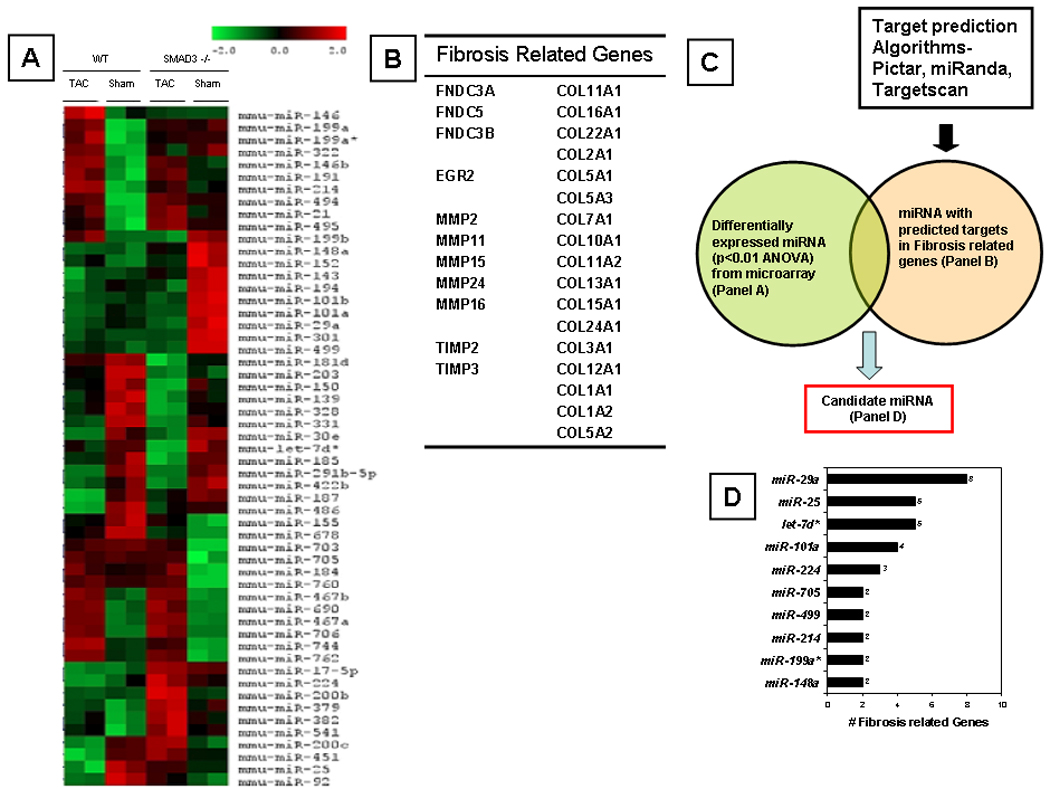
MicroRNA microarray in sham operated and banded mice. Panel A: Clustering graph of microRNA levels in littermate (LM) and SMAD3 −/− subjected to sham operation (day 10) or TAC (day 10). The columns depict the data for a given group (i.e., littermate and SMAD3 −/− sham operated and banded [TAC], whereas the rows depict the 55 microRNAs that were expressed differentially. Red depicts increased levels whereas green depicts decreased levels of the microRNA relative to the average of the absolute values for microRNA values for all 4 groups. Panel B: Table of genes that regulate expression of the extracellular matrix based on a survey of the extant literature (see methods). Panel C: Schematic of algorithm used to select "candidate microRNAs" potentially related to fibrosis. Panel D: List of candidate microRNAs with corresponding number of potential gene targets (from Panel B).
Selection of Candidate miRNAs
To determine whether any of the 55 miRNAs that were differentially expressed may have contributed to the decreased myocardial fibrosis observed in SMAD3 −/− mice, we first generated a list of 28 potential genes (Figure 5B) that have been implicated in the development of myocardial fibrosis, based upon a review of the extant literature.23,2413322}25,26 For the 28 potential gene targets, we then cross-referenced known or predicted miRNA interactions for these genes using three target-prediction algorithms: (I) Targetscan (http://www.targetscan.org), (ii) miRbase (http://micronrna.sanger.ac.uk) and (iii) PicTar (http://pictar.bio.nyu.edu). As shown by Figure 5D there were 10 candidate microRNAs that were significantly up- or down-regulated in the banded SMAD3 −/− mice and/or littermate control mice (relative to sham operated mice) that were predicted to bind to genes that regulate the extracellular cellular matrix (ECM). Of these 10 candidate microRNAs, 5 microRNAs had 4 or more predicted targets, of which miR-25 and miR-29a had the most predicted ECM targets (we did not include Let-7d* due to low expression levels in cardiac tissue). As shown in the online data supplement miR-25 was decreased by 40% in the banded littermate control mice and increased 15% in the banded SMAD3 −/− mice relative to their respective sham-operated controls, whereas miR-29a was decreased 8% in the banded littermate controls and decreased 36% decreased in the banded SMAD3 −/− mice relative to sham-operated mice. Based on the number of predicted ECM gene targets for these two microRNAs, as well as the differential expression of miR-25 and miR-29a in littermate control and SMAD3 null mice, we selected to study the effects of these two miRs in isolated cardiac fibroblasts.
Effects of miR-25 and miR-29a on Collagen mRNA Levels
Accumulation of collagen in the interstitium is secondary to an imbalance between collagen synthesis and degradation. The predominant collagens in the heart are types I, III and IV, of which type I and III are almost exclusively secreted by cardiac fibroblasts. Type I collagen is composed of two alpha-1 chains and one alpha-2 chain, the product of collagen genes COL1A1 and COL1A2 respectively. Type III collagen is comprised of two alpha-1 chains from COL3A1 gene.27 Figures 6A–C depict representative photomicrographs of Cy3-tagged pre-miR microRNAs that were transfected into cardiac fibroblasts (48 hours), and show that the microRNAs were predominately localized in the cytoplasm of the cardiac fibroblasts. As shown in Figure 6D, transfection of pre-miR-25 and pre-miR-29a resulted in decreased collagen-1 (COL1A1 and COL1A2) and collagen-3 (COL3A1) gene expression at 48 hours. The changes in COL1A2 gene expression were significant statistically (p <0.05) for miR-25 and miR-29a, whereas miR-29a resulted in significantly (p <0.05) decreased expression of COL1A1 and COL3A1 mRNA levels. Importantly, transfection of a pre-miR negative control had no effect on COL1A1, COL1A2 or COL3A1 mRNA levels.
Figure 6.
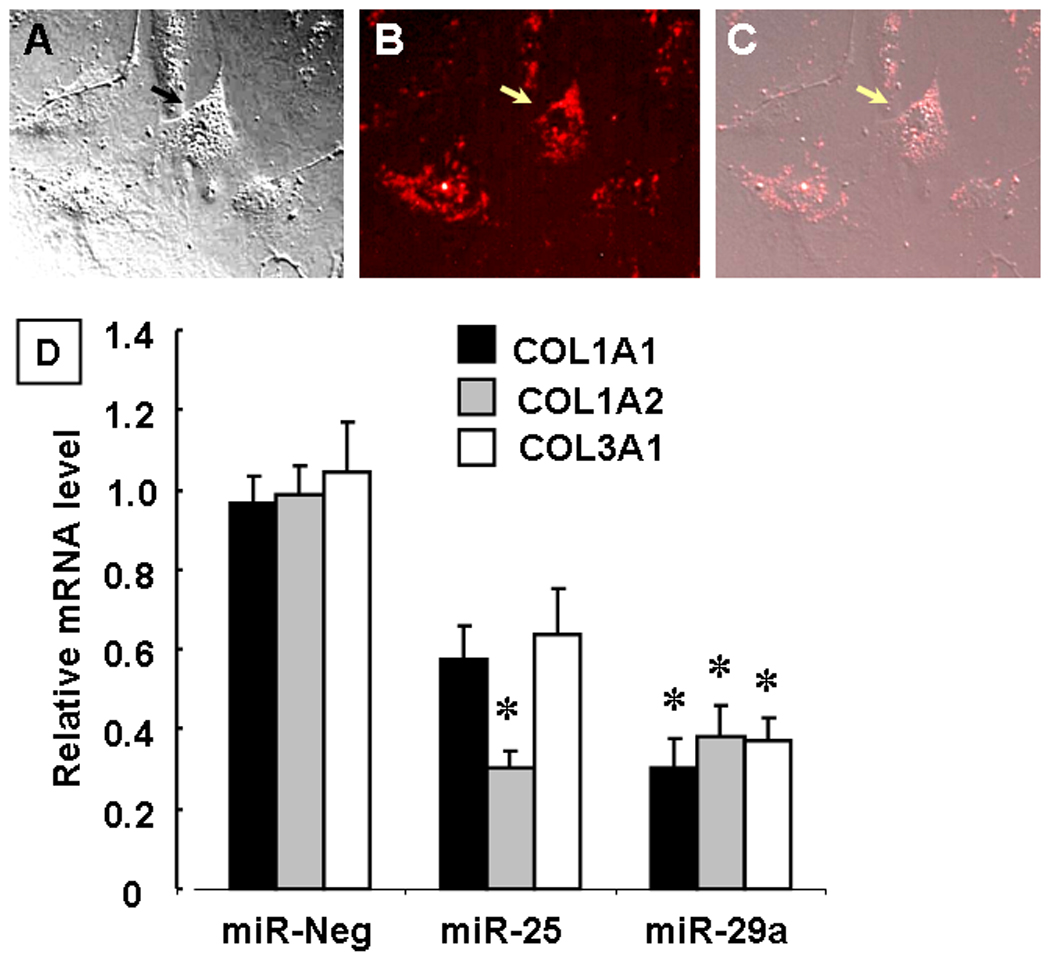
Effect of microRNAs on collagen gene expression in isolated cardiac fibroblasts. Panels A–C show respectively, brightfield images of isolated cardiac fibroblast, fluorescent images of Cy3 (red)-labeled pre-miR constructs transfected into cardiac fibroblasts and a merged image showing microRNA localization the cytoplasm of fibroblasts. Panel D: Collagen mRNA levels for collagen I (COL1A1 and COL1A2) and collagen III (COL3A1) gene expression in fibroblasts treated with miR 25 and 29a (48 hours) compared to the negative control microRNA C. elegans). Data are expressed relative to mRNA levels in diluent treated fibroblasts (* = p < 0.05 vs mRNA levels obtained in cardiac fibroblasts treated with the negative control miRNA.)
DISCUSSION
The study, in which we examined the effects of a transaortic constriction (TAC) in SMAD3 null mice (SMAD3 −/−), suggests that SMAD3 signaling plays both beneficial and deleterious roles in the heart in response to superimposed hemodynamic pressure overloading. Three major lines of evidence support this statement. First, there was a significant and striking increase in mortality in the SMAD3 −/− mice following TAC (Figure 1A). Although the mechanism for the increase in mortality in the SMAD3 −/− mice was not determined, it was likely related to an arrhythmia insofar as there was no evidence of increased pulmonary congestion (Figure 1B), systemic congestion, nor LV dysfunction (Table 2) in the SMAD3 −/− mice. Second, there was a significant increase in concentric hypertrophy in the SMAD3 −/− mice, as shown by the increase in the heart-weight-to-tibial length ratio (Figure 2B), increased LV wall thickness (Figure 2C), decreased r/h ratio (Figure 2D) and increased myocyte cross-sectional area in these mice. Third, there was a significant decrease in myocardial fibrosis in the SMAD3 −/− mice, consistent with the known role of SMAD3 in modulating fibrosis.13,28–31,32 The decrease in myocardial fibrosis in the SMAD3 null mice did not appear to be secondary to decreased inflammation, in that the inflammatory scores were overlapping in the SMAD3 −/− mice and littermate control mice (Figure 4D).
Given the recent insight the SMAD dependent signaling regulates the processing of mature microRNAs,22 as well role that microRNAs play a role in modulating stress signals in the heart,21 including the regulation of myocardial fibrosis, we performed a microarray analysis of microRNAs in the littermate control and SMAD3 −/− mice with the intent of identifying microRNAs that targeted genes that govern remodeling of the extracellular matrix. As shown, in Figure 5D there were 10 "candidate" microRNAs that were differentially regulated in the banded littermate control and SMAD3 −/− mice that were predicted to bind to genes that regulate the extracellular matrix, including miR-25, 29a, 101a, 199a, 148a, 214, 224, 499, 705 and let-7D. Transfection of two of these candidate microRNAs (miR-25 and miR-29a) that had COL1 and COL3 as predicted gene targets resulted in decreased collagen gene expression in isolated cardiac fibroblasts in vitro. Of note, miR-29a has been shown to be upregulated in experimental and clinical models of heart failure33 and down-regulated in the border zones of infarcts in mice.34 Further in vivo studies will be necessary to confirm the role of miR-25 and miR-29a in terms of mediating SMAD3 dependent fibrosis in the adult heart. Viewed together, these results suggest that SMAD3 signaling plays a beneficial role in the heart by delimiting hypertrophic growth and conferring cytoprotection, as well as a deleterious role by modulating myocardial fibrosis.
SMAD3 Signaling in the Heart
The decreased pressure overload induced-fibrosis in the SMAD3−/− mice relative to banded littermate control mice (Figure 3) is consistent with prior studies that have shown decreased fibrosis in SMAD3 null mice following acute myocardial infarction,13 bleomycin-induced lung injury,28 vascular injury,29,30 gamma-radiation injury,31 and CC14-induced hepatic injury.32 With that said, the results of the present study expand upon prior studies by demonstrating that several micro-RNAs, including miR 25, 29a, 499, 705, 224, 199a and 148 are differentially regulated in the SMAD3 null mice relative to littermate controls following TAC. Our findings with respect to decreased relative expression levels of miR-29a in the banded SMAD3 null mice are consistent with the recent findings reported by Olson and colleagues,34 who observed that both miR-29a and miR-29b were down-regulated in the border zone infarcts in mice, and that down-regulation of Mir-29b with anti-miRs induced collagen expression both in vitro and in vivo, whereas over-expression of a miR-29b mimic in isolated cardiac fibroblasts reduced collagen transcripts. They further reported that stimulation of isolated cardiac fibroblasts with TGF-∃ significantly decreased miR-29a–c expression. Our results confirm and expand upon these important findings in two ways. First, our data suggest SMAD3-dependent signaling may be responsible for “fine tuning” the expression levels of different pro-fibrotic microRNA transcripts. Indeed, our data show that the changes in miR29 expression levels in the banded SMAD3−/− mice were greater for miR-29a (p < 0.007), than for miR-29c (p < 0.06) and miR-29b (p < 0.22). Given that both miR-29a and miR-29b are co-expressed as part of the same bicistronic unit, it is possible that SMAD-dependent signaling may allow for the differential expression of one or more pro-fibrotic microRNAs at different times. In this regard it is interesting to note that SMAD-dependent signaling has recently been shown to lead to increased levels of mature microRNAs by interacting with the DROSHA microprocessor complex.22 Accordingly, in future studies it will be important to determine whether the changes in the expression levels of the profibrotic microRNAs identified in this study are regulated by SMAD-dependent interaction with the DROSHA processing complex (e.g. through inhibitory microRNAs). Second, our data suggest that multiple microRNAs are likely involved in fibrosis, either singly or in combination. For example, our data suggest a potential inhibitory role for miR-25 in terms of COL1A2, which was not identified in the screen by Olson and colleagues. The observation the relative expression levels of miR-25 were increased in the banded SMAD3−/− mice suggests that the effects of miR-25 are either indirect or the result of suppression of an inhibitory microRNA.
Although the focus of this study was on delineating the role of SMAD3 dependent signaling on myocardial fibrosis and pathological remodeling, the observed increase in pressure overload induced hypertrophic growth in the SMAD3 null mice (Figure 2) warrants further discussion, particularly in view the plethora of studies that have suggested an important role for TGF-∃ signaling in terms of mediating hypertrophic growth in the heart.6–11 This statement notwithstanding, the results of the present study are consistent with two recent lines of evidence which suggest that SMAD-dependent signaling inhibits rather than stimulates hypertrophic growth in the mammalian heart. First, recent studies of growth and differentiation factor 15 (GDF15), a 12-kDa secreted protein that signals through SMAD2/3 in the heart,5 show that mice with cardiac restricted overexpression of GDF15 were partially resistant to pressure overload-induced hypertrophy, whereas GDF15 gene-targeted mice showed enhanced cardiac hypertrophic growth following superimposition of a hemodynamic pressure overload.5 Second cardiac restricted deletion of SMAD4 (which would inhibit SMAD1,2,3,6,8 signaling) resulted in increased cardiac hypertrophy, decreased contractility and increased mortality,35 consistent with that point of view that SMAD-dependent pathways inhibit hypertrophic growth in the heart. Lastly, it should be recognized that the results are this and prior studies5,35 are consistent with the observation that SMAD-dependent signaling is growth inhibitory in non-myocyte cell types, and that loss of SMAD dependent signaling results in tumorigenesis.36,37
SMAD3 Signaling in the Heart
Given that both gain of function and loss of function studies have suggested an important role for TGF-∃ signaling with respect to mediating hypertrophic growth in the heart,6–11 the increase in hypertrophic growth in the SMAD3 null mice following TAC was somewhat surprising. This statement notwithstanding, the results of the present study are consistent with two recent lines of evidence which suggest that SMAD-dependent signaling inhibits rather than stimulates hypertrophic growth in the mammalian heart. First, loss of receptor SMAD signaling resulted in increased cardiac hypertrophy, decreased contractility and an increased mortality in mice with cardiac restricted deletion of SMAD4,35 suggesting that SMAD-dependent pathways inhibit hypertrophic growth in the heart. Second, recent studies of growth and differentiation factor 15 (GDF15), a 12-kDa secreted protein that signals through SMAD2/3 in the heart,5 show that mice with cardiac restricted overexpression of GDF15 were partially resistant to pressure overload-induced hypertrophy, whereas GDF15 gene-targeted mice showed enhanced cardiac hypertrophic growth following superimposition of a hemodynamic pressure overload.5 Lastly, it should be recognized that the results of this and prior studies5,35 are consistent with the observation that SMAD-dependent signaling is growth inhibitory in non-myocyte cell types, and that loss of SMAD dependent signaling results in tumorigenesis.36,37
If SMAD dependent signaling is anti-hypertrophic in the heart, how can the results of this and other studies be reconciled with the known deleterious effects of TGF-∃ signaling?
Although the answer to this question is not known, there are two possibilities that warrant discussion. One possibility is that the pro-hypertrophic effects of TGF-∃ signaling are conveyed through SMAD-independent pathways, whereas the cardioprotective effects of TGF-∃ are mediated via SMAD-dependent signaling. Support for this point of view is provided by a study which suggests that TAK-1, which is rapidly activated by TGF-∃, is sufficient to provoke cardiac hypertrophy when selectively overexpressed in the heart.12 A second possibility is that a SMAD independent signaling kinases that have been implicated in hypertrophic growth, such as the the Ras-MEK-Erk pathway, negatively modulate TGF-∃ signaling by increasing the amount of the TGF-∃ interacting factor, a co-repressor of SMAD signaling. Activation of these SMAD independent signaling would reduce SMAD accumulation in the nucleus, thereby suppressing the anti-hypertrophic effects of SMAD3 dependent signaling.38
The decreased pressure overload induced-fibrosis in the SMAD3−/− mice relative to banded littermate control mice (Figure 3) is consistent with prior studies that have shown decreased fibrosis in SMAD3 null mice following acute myocardial infarction,13 bleomycin-induced lung injury,28 vascular injury,29,30 gamma-radiation injury,31 and CC14-induced hepatic injury.32 The results of the present study expand upon prior studies by showing that several micro-RNAs, including miR 25, 29a, 499, 705, 224, 199a and 148 are differentially regulated in the SMAD3 null mice relative to littermate controls following TAC. Our findings with respect miR-29a are consistent with the recent findings reported by Olson and colleagues,34 who observed that both miR-29a and miR-29b were downregulated in the border zone infarcts in mice, and that downregulation of miR-29b with anti-miRs induced collagen expression both in vitro and in vivo, whereas overexpression of a miR-29b mimic in isolated cardiac fibroblasts reduced collagen transcripts. They further reported that stimulation of isolated cardiac fibroblasts with TGF-∃ significantly decreased miR-29a–c expression. Our results confirm and expand upon these important findings in two ways. First, our data suggest that multiple miRs are likely involved in fibrosis, either singly or in combination. For example, our data suggest a potential inhibitory role for miR-25 in terms of COL1A2, which was not identified in the screen by Olson and colleagues. Second, our data suggest SMAD3-dependent signaling may regulate the expression levels of pro-fibrotic microRNA transcripts differentially. For example, our data show that the changes in miR-29 expression levels in the banded SMAD3−/− mice were greater for miR-29a (p < 0.007), than for miR-29c (p < 0.06) and miR-2b (p < 0.22). Germane to this discussion is the recent observation that SMAD-dependent signaling is responsible for increased levels of mature microRNAs by interacting with the DROSHA microprocessor complex.22 Accordingly, in future studies it will be important to determine whether the changes in the expression levels of the profibrotic microRNAs identified in this study are regulated by SMAD-dependent interaction with the DROSHA processing complex (e.g. through inhibitory microRNAs).
Conclusion
In summary, the results of the present study suggest that SMAD3 mediated signaling plays dual roles in the heart: one that is beneficial, by delimiting hypertrophic growth and providing cardioprotective response in the heart following hemodynamic overloading, and a second role that is maladaptive, by mediating progressive myocardial fibrosis that can lead to increased myocardial stiffness, contractile dysfunction, and increased cardiac arrhythmias. Given that SMAD dependent signaling is highly conserved in nature,39 and has been observed in both vertebrates and invertebrate species, the provocative question that arises from these conflicting observations is why nature has retained both adaptive and maladaptive pathways in the adult mammalian heart? Although the immediate answer to this important question is not obvious, one potential explanation (albeit speculative) is that cross-talk between the SMAD dependent and SMAD independent signaling pathways may ultimately determine whether signaling by the TGF-∃ superfamily of ligands becomes adaptive or maladaptive in the heart. That is, whereas the immediate role for TGF-∃ signaling is to confer cytoprotective responses and to delimit hypertrophic growth, sustained signaling through SMAD dependent and SMAD independent signaling results in myocardial fibrosis and hypertrophic growth, respectively. In this regard, it will also be interesting to determine whether SMAD-induced accumulation of one or more microRNAs in the heart contributes to the transition from adaptive hypertrophy to decompensated cardiac failure.
Supplementary Material
Acknowledgments
Funding Source: This research was supported by research funds from the N.I.H. (UO1 HL084890-01 and RO1 HL58081, RO1 HL61543, HL-42250).
Footnotes
DISCLOSURES
The authors have no conflicts to disclose.
REFERENCES
- 1.Ruiz-Ortega M, Rodriguez-Vita J, Sanchez-Lopez E, Carvajal G, Egido J. TGF-beta signaling in vascular fibrosis. Cardiovasc Res. 2007;74:196–206. doi: 10.1016/j.cardiores.2007.02.008. [DOI] [PubMed] [Google Scholar]
- 2.Euler-Taimor G, Heger J. The complex pattern of SMAD signaling in the cardiovascular system. Cardiovasc Res. 2006;69:15–25. doi: 10.1016/j.cardiores.2005.07.007. [DOI] [PubMed] [Google Scholar]
- 3.Derynck R, Zhang YE. Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature. 2003;425:577–584. doi: 10.1038/nature02006. [DOI] [PubMed] [Google Scholar]
- 4.Massague J, Wotton D. Transcriptional control by the TGF-beta/Smad signaling system. EMBO J. 2000;19:1745–1754. doi: 10.1093/emboj/19.8.1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Xu J, Kimball TR, Lorenz JN, Brown DA, Bauskin AR, Klevitsky R, Hewett TE, Breit SN, Molkentin JD. GDF15/MIC-1 functions as a protective and antihypertrophic factor released from the myocardium in association with SMAD protein activation. Circ Res. 2006;98:342–350. doi: 10.1161/01.RES.0000202804.84885.d0. [DOI] [PubMed] [Google Scholar]
- 6.Nakajima H, Nakajima HO, Salcher O, Dittie AS, Dembowsky K, Jing S, Field LJ. Atrial but not ventricular fibrosis in mice expressing a mutant transforming growth factor-beta(1) transgene in the heart. Circ Res. 2000;86:571–579. doi: 10.1161/01.res.86.5.571. [DOI] [PubMed] [Google Scholar]
- 7.Brooks WW, Conrad CH. Myocardial fibrosis in transforming growth factor beta(1)heterozygous mice. J Mol Cell Cardiol. 2000;32:187–195. doi: 10.1006/jmcc.1999.1065. [DOI] [PubMed] [Google Scholar]
- 8.Schultz JJ, Witt SA, Glascock BJ, Nieman ML, Reiser PJ, Nix SL, Kimball TR, Doetschman T. TGF-beta1 mediates the hypertrophic cardiomyocyte growth induced by angiotensin II. J Clin Invest. 2002;109:787–796. doi: 10.1172/JCI14190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sanderson N, Factor V, Nagy P, Kopp J, Kondaiah P, Wakefield L, Roberts AB, Sporn MB, Thorgeirsson SS. Hepatic expression of mature transforming growth factor beta 1 in transgenic mice results in multiple tissue lesions. Proc Natl Acad Sci U S A. 1995;92:2572–2576. doi: 10.1073/pnas.92.7.2572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rosenkranz S, Flesch M, Amann K, Haeuseler C, Kilter H, Seeland U, Schluter KD, Bohm M. Alterations of beta-adrenergic signaling and cardiac hypertrophy in transgenic mice overexpressing TGF-beta(1) Am J Physiol Heart Circ Physiol. 2002;283:H1253–H1262. doi: 10.1152/ajpheart.00578.2001. [DOI] [PubMed] [Google Scholar]
- 11.Lim JY, Park SJ, Hwang HY, Park EJ, Nam JH, Kim J, Park SI. TGF-beta1 induces cardiac hypertrophic responses via PKC-dependent ATF-2 activation. J Mol Cell Cardiol. 2005;39:627–636. doi: 10.1016/j.yjmcc.2005.06.016. [DOI] [PubMed] [Google Scholar]
- 12.Zhang D, Gaussin V, Taffet GE, Belaguli NS, Yamada M, Schwartz RJ, Michael LH, Overbeek PA, Schneider MD. TAK1 is activated in the myocardium after pressure overload and is sufficient to provoke heart failure in transgenic mice. Nat Med. 2000;6:556–563. doi: 10.1038/75037. [DOI] [PubMed] [Google Scholar]
- 13.Bujak M, Ren G, Kweon HJ, Dobaczewski M, Reddy A, Taffet G, Wang XF, Frangogiannis NG. Essential role of Smad3 in infarct healing and in the pathogenesis of cardiac remodeling. Circulation. 2007;116:2127–2138. doi: 10.1161/CIRCULATIONAHA.107.704197. [DOI] [PubMed] [Google Scholar]
- 14.Baumgarten G, Knuefermann P, Kalra D, Gao F, Taffet GE, Michael L, Blackshear PJ, Carballo E, Sivasubramanian N, Mann DL. Load-dependent and -independent regulation of proinflammatory cytokine and cytokine receptor gene expression in the adult mammalian heart. Circulation. 2002;105:2192–2197. doi: 10.1161/01.cir.0000015608.37608.18. [DOI] [PubMed] [Google Scholar]
- 15.Sivasubramanian N, Coker ML, Kurrelmeyer K, DeMayo F, Spinale FG, Mann DL. Left ventricular remodeling in transgenic mice with cardiac restricted overexpression of tumor necrosis factor. Circulation. 2001;2001:826–831. doi: 10.1161/hc3401.093154. [DOI] [PubMed] [Google Scholar]
- 16.Sakata Y, Chancey AL, Divakaran VG, Sekiguchi K, Sivasubramanian N, Mann DL. Transforming growth factor-beta receptor antagonism attenuates myocardial fibrosis in mice with cardiac-restricted overexpression of tumor necrosis factor. Basic Res Cardiol. 2007 doi: 10.1007/s00395-007-0689-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bozkurt B, Kribbs S, Clubb FJ, Jr, Michael LH, Didenko VV, Hornsby PJ, Seta Y, Oral H, Spinale FG, Mann DL. Pathophysiologically relevant concentrations of tumor necrosis factor-α promote progressive left ventricular dysfunction and remodeling in rats. Circulation. 1998;97:1382–1391. doi: 10.1161/01.cir.97.14.1382. [DOI] [PubMed] [Google Scholar]
- 18.Kanda T, Mcmanus JEW, Nagai R, Imai S, Suzuki T, Yang DC, McManus BM, Kobayashi I. Modification of viral myocarditis in mice by interleukin-6. Circ Res. 1996;78:848–856. doi: 10.1161/01.res.78.5.848. [DOI] [PubMed] [Google Scholar]
- 19.Sayed D, Hong C, Chen IY, Lypowy J, Abdellatif M. MicroRNAs play an essential role in the development of cardiac hypertrophy. Circ Res. 2007;100:416–424. doi: 10.1161/01.RES.0000257913.42552.23. [DOI] [PubMed] [Google Scholar]
- 20.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(T)(-Delta Delta C) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 21.van Rooij E, Olson EN. MicroRNAs: powerful new regulators of heart disease and provocative therapeutic targets. J Clin Invest. 2007;117:2369–2376. doi: 10.1172/JCI33099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Davis BN, Hilyard AC, Lagna G, Hata A. SMAD proteins control DROSHA-mediated microRNA maturation. Nature. 2008;454:56–61. doi: 10.1038/nature07086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li YY, McTiernan CF, Feldman AM. Interplay of matrix metalloproteinases, tissue inhibitors of metalloproteinases and their regulators in cardiac matrix remodeling. Cardiovasc Res. 2000;46:214–224. doi: 10.1016/s0008-6363(00)00003-1. [DOI] [PubMed] [Google Scholar]
- 24.Corda S, Samuel J-L, Rappaport L. Extracellular matrix and growth factors during heart growth. Heart Fail Rev. 2001;5:119–130. doi: 10.1023/A:1009806403194. [DOI] [PubMed] [Google Scholar]
- 25.Manabe I, Shindo T, Nagai R. Gene expression in fibroblasts and fibrosis: involvement in cardiac hypertrophy. Circ Res. 2002;91:1103–1113. doi: 10.1161/01.res.0000046452.67724.b8. [DOI] [PubMed] [Google Scholar]
- 26.Heeneman S, Cleutjens JP, Faber BC, Creemers EE, van Suylen RJ, Lutgens E, Cleutjens KB, Daemen MJ. The dynamic extracellular matrix: intervention strategies during heart failure and atherosclerosis. J Pathol. 2003;200:516–525. doi: 10.1002/path.1395. [DOI] [PubMed] [Google Scholar]
- 27.Kadler KE, Holmes DF, Trotter JA, Chapman JA. Collagen fibril formation. Biochem J. 1996;316(Pt 1):1–11. doi: 10.1042/bj3160001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhao J, Shi W, Wang YL, Chen H, Bringas P, Jr, Datto MB, Frederick JP, Wang XF, Warburton D. Smad3 deficiency attenuates bleomycin-induced pulmonary fibrosis in mice. Am J Physiol Lung Cell Mol Physiol. 2002;282:L585–L593. doi: 10.1152/ajplung.00151.2001. [DOI] [PubMed] [Google Scholar]
- 29.Kobayashi K, Yokote K, Fujimoto M, Yamashita K, Sakamoto A, Kitahara M, Kawamura H, Maezawa Y, Asaumi S, Tokuhisa T, Mori S, Saito Y. Targeted disruption of TGF-beta-Smad3 signaling leads to enhanced neointimal hyperplasia with diminished matrix deposition in response to vascular injury. Circ Res. 2005;96:904–912. doi: 10.1161/01.RES.0000163980.55495.44. [DOI] [PubMed] [Google Scholar]
- 30.Wang WS, Huang XR, Canlas E, Oka K, Truong LD, Deng CX, Bhowmick NA, Ju WJ, Bottinger EP, Lan HY. Essential role of Smad3 in angiotensin II-induced vascular fibrosis. Circ Res. 2006;98:1032–1039. doi: 10.1161/01.RES.0000218782.52610.dc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Flanders KC, Sullivan CD, Fujii M, Sowers A, Anzano MA, Arabshahi A, Major C, Deng C, Russo A, Mitchell JB, Roberts AB. Mice lacking Smad3 are protected against cutaneous injury induced by ionizing radiation. Am J Pathol. 2002;160:1057–1068. doi: 10.1016/S0002-9440(10)64926-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yuan W, Varga J. Transforming growth factor-beta repression of matrix metalloproteinase-1 in dermal fibroblasts involves Smad3. J Biol Chem. 2001;276:38502–38510. doi: 10.1074/jbc.M107081200. [DOI] [PubMed] [Google Scholar]
- 33.Divakaran V, Mann DL. The emerging role of microRNAs in cardiac remodeling and heart failure. Circ Res. 2008;103:1072–1083. doi: 10.1161/CIRCRESAHA.108.183087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.van Rooij E, Sutherland LB, Thatcher JE, DiMaio M, Naseem RH, Marshall WS, Hill JA, Olson EN. Dysregulation of microRNAs after myocardial infarction reveals a role of miR-29 in cardiac fibrosis. Proc Natl Acad Sci U S A. 2008;105:13027–13032. doi: 10.1073/pnas.0805038105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang J, Xu N, Feng X, Hou N, Zhang J, Cheng X, Chen Y, Zhang Y, Yang X. Targeted disruption of Smad4 in cardiomyocytes results in cardiac hypertrophy and heart failure. Circ Res. 2005;97:821–828. doi: 10.1161/01.RES.0000185833.42544.06. [DOI] [PubMed] [Google Scholar]
- 36.Tang B, de CK, Barnes HE, Parks WT, Stewart L, Bottinger EP, Danielpour D, Wakefield LM. Loss of responsiveness to transforming growth factor beta induces malignant transformation of nontumorigenic rat prostate epithelial cells. Cancer Res. 1999;59:4834–4842. [PubMed] [Google Scholar]
- 37.Tang B, Vu M, Booker T, Santner SJ, Miller FR, Anver MR, Wakefield LM. TGF-beta switches from tumor suppressor to prometastatic factor in a model of breast cancer progression. J Clin Invest. 2003;112:1116–1124. doi: 10.1172/JCI18899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Massague J. Integration of Smad and MAPK pathways: a link and a linker revisited. Genes Dev. 2003;17:2993–2997. doi: 10.1101/gad.1167003. [DOI] [PubMed] [Google Scholar]
- 39.Newfeld SJ, Wisotzkey RG, Kumar S. Molecular evolution of a developmental pathway: phylogenetic analyses of transforming growth factor-beta family ligands, receptors and Smad signal transducers. Genetics. 1999;152:783–795. doi: 10.1093/genetics/152.2.783. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.