

Abstract
In our ongoing study of the desmosdumotin C (1) series, twelve new analogues, 21–32, mainly with structural modifications in ring-A, were prepared and evaluated for in vitro antiproliferative activity against several human tumor cell lines. Among them, the 4′-iodo-3,3,5-tripropyl-4-methoxy analogue (31) showed significant antiproliferative activity against multiple human tumor cell lines with ED50 values of 1.1–2.8 μM. Elongation of the C-3 and C-5 carbon chains reduced activity relative to propyl substituted analogues; however, activity was still better than that of natural compound 1. Among analogues with various ether groups on C-4, compounds with methyl (2) and propyl (26) ethers inhibited cell growth of multiple tumor cells lines, while 28 with an isobutyl ether showed selective antiproliferative activity against lung cancer A549 cells (ED50 1.7 μM). The gene expression profiles showed that 3 may modulate the spindle assembly checkpoint (SAC) and chromosome separation, and thus, arrest cells at the G2/M-phase.
Keywords: Desmosdumotin C, Antiproliferative activity, Human tumor cell lines, Microarray
1. Introduction
Desmosdumotin C (1), isolated from the roots of Desmos dumosus,1 has a distinctive chalcone skeleton with an unusual non-aromatic A-ring possessing a gem-dimethyl group on C-3 and methyl group on C-5. This compound showed significant and selective antiproliferative activity against 1A9 (ovarian cancer) and A549 (human lung carcinoma) cell lines with ED50 values of 3.5 μg/mL (11.2 μM).1 In addition, it was more active against KB-VIN [vincristine-resistant KB, overexpressing P-glycoprotein (P-gp)] cells than against the parent KB (epidermoid nasopharyngeal carcinoma) cell line. We previously established the first total synthesis of 1.2 Based on our synthetic methodology, the A-ring was modified with triethyl and tripropyl groups at C-3 and -5 positions and various substituted aromatic B-rings were also incorporated.3 From the preliminary data, analogues with tripropyl substitution at the C-3 and C-5 positions (i.e., 2) showed better activity than analogues with triethyl and trimethyl groups. Furthermore, addition of a bromophenyl B-ring (bromide at C-4′) enhanced cell growth inhibition against all tested tumor cell lines. As a result, 3,5,5-tripropyl-4′-bromo analogue 3 possessed the most potent activity against A549, HCT-8 (colon adenocarcinoma), 1A9, PC-3 (prostate cancer), KB and KB-VIN with ED50 values of 0.87–2.25 μg/mL (1.8–2.6 μM).
Subsequent modifications of 1 focused on the A-ring and included further elongation of the C-3 and C-5 carbon side chains as well as introduction of various alkoxy groups at C-4′. Naturally occurring C-prenylated flavonoids, including chalcones,4 have shown interesting bioactivities, such as antimalarial, antitumor, anti-HIV and anti-oxidant effects.4,5 Thus, the insertion of prenyl groups at the C-3 and -5 positions was another design target. Because we did not include C-4′ iodinated compounds in our prior study, 4'-iodo analogues were also prepared. Herein, we describe the synthesis of 1-derivatives and evaluation of newly synthesized analogues against seven human tumor cell lines, A549, HCT-8, MCF-7 (breast cancer), 1A9, PC-3, HepG2 (liver cancer), KB and KB-VIN.
2. Chemistry
Trialkylated intermediates 5–8 with propyl, butyl, isobutyl, and isopentyl (R = Pr, Bu, iBu, and iPen, respectively) at the C-3 and C-5 positions were obtained by reacting 2,4,6-trihydroxyacetophenone (4) with the appropriate alkyl halide and sodium methoxide at reflux temperature. However, attempted tri-prenylation of 4 under the same conditions gave a complex mixture. Triprenylated 9 (R = prenyl) was finally obtained by treatment of 4 with prenyl bromide and KOH in water.6 Target compounds 21–24 were produced by methylation of the 4-OH of 5–9 using TMSCHN2 followed by Claisen-Schmidt condensation of the resulting 10–14 with benzaldehyde.
Regioselective mono-alkylation of the 4-OH group of tripropyl 5 (R = Pr) was accomplished by using various alkyl halides (EtI, PrI, BuI, iBuI, and iPenI) in the presence of potassium carbonate, rather than TMSCHN2, to obtain 15–19 (R = Pr; R′ = Et, Pr, Bu, iBu, and iPen). Under the same reaction conditions, use of prenyl bromide resulted in 20, in which the prenyl is attached to the carbon at the C-5 position rather than on the hydroxy group. Compounds 15–20 were condensed with benzaldehyde to obtain the target compounds 25–30. Claisen-Schmidt condensation of 10 (R = Pr; R′ = Me) and 12 (R = Pr; R′ = Pr) with 4-iodobenzaldehyde produced 31, and 32, respectively. Compounds 1–3 were previously synthesized.2,3 All final compounds exist as a mixture of two tautomeric isomers, as discussed in our prior papers.3
3. Results and discussion
Newly synthesized analogues 21–32 were evaluated for in vitro antiproliferative activity against the cell line panel described above. The average ED50 values (μM) are listed in Table 1, together with those of 1–3 as references. All analogs, except for 23, inhibited tumor cell growth, with greater or similar potency to that of the parent compound (1).
Table 1.
| Compound | ED50 (μM)a | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| R | R′ | X | A549 | HCT-8 | MCF-7 | 1A9 | PC-3 | HepG2 | KB | KB-V | |
| 1 | Me | Me | H | 11.2 | 11.8 | NTb | 11.2 | 35.6 | NT | 12.8 | 9.6 |
| 2 | Pr | Me | H | 3.6 | 3.5 | NT | 3.7 | 8.2 | NT | 4.6 | 3.4 |
| 3 | Pr | Me | Br | 2.0 | 2.6 | NT | 1.9 | 4.7 | NT | 1.8 | 1.8 |
| 21 | Bu | Me | H | 6.4 | 7.8 | 4.4 | 5.9 | 13.0 | 23.9 | 7.0 | 8.6 |
| 22 | iBu | Me | H | 4.4 | 7.5 | 3.7 | 8.0 | 12.4 | 14.5 | 7.9 | 8.2 |
| 23 | iPen | Me | H | 18.4 | 13.0 | 25.2 | 16.3 | NAc | NA | 17.5 | 20.3 |
| 24 | Prenyl | Me | H | 10.3 | 13.7 | 8.2 | NT | 12.2 | 11.2 | 13.3 | 8.9 |
| 25 | Pr | Et | H | 4.6 | 5.6 | 2.6 | 4.3 | 12.5 | 11.2 | 6.4 | 3.5 |
| 26 | Pr | Pr | H | 1.9 | 2.6 | 1.6 | 2.0 | 8.6 | 8.3 | 2.7 | 2.4 |
| 27 | Pr | Bu | H | 7.1 | 8.7 | 5.3 | NT | 9.6 | 8.0 | 8.0 | 8.9 |
| 28 | Pr | iBu | H | 1.7 | 8.0 | 4.4 | 6.9 | 12.2 | 13.4 | 4.8 | 4.1 |
| 29 | Pr | iPen | H | 11.3 | 15.3 | 9.1 | NT | 17.0 | 15.5 | 10.6 | 19.2 |
| 30 | – | – | – | 7.1 | 6.7 | 5.8 | NT | 8.4 | 6.4 | 8.2 | 7.3 |
| 31 | Pr | Me | I | 1.4 | 1.1 | 1.8 | 1.8 | 2.8 | 2.4 | 1.5 | 1.4 |
| 32 | Pr | Pr | I | 8.2 | 7.8 | 5.1 | NT | 9.6 | 7.3 | 6.4 | 6.2 |
| Paclitaxel (nM) | 2.56 | >100 | >100 | 2.09 | 8.87 | >100 | 2.87 | >100 | |||
| Vincristine (nM) | 4.8 | 24.2 | 12.1 | NT | 12.1 | 2.9 | 2.4 | >100 | |||
Antiproliferative activity as ED50 values for each cell line, the concentration of compound that caused 50% reduction in absorbance at 562 nm relative to untreated cells using the sulforhodamine B assay. Human lung carcinoma (A549), colon adenocarcinoma (HCT-8), breast cancer (MCF-7), ovarian carcinoma (1A9), prostate cancer (PC-3), liver cancer (HepG2), epidermoid carcinoma of the nasopharynx (KB), and MDR expressing P-glycoprotein (KB-VIN).
NT, not tested.
NA, not active.
Test compound (20 μg/mL) did not reach 50% inhibition.
Consistent with our prior findings,3 2 (3,3,5-tripropyl) was again more potent than 1 (3,3,5-trimethyl). Further elongation of the C-3 and C-5 alkyl chains to Bu, iBu, iPen, and prenyl decreased the potency. The rank order of potency based on C-3 and -5 alkyl substitutents was Pr (2) > iBu (22) ≈ Bu (21) > Et3b > Me (1) ≈ Prenyl (24) > iPen (23).
Our next interest was to determine how the alkoxy group (OR′) at C-4 affected activity. From comparison of data in Table 1, the rank order of activity was OPr (26) > OMe (2) > OEt (25) > OBu (27) > OiBu (28) > OiPen (29). Analogue 26 showed good in vitro antiproliferative activity against all tested cell lines with ED50 values of 1.9–8.6 μM. While 2 and 25–27 did not show significant cell line selectivity, isobutoxy analogue 28 displayed selective activity against A549 with an ED50 value of 1.7 μM. Isopentoxy analogue 29 exhibited the lowest activity against all tested cell lines.
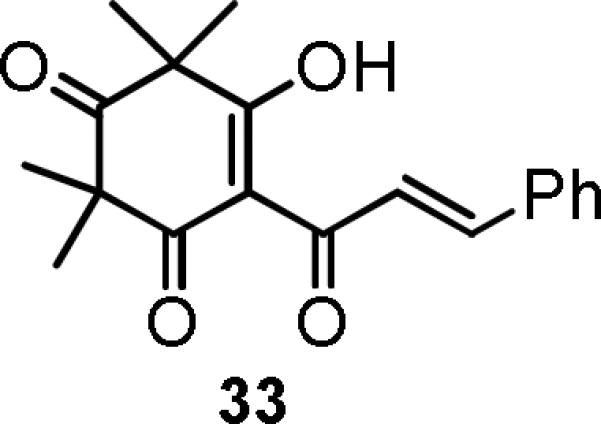
Analogue 30, which has a tetraalkyl, rather than trialkyl, substitution pattern at C-3 (gem-propyl) and C-5 (propyl and prenyl), showed significant tumor cell growth inhibition, while tetramethyl analogue 333b (Figure 2) did not show cytotoxic activity. These results suggested that the alkyl or alkoxy groups on ring-A can strongly affect the activity.
Figure 2.
As previously reported,3 analogue 3 with tripropyl substitution on ring-A and bromide at C-4′ on ring-B showed 5- to 7-fold greater activity than 1. Replacement of bromide with iodide led to slightly higher potency. Thus, the 4′-iodo-3,3,5-tripropyl substituted analogue 31 exhibited significant antiproliferative activity against all tested cell lines (ED50 1.1–2.8 μM), and was 6- to 13-fold more potent than 1. Interestingly, 31 was active even against PC-3 and HepG2 (ED50 2.8 and 2.4 μM, respectively) cell lines, which were generally less sensitive than the other cell lines to 1-analogues. We postulated that analogue 32, which has tripropyl substitution at C-3 and C-5 as well as propoxy at C-4 and iodide at C-4′, would exhibit greater inhibitory potency against tumor cell growth. Unexpectedly, 32 was less active than 31 (methoxy at C-4) or 26 (hydrogen at C-4′).
The cell growth of KB-VIN cancer cells, which express P-gp and selected for resistance to vincristine, was inhibited by analogues at the same or greater level than the parent KB cells, supporting the idea that all analogues are not affected by P-gp-related multidrug resistance (MDR).
Analogue 3 also showed potent antiproliferative activity against the highly invasive non-small-cell lung cancer cell line CL1-5 with an ED50 value of 0.11 μM. To determine which genes were differentially expressed upon CL1-5 treatment with analogue 3, the genome-wide mRNA expression profiles of 3-treated cells and control cells were determined using Affymetrix human genome U133 plus 2.0 GeneChip according to the Manufacturer's protocols (Santa Clara, CA, http://www.affymetrix.com) by the Microarray Core Facility of National Research Program for Genomic Medicine of National Science Council in Taiwan as previously described.7 This Affymetrix GeneChip contains 54,675 probe sets to analyze the expression levels of 47,400 transcripts and variants, including 38,500 well-characterized human genes. GeneChips from the hybridization experiments were read by the Affymetrix GeneChip scanner 3000 7G, and raw data were processed using GC-RMA algorithm. The raw data were then analyzed by GeneSpring GX software version 11.01.8 2.5 × 105 CL1-5 cells were treated for 24 h with 3 at a concentration of 0.05 μg/mL, and then total RNA was extracted by TRI zol (Life Technologies, Gaithersburg, MD) RNA from non-treated CL1-5 cells was used as a control. A total of 2,838 genes showed at least two-fold changes in expression levels between the CL1-5 treated with 3 and CL1-5 DMSO control. Analogue 3 up-regulated the expression of 1,112 genes and down-regulate 1,726 genes. The differentially expressed genes were analyzed for GeneGo canonical pathway maps by using MetaCore Analytical Suite (GeneGo Inc., St Joseph, MI). The top ten pathways involved in analog 3 affected genes were shown in Table 2. Seven pathways are cell cycle-related pathways and three are DNA damage-related. For example, in the spindle assembly checkpoint (SAC) or chromosome segregation pathway, the genes altered by the treatment with 3 encoded mitotic kinases (e.g., CDK1-cyclin B, Aurora A, Aurora B, and NEK2A), SAC proteins (e.g., MAD1, MAD2, securin, and separase), and motor proteins (e.g., dynein1, dynein activator complex dynactin, and KNSL1) (Figure 3). The cyclin dependent kinase, Aurora kinases, and NEK2A kinases are critical for mitotic progression, through phosphorylation of their numerous substrates. NEK2A or Aurora kinases are required for spindle formation at the onset of mitosis or chromosome segregation and cytokinesis, respectively. The SAC proteins, such as MAD2, activate spindle checkpoint and inhibit securin degradation, until all chromosomes are aligned at the metaphase plate. When chromosomes are aligned correctly, the E3 ubiquitin ligase anaphase-promoting complex/cyclosome (APC/C) inhibitor MAD2 is dissociated from the APC/C and removed from the attached kinetochore by dynein. Subsequently, APC/C is activated by CDC20 or CDH1.9 APC/C-CDC20 or -CDH1 recognizes substrates such as cyclins, NEK2A, and securin or Aurora kinases and cyclins, respectively. At the onset of anaphase, the separase inhibitor securin is poly-ubiquitinated by activated APC/C followed by digestion by the proteasome. Subsequently, activated separase cleaves thecohesin complex, resulting in separation of the sister chromatids. All of these proteins are expressed in a cell cycle-dependent manner.10 In our oligonucleotide microarray studies, genes encoding these proteins were up-regulated by the treatment with 3. The up-regulation of MAD2L1 transcript was confirmed by semi-quantitative RT-PCR (Figure 4). Therefore, we assume that 3 may modulate SAC and chromosome separation, and conclude that 3 induces cell cycle arrest mainly in the G2/M-phase. Because oligonucleotide microarray data are quite complicated and can be contradictory, we will need to conduct additional experiments, such as real-time qPCR, to verify our results.
Table 2.
| Statistically significant pathways | p Value |
|---|---|
| Cell cycle_The metaphase checkpoint | 1.28E-23 |
| Cell cycle_Role of APC in cell cycle regulation | 2.09E-19 |
| Cell cycle_Start of DNA replication in early S phase | 1.62E-16 |
| Cell cycle_Spindle assembly and chromosome separation | 3.87E-16 |
| Cell cycle_Chromosome condensation in prometaphase | 1.39E-15 |
| Cell cycle_Transition and termination of DNA replication | 1.35E-12 |
| Cell cycle_Role of Nek in cell cycle regulation | 2.08E-11 |
| DNA damage_ATM / ATR regulation of G2 / M checkpoint | 1.05E-10 |
| DNA damage_ATM/ATR regulation of G1/S checkpoint | 3.56E-09 |
| DNA damage_Role of Brca1 and Brca2 in DNA repair | 1.51E-08 |
Figure 3.

Figure 4.

CL1-5 cells were treated for 24 h with DMSO (w/o) or 0.05 μg/mL of 3, followed by RNA isolation and RT-PCR for MAD2L1 (arrow in upper panel). The Gβ-like was used as control (lower panel). Asterisk in upper panel shows non-specific amplification by primer-dimer.
In summary, among the tested compounds, tripropyl substitution at C-3 and -5 (R = Pr) was optimal for tumor cell growth inhibition. A methoxy or propoxy group at C-4 (OR′ = OMe or OPr) was generally preferred over other alkyl ether groups. Finally, the combination of a 3,3,5-tripropyl-4-methoxy A-ring and a 4′-bromo- or 4′-iodo-phenyl B-ring (3 and 31) led to the greatest tumor cell growth inhibition. Isobutoxy analog 28 selectively inhibited the A549 lung tumor cell line. Oligonucleotide microarray studies showed that 3 may modulate SAC and chromosome separation and arrest cells mainly in the G2/M-phase. Further modifications of 28 as selective anti-lung tumor agents as well as further investigations to verify oligonucleotide microarray data are currently undergoing, and will be reported in the future.
4. Experimental section
All chemicals and solvents were used as purchased. All melting points were measured on a Fisher-Johns melting point apparatus without correction. 1H-NMR spectra were recorded on a Varian Gemini 300 (300 MHz) spectrometer with TMS as the internal standard. All chemical shifts are reported in ppm. 13C-NMR spectra were recorded on a Varian Inova 400 (400MHz) spectrometer, referenced to the residual solvent peak. Mass spectroscopic data were obtained on a TRIO 1000 mass spectrometer. Analytical thin-layer chromatography (TLC) was carried out on Merck precoated aluminum silica gel sheets (Kieselgel 60 F-254). Final target compounds were characterized by 1H-NMR and HRMS analyses, and others were characterized by 1H-NMR. The purities of the final targets were >90% determined by 1H-NMR and HPLC analyses.
2-Acetyl-4,4,6-tributyl-3,5-dihydroxycyclohexa-2,5-dienone (6)
A solution of 2,4,6-trihydroxyacetophenone (4, 595 mg, 3.5 mmol), sodium methoxide (2.5 mL, 11.6 mmol, 25% MeOH solution) and butyl iodide (1.2 mL, 10.6 mmol) in anhydrous MeOH (3 mL) was refluxed overnight. The reaction mixture was cooled to 0 °C and acidified with 1N aqueous HCl solution, then extracted three times with EtOAc. The combined organic layers were dried over Na2SO4 and concentrated under vacuum. The residue was chromatographed on silica gel with EtOAc-hexane (1:9 to 1:4, v/v) as an eluent to obtain 6 (516 mg, 44%) as colorless solid, which was used directly in the next reaction without recrystallization. 1H NMR (300 MHz, CDCl3): δ 19.03 and 18.40 (2:1, each s, 1H, chelated-OH), 5.95 and 5.38 (2:1, each s, 1H, OH), 2.72 and 2.62 (1:2, each s, 3H, COCH3), 2.48–2.38 (m, 2H), 2.04–1.87 (m, 2H), 1.80–1.67 (m, 2H), 1.52–1.32 (m, 4H), 1.29–1.13 (m, 4H), 1.06–0.90 (m, 8H), 0.86–0.78 (m, 6H). MS (ESI, m/z) 337 [M+H]+.
2-Acetyl-4,4,6-triisobutyl-3,5-dihydroxycyclohexa-2,5-dienone (7)
Compound 4 (412 mg, 2.5 mmol), sodium methoxide (1.8 mL, 8.3 mmol, 25% MeOH solution), and isobutyl iodide (0.9 mL, 7.8 mmol) in anhydrous MeOH (3 mL) were treated similarly to the above procedure to obtain 7 (174 mg, 21%) as colorless solid, which was used directly in the next reaction without recrystallization. 1H NMR (300 MHz, CDCl3): δ 19.02 and 18.30 (3:1, each s, 1H, chelated-OH), 6.07 and 5.40 (3:1, each s, 1H, OH), 2.71 and 2.61 (1:3, each s, 3H, COCH3), 2.32 and 2.29 [3:1, each d, 2H, J = 7.4 Hz, 6-CH2CH(CH3)2], 2.16–1.81 (m, 3H), 1.76–1.67 (m, 2H), 1.48–1.36 (m, 2H, 4-CH2CH(CH3)2×2], 0.96 and 0.95 [3:1, each d, 6H, J = 6.6 Hz, CH2CH(CH3)2], 0.81 and 0.80 [1:3, each d, 6H, J = 6.6 Hz, CH2CH(CH3)2], 0.72 and 0.71 [1:3, each d, 6H, J = 6.6 Hz, CH2CH(CH3)2]. MS (ESI, m/z) 337 [M+H]+.
2-Acetyl-4,4,6-triisopentyl-3,5-dihydroxycyclohexa-2,5-dienone (8)
Compound 4 (510 mg, 3.0 mmol), sodium methoxide (2.2 mL, 10.2 mmol, 25% MeOH solution), and isobutyl iodide (1.3 mL, 9.9 mmol) in anhydrous MeOH (2 mL) were treated as described above to obtain 8 (317 mg, 28%) as colorless solid, which was used directly in the next reaction without recrystallization. 1H NMR (300 MHz, CDCl3): δ 19.01 and 18.30 (3:1, each s, 1H, chelated-OH), 5.82 and 5.22 (3:1, each s, 1H, OH), 2.73 and 2.70 (1:3, each s, 3H, COCH3), 2.48–2.38 (m, 2H), 2.04–1.90 (m, 4H), 1.79–1.56 (m, 3H), 1.46–1.30 (m, 6H), 0.96 [d, 6H, J = 6.6 Hz, CH2CH2CH(CH3)2], 0.81 and 0.80 [1:3, each d, 12H, J = 6.6 Hz, CH2CH2CH(CH3)2×2]. MS (ESI, m/z) 379 [M+H]+.
2-Acetyl-4,4,6-triprenyl-3,5-dihydroxycyclohexa-2,5-dienone (9)
To a solution of 4 (835 mg, 4.5 mmol) and KOH (572 mg, 10.2 mmol) in H2O (5.7 mL) at 0 °C under an argon atmosphere was added prenyl bromide (1.2 mL, 10.3 mmol) dropwise over five min. The resulting mixture was stirred at 0 °C for 1 h. Subsequently, KOH (255 mg, 4.6 mmol) in H2O (0.25 mL) and prenyl bromide (0.55 mL, 4.7 mmol) were added at 0 °C. After stirring for 15 min at 0 °C, the mixture was allowed to warm to room temperature and stirred for 1 h. The reaction mixture was quenched with aqueous HCl to pH 1 and extracted three times with EtOAc. The combined organic layers were washed with brine, dried over anhydrous magnesium sulfate, filtered, and concentrated under vacuum. Purification on silica gel (hexane: EtOAc) provided 9 (269 mg, 16%) as brown oil, tetraprenyl compound (100 mg, 5%) and diprenyl compound (327 mg, 24%).
2-Acetyl-4,4,6-tripropyl-5-ethoxy-3-hydroxycyclohexa-2,5-dienone (15)
To a solution of 5 (102 mg, 0.35 mmol) in anhydrous acetone (2 mL), potassium carbonate (1045 mg, 7.6 mmol) and ethyl iodide (0.13 mL, 1.6 mmol) were added, and the mixture was stirred for 2 days. After filtration, the solvent was removed under vacuum. The residue was purified by column chromatography with EtOAc-hexane as an eluent to obtain 15 (50 mg, 44%) as brown oil, along with recovered starting material (39 mg, 38%). 1H NMR (300 MHz, CDCl3): δ 19.00, 18.31 and 18.14 (2:1:1, each s, 1H, chelated-OH), 4.19 and 4.07 (2:1, each q, 2H, J = 7.0 Hz, OCH2CH3), 2.69 and 2.60 (1:2, each s, 3H, COCH3), 2.55–2.37 (m, 2H), 1.93–1.64 (m, 8H), 1.58–1.34 (m, 3H), 1.30–1.10 (m, 2H), 1.10–0.92 (m, 3H), 0.90–0.73 (m, 6H). MS (ESI, m/z) 323 [M+H]+.
2-Acetyl-4,4,6-tripropyl-5-propoxy-3-hydroxycyclohexa-2,5-dienone (16)
Compound 5 (95 mg, 0.32 mmol), potassium carbonate (940 mg, 6.8 mmol), and propyl iodide (0.5 mL, 2.1 mmol) were treated as described above for 15 to obtain 16 (67 mg, 62%) as brown oil. 1H NMR (300 MHz, CDCl3): δ 19.00, 18.38 and 18.14 (2:1:1, each s, 1H, chelated-OH), 4.09 and 3.96 (2:1, each t, 2H, J = 6.4 Hz, OCH2CH2CH3), 2.69, 2.62 and 2.60 (1:2:1, each s, 3H, COCH3), 2.50–2.36 (m, 2H), 1.90–1.65 (m, 8H), 1.58–1.38 (m, 2H), 1.30–1.10 (m, 2H), 1.10–0.92 (m, 6H), 0.90–0.74 (m, 6H). MS (ESI, m/z) 337 [M+H]+.
2-Acetyl-4,4,6-tripropyl-5-isobutoxy-3-hydroxycyclohexa-2,5-dienone (18)
Compound 5 (110 mg, 0.37 mmol), potassium carbonate (1272 mg, 9.2 mmol) and isobutyl iodide (0.4 mL, 3.5 mmol) were treated as described above for 15 to obtain 18 (36 mg, 28%) as brown oil along with the recovery of starting material (77 mg, 70%). 1H NMR (300 MHz, CDCl3): δ 19.00, 18.38 and 18.14 (2:1:1, each s, 1H, chelated-OH), 3.90 and 3.77 [2:1, each d, 2H, J = 6.4 Hz, OCH2CH(CH3)2], 2.69, 2.62 and 2.61 (1:2:1, each s, 3H, COCH3), 2.51–2.38 (m, 2H), 2.10–1.65 (m, 8H), 1.56–1.40 (m, 2H), 1.25–1.10 (m, 2H), 1.10–0.92 (m, 9H), 0.90–0.75 (m, 6H). MS (ESI, m/z) 351 [M+H]+.
2-Acetyl-4,4,6-tripropyl-5-isopentoxy-3-hydroxycyclohexa-2,5-dienone (19)
Compound 5 (154 mg, 0.52 mmol), potassium carbonate (1840 mg, 13.3 mmol) and iso-pentyl iodide (0.7 mL, 5.3 mmol) were treated as described above for 15 to obtain 19 (94 mg, 26%) as brown oil.
4-Acetyl-2-(3-methylbut-2-en-1-yl)-2,6,6-tripropyl-5-hydroxycyclohex-4-ene-1,3-dione (20)
Compound 5 (157 mg, 0.53 mmol), potassium carbonate (1030 mg, 7.5 mmol) and prenyl bromide (0.15 mL, 1.3 mmol) were treated as described above for 15 to obtain 20 (84 mg, 44%) as brown oil. 1H NMR (300 MHz, CDCl3): δ 19.00 (s, 1H, chelated-OH), 6.98 (s, 1H, OH), 5.20–5.09 [m, 1H, CH2CHC(CH3)2], 4.86–4.75 [m, 2H, CH2CHC(CH3)2×2], 3.21–3.13 [m, 2H, CH2CHC(CH3)2], 2.70–2.46 [m, 4H, CH2CHC(CH3)2×2], 1.82–1.74 (m, 6H), 1.65–1.50 (m, 12H). MS (ESI, m/z) 363 [M+H]+.
General Procedures for Aldol Reactions
A solution of acetyl compound (10–19) in EtOH–50% aq. KOH (1:1, v/v) and an appropriate aldehyde (excess) was stirred at room temperature. After the reaction was complete by TLC analysis, the mixture was poured into ice-cold 1N HCl, then extracted with CH2Cl2. The extract was washed with brine, dried over Na2SO4, and concentrated under vacuum. The residue was chromatographed on silica gel with CH2Cl2-hexane as eluent to afford the target compound, which was crystallized from CH2Cl2-hexane.
3,3,5-Tributyldesmosdumotin C (21)
56% Yield. Yellow oil. 1H NMR (300 MHz, CDCl3): δ 19.30 and 18.90 (2:1, each s, 1H, chelated-OH), 8.51 and 8.42 (1:2, each d, 1H, J = 15.6 Hz, olefin), 7.96 and 7.93 (1:2, each d, 1H, J = 15.6 Hz, olefin), 7.73–7.65 (m, 2H, Ar-H), 7.44–7.36 (m, 3H, Ar-H), 4.00 and 3.92 (2:1, each s, 3H, OCH3), 2.59–2.48 (m, 2H), 2.00–1.70 (m, 4H), 1.58–1.37 (m, 4H), 1.30–1.14 (m, 4H), 1.10–0.92 (m, 6H), 0.86–0.78 (m, 6H). HRMS: Calcd. For C28H39O4 439.2848 [M+H]+, Found 439.2876.
3,3,5-Triisobutyldesmosdumotin C (22)
36% Yield. Yellow oil. 1H NMR (300 MHz, CDCl3): δ 19.36 and 18.86 (2:1, each s, 1H, chelated-OH), 8.48 and 8.44 (1:2, each d, 1H, J = 15.6 Hz, olefin), 7.96 and 7.93 (1:2, each d, 1H, J = 15.6 Hz, olefin), 7.73–7.64 (m, 2H, Ar-H), 7.43–7.36 (m, 3H, Ar-H), 4.03 and 3.94 (2:1, each s, 3H, OCH3), 2.57–2.50 (m, 2H), 1.97–1.84 (m, 4H), 1.80–1.71 (m, 1H), 1.54–1.39 (m, 2H), 0.94 and 0.93 [2:1, d, 6H, J = 6.6 Hz, CH2CH(CH3)2], 0.83 and 0.82 [1:2, d, 6H, J = 6.6 Hz, CH2CH(CH3)2], 0.73 and 0.72 [2:1, d, 6H, J = 6.6 Hz, CH2CH(CH3)2]. HRMS: Calcd. For C28H39O4 [M+H]+439.2848, Found 439.2879.
3,3,5-Triisopentyldesmosdumotin C (23)
43% Yield. Yellow oil. 1H NMR (300 MHz, CDCl3): δ 19.29 and 18.89 (2:1, each s, 1H, chelated-OH), 8.50 and 8.41 (1:2, each d, 1H, J = 15.6 Hz, olefin), 7.96 and 7.93 (1:2, each d, 1H, J = 15.6 Hz, olefin), 7.74–7.66 (m, 2H, Ar-H), 7.45–7.37 (m, 3H, Ar-H), 4.00 and 3.93 (2:1, each s, 3H, OCH3), 2.60–2.46 (m, 2H), 1.98–1.71 (m, 5H), 1.70–1.60 (m, 1H), 1.51–1.36 (m, 4H), 0.98 and 0.97 [2:1, d, 6H, J = 6.6 Hz, CH2CH(CH3)2], 0.94–0.80 (m, 15H). HRMS: Calcd. For C31H45O4 481.3318 [M+H]+, Found 481.3342.
3,3,5-Triprenyldesmosdumotin C (24)
63% Yield. Yellow oil. 1H NMR (300 MHz, CDCl3): δ 18.82 (s, 1H, chelated-OH), 8.53 and 8.38 (1:2, each d, 1H, J = 15.6 Hz, olefin), 7.94 and 7.92 (1:2, each d, 1H, J = 15.6 Hz, olefin), 7.74–7.62 (m, 2H, Ar-H), 7.45–7.37 (m, 3H, Ar-H), 5.11–5.02 [m, 1H, CH2CH=C(CH3)2], 4.85–4.74 [m, 2H, CH2CH=C(CH3)2×2], 3.95 and 3.88 (2:1, each s, 3H, OCH3), 3.28–3.16 [m, 2H, CH2CH=C(CH3)2], 2.80–2.50 [m, 4H, CH2CH=C(CH3)2×2], 1.76–1.69 (m, 6H), 1.62–1.56 (m, 12H). HRMS: Calcd. For C31H37O4 473.2692 [M+H]+, Found 473.2731.
4-Ethoxy-3,3,5-tripropyldesmosdumotin C (25)
41% Yield. Yellow oil. 1H NMR (300 MHz, CDCl3): δ 19.32 and 18.89 (2:1, each s, 1H, chelated-OH), 8.50 and 8.42 (1:2, each d, 1H, J = 15.6 Hz, olefin), 7.95 and 7.92 (1:2, each d, 1H, J = 15.6 Hz, olefin), 7.74–7.64 (m, 2H, Ar-H), 7.45–7.34 (m, 3H, Ar-H), 4.20 and 4.10 (2:1, each q, 2H, J = 6.9 Hz, OCH2CH3), 2.54–2.40 (m, 2H), 2.00–1.66 (m, 4H), 1.61–1.48 (m, 2H), 1.48–1.37 (m ,3H), 1.19–0.95 (m, 5H), 0.87–0.79 (m, 6H). HRMS: Calcd. for C26H35O 411.2535 [M+H]+, Found 411.2573.
4-Propoxy-3,3,5-tripropyldesmosdumotin C (26)
29% Yield. Yellow oil. 1H NMR (300 MHz, CDCl3): δ 19.33 and 18.84 (2:1, each s, 1H, chelated-OH), 8.50 and 8.42 (1:2, each d, 1H, J = 15.6 Hz, olefin), 7.94 and 7.92 (1:2, each d, 1H, J = 15.6 Hz, olefin), 7.73–7.64 (m, 2H, Ar-H), 7.44–7.34 (m, 3H, Ar-H), 4.10 and 3.99 (2:1, each t, 2H, J = 6.9 Hz, OCH2CH2CH3), 2.54–2.42 (m, 2H), 2.00–1.64 (m, 6H), 1.52–1.47 (m, 2H), 1.15–0.95 (m, 10H), 0.92–0.78 (m, 6H). 13C NMR (400 MHz, CDCl3): δ 200.78, 198.22, 192.81, 189.20, 187.08, 186.47, 174.23, 167.42, 145.40, 144.82, 135.57, 135.47, 130.83, 130.67, 129.24, 129.12, 129.05, 129.02, 124.22, 124.08, 120.66, 112.02, 109.24, 76.15, 75.82, 59.82, 55.48, 42.38, 41.08, 39.04, 38.09, 26.70, 26.23, 24.10, 24.05, 23.17, 23.00, 18.48, 18.42, 18.32, 18.18, 14.75, 14.68, 14.61, 14.56, 14.39, 10.63, 10.59. HRMS: Calcd. for C27H35O4 423.2535 [M-H]+, Found 423.2559.
4-Butoxy-3,3,5-tripropyldesmosdumotin C (27)
67% Yield. Yellow oil. 1H NMR (300 MHz, CDCl3): δ 19.33 and 18.90 (2:1, each s, 1H, chelated-OH), 8.50 and 8.42 (1:2, each d, 1H, J = 15.6 Hz, olefin), 7.94 and 7.92 (1:2, each d, 1H, J = 15.6 Hz, olefin), 7.73–7.64 (m, 2H, Ar-H), 7.44–7.35 (m, 3H, Ar-H), 4.15 and 4.02 [2:1, each t, 2H, J = 6.4 Hz, OCH2(CH)2CH3], 2.00–1.81 (m, 2H), 1.80–1.69 (m, 2H). 1.60–1.42 (m, 6H), 1.15–0.96 (m, 10H), 0.89–0.77 (m, 6H). HRMS: Calcd. for C28H39O4 439.2848 [M+H]+, Found 439.2880.
4-Isobutoxy-3,3,5-tripropyldesmosdumotin C (28)
32% Yield. Yellow oil. 1H NMR (300 MHz, CDCl3): δ 19.34 and 18.83 (2:1, each s, 1H, chelated-OH), 8.50 and 8.42 (1:2, each d, 1H, J = 15.6 Hz, olefin), 7.94 and 7.92 (1:2, each d, 1H, J = 15.6 Hz, olefin), 7.73–7.64 (m, 2H, Ar-H), 7.45–7.35 (m, 3H, Ar-H), 3.92 and 3.79 [2:1, each d, 2H, J = 6.4 Hz, OCH2CH(CH3)2], 2.56–2.42 (m, 2H), 2.11–1.70 (m, 6H), 1.60–1.48 (m, 2H), 1.18–0.95 (m, 10H), 0.90–0.78 (m, 6H). HRMS: Calcd. for C28H39O4 439.2848 [M+H]+, Found 439.2878.
4-Isopentoxy-3,3,5-tripropyldesmosdumotin C (29)
52% Yield. Yellow oil. 1H NMR (300 MHz, CDCl3): δ 19.31 and 18.87 (2:1, each s, 1H, chelated-OH), 8.50 and 8.42 (1:2, each d, 1H, J = 15.6 Hz, olefin), 7.94 and 7.92 (1:2, each d, 1H, J = 15.6 Hz, olefin), 7.74–7.65 (m, 2H, Ar-H), 7.44–7.35 (m, 3H, Ar-H), 4.17 and 4.05 [2:1, each t, 2H, J = 6.4 Hz, OCH2CH2CH (CH3)2], 2.56–2.42 (m, 2H), 1.98–1.46 (m, 8H), 1.14–0.94 (m, 10H), 0.86–0.78 (m, 6H). HRMS: Calcd. for C29H39O4 451.2848 [M+H]+, Found 451.2886.
3,3,5-Tripropyl-5-prenyldesmosdumotin C (30)
32% Yield. Yellow oil. 1H NMR (300 MHz, CDCl3): δ 18.49 and 18.36 (1:1, each s, 1H, chelated-OH), 8.06–7.94 (m, 2H), 7.71–7.61 (m, 2H), 7.49–7.36 (m, 3H), 5.00–4.87 [m, 1H, CH2CH=C(CH3)2], 2.70–2.34 (m, 2H), 1.90–1.64 (m, 6H), 1.64–1.52 (m, 6H), 1.36–1.10 (m, 6H), 0.94–0.80 (m, 9H). HRMS: Calcd. For C29H37O4 449.2692 [M+H]+, Found 449.2725.
4′-Iodo-3,3,5-tripropyldesmosdumotin C (31)
23% Yield. Yellow prisms. mp. 123–124°C (Hexane). 1H NMR (300 MHz, CDCl3): δ 19.27 and 18.82 (2:1, each s, 1H, chelated-OH), 8.49 and 8.41 (1:2, each d, 1H, J = 15.6 Hz, olefin), 7.84 and 7.81 (1:2, each d, 1H, J = 15.6 Hz, olefin), 7.77–7.70 (m, 2H, Ar-H), 7.44–7.37 (m, 2H, Ar-H), 4.00 and 3.92 (2:1, each s, 3H, OCH3), 2.55–2.43 (m, 2H), 1.99–1.67 (m, 4H), 1.63–1.46 (m, 2H), 1.14–0.97 (m, 7H), 0.87–0.79 (m, 6H). 13C NMR (400 MHz, CDCl3): δ 198.22, 192.85, 186.86, 175.05, 144.03, 143.56, 138.30, 138.25, 135.00, 130.57, 130.50, 126.76, 124.88, 124.70, 121.54, 109.39, 62.35, 59.95, 55.58, 42.15, 40.89, 26.62, 26.14, 23.03, 22.88, 18.46, 18.35, 14.78, 14.64, 14.54, 14.36. HRMS : Calcd. for C25H32IO4 523.1345 [M+H]+, Found 523.1363.
4′-Iodo-4-propoxy-3,3,5-tripropyldesmosdumotin C (32)
9% Yield. Yellow oil. 1H NMR (300 MHz, CDCl3): δ 19.31 and 18.81 (2:1, each s, 1H, chelated-OH), 8.49 and 8.42 (1:2, each d, 1H, J = 15.6 Hz, olefin), 7.84 and 7.80 (1:2, each d, 1H, J = 15.6 Hz, olefin), 7.76–7.69 (m, 2H, Ar-H), 7.44–7.36 (m, 2H, Ar-H), 4.11 and 3.99 (2:1, each t, 2H, J = 6.9 Hz, OCH2CH2CH3), 2.55–2.42 (m, 2H), 2.00–1.68 (m, 6H), 1.60–1.42 (m, 2H), 1.12–0.95 (m, 10H), 0.86–0.77 (m, 6H). HRMS: Calcd. for C27H36IO4 551.1658 [M+H]+, Found 551.1691.
Antiproliferative Activity Assay
All stock cultures were grown in T-25 flasks. Freshly trypsinized cell suspensions were seeded in 96-well microtitre plates at densities of 1500–7500 cells per well with compounds added from DMSO-diluted stock. After three days in culture, attached cells were fixed with cold 50% trichloroacetic acid and then stained with 0.4% sulforhodamine B (SRB). The absorbency at 562 nm was measured using a microplate reader after solubilizing the bound dye. The mean ED50 is the concentration of agent that reduces cell growth by 50% under the experimental conditions and is the average from at least three independent determinations that were reproducible and statistically significant. The following human tumor cell lines were used in the assay: A549 (human lung carcinoma), 1A9 (human ovarian carcinoma), HCT-8 (colon adenocarcinoma), PC-3 (prostate cancer), KB (nasopharyngeal carcinoma), KB-VIN (vincristine resistant KB subline), HUVEC (human umbilical vein endothelial cell). All cell lines were obtained from the Lineberger Cancer Center (UNC-CH) or from ATCC (Rockville, MD) and were cultured in RPMI-1640 medium supplemented with 25 mM HEPES, 0.25% sodium bicarbonate, 10% fetal bovine serum, and 100 μg/mL kanamycin.
RT-PCR
Freshly trypsinized CL1-5 cell suspensions were seeded in 60 mm cell culture dishes at density of 2.5×105 and cultured for 48 h in RPMI-164 medium supplemented with 10% fetal bovine serum (Gibco-BRL).11 Cells were incubated in 5% CO2 and 95% air at 37°C. Cells were treated with compound and continued cultivation for 24 h followed by the total RNA extraction by TRIzol (Invirtogen). The cDNAs were synthesized from 1 μg total RNA using random hexamer primers and Superscript III reverse transcriptase (Invitrogen). The MAD2L1 was amplified from cDNA pool (1:10 diluted) by PCR (30 cycles) using DyNAzyme II DNA polymerase (Finnzymes) with forward primer 5'-AGGCAGCGCTGAGCTTGTGG-3' and reverse primer 5'-AGGCAGTCTCCAGCAGGGGT-3'. The Gβ-like was amplified from same cDNA pool by PCR (25 cycles) using forward primer 5'-GTATGGAACCTGGCTAACTG-3' and reverse primer 5'-TACTGATAACTTCTTGCTTC-3'. The PCR products were separated by agarose gel and stained by ethidium bromide.
Scheme 1. Syntheses of Desmosdumotin C Derivatives.

Reagents: a) Prenyl Br, KOH, water for R = Prenyl; RI, NaOMe, MeOH, reflux for others; b) TMSCHN2 for R' = Me; R'I, K2CO3, acetone, reflux for others; c) 50% aq. KOH, EtOH, ArCHO, rt; d) Prenly Br, K2CO3, acetone reflux
Figure 1.

Desmosdumotin C and its analogs
Acknowledgements
This study was supported by grant CA-17625 from the National Cancer Institute, NIH, awarded to K. H. L and by a grant from the University Research Council, awarded to K.N. G. We thank the technical assistance by the research assistants at Microarray Core Facility of National Research Program for Genomic Medicine of National Science Council in Taiwan.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Wu JH, McPhail AT, Bastow KF, Shiraki H, Ito J, Lee KH. Tetrahedron Lett. 2002;43:1391. [Google Scholar]
- 2.Nakagawa-Goto K, Wu JH, Lee KH. Syn. Commun. 2005;35:1735. [Google Scholar]
- 3.a) Nakagawa-Goto K, Wu JH, Bastow KF, Wu CC, Lee KH. Antitumor agents 243. Bioorg. Med. Chem. 2005;13:2325. doi: 10.1016/j.bmc.2004.12.040. [DOI] [PubMed] [Google Scholar]; b) Nakagawa-Goto K, Chen T-H, Peng C-Y, Bastow KF, Wu JH, Lee KH. J. Med. Chem. 2007;50:3354. doi: 10.1021/jm0702534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.For Review: Barron D, Ibrahim RK. Phytochemistry. 1996;43:921.; Zanoli P, Zavatti M. J. Ethnopharmacology. 2008;116:383. doi: 10.1016/j.jep.2008.01.011..
- 5.Recent reports: Harikumar K, Kunnumakkara AB, Ahn KS, Anand P, Krishnan S, Guha S, Aggarwal BB. Blood. 2009;113:2003. doi: 10.1182/blood-2008-04-151944.; Rao GV, Swamy BN, Chandregowda V, Reddy GC. Eur. J. Med. Chem. 2009;44:2239. doi: 10.1016/j.ejmech.2008.05.032.; Jamil S, Sirat HM, Jantan I, Aimi N, Kitajima M. J. Nat. Med. 2008;62:321. doi: 10.1007/s11418-008-0226-3.; Vogel S, Heilmann J. J. Nat. Prod. 2008;71:1237. doi: 10.1021/np800188b.; Vogel S, Ohmayer S, Brunner G, Heilmann J. Bioorg. Med. Chem. 2008;16:4286. doi: 10.1016/j.bmc.2008.02.079.
- 6.a) Xiao L, Tan W, Li Y. Synth. Commun. 1998;28:2861. [Google Scholar]; b) Qi J, Porco JA., Jr. J. Am. Chem. Soc. 2007;129:12682. doi: 10.1021/ja0762339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chao YC, Pan SH, Yang SC, Yu SL, Che TF, Lin CW, Tsai MS, Chang GC, Wu CH, Wu YY, Lee YC, Hong TM, Yang PC. Am. J. Respir. Crit. Care Med. 2009;179:123. doi: 10.1164/rccm.200803-456OC. [DOI] [PubMed] [Google Scholar]
- 8.Silicon Genetics, Redwood City, CA. The original data have been deposited to NCBI database, and the GEO series number is GSE24584.
- 9.Zich J, Hardwick KG. Trends Biochem. Sci. 2010;35:18. doi: 10.1016/j.tibs.2009.09.002. [DOI] [PubMed] [Google Scholar]
- 10.Manchado E, Eguren M, Malumbres M. Biochem. Soc. Trans. 2010;38:65. doi: 10.1042/BST0380065. [DOI] [PubMed] [Google Scholar]
- 11.Chu YW, Yang PC, Yang SC, Shyu YC, Hendrix MJ, Wu R, Wu CW. Am J Respir Cell Mol Biol. 1997;17:353. doi: 10.1165/ajrcmb.17.3.2837. [DOI] [PubMed] [Google Scholar]