

Abstract
Acute oxygen sensing in the heart is thought to occur through redox regulation and phosphorylation of membrane channels. Here we report a novel O2-sensing mechanism involving the C-terminus of the L-type Ca2+ channel and regulated by PKA phosphorylation. In patch-clamped myocytes, oxygen deprivation decreased ICa within 40 s. The suppressive effect of anoxia was relieved by PKA-mediated phosphorylation only when Ca2+ was the charge carrier, whereas phosphorylated IBa remained sensitive to O2 withdrawal. Suppression of Ca2+ release by thapsigargin did not alter the response of ICa to anoxia, suggesting a mandatory role for Ca2+ influx and not Ca2+-induced Ca2+ release (CICR) in O2 regulation of the channel. Consistent with this idea, mutation of 80 amino acids in the Ca2+/CaM-binding domain of the recombinant α1C subunit that removes Ca2+ dependent inactivation (CDI) abolished O2 sensitivity of the channel. Our findings suggest that the Ca2+/CaM binding domain of the L-type Ca2+ may represent a molecular site for O2 sensing of the heart.
Keywords: cardiac oxygen sensor, L-type calcium channel, C-terminus, PKA phosphorylation
Introduction
Voltage-gated calcium channels are widely expressed in the animal kingdom and in particular in the mammalian heart, brain, and muscle. All members of this family have a high selectivity for transport of calcium over other mono- or divalent cations. Although different types of voltage-gated Ca2+ channels have specific differences in their gating, voltage dependence, kinetics of activation and inactivation, pharmacology, and regulation by permeating Ca2+ and phosphorylation, they all transport Ca2+ efficiently, can be blocked by Ni2+, Cd2+, and La3+, and share many overlapping properties. It is unclear as yet the evolutionary need for the development of this large family of voltage-gated Ca2+ channels, especially since Ca2+ serves mostly as a messenger signaling molecule. There are numerous reports that some members of this family, in addition to having a Ca2-transporting function, may directly gate the release of Ca2+ from the ryanodine receptors of sarcoplasmic reticulum (SR) in skeletal muscle,1 modulate CICR in cardiac muscle via specific domains of the C-terminus tail of the L-type Ca2+ channel,2 or directly signal the nuclear transcription factors.3
The energy required by the heart to carry out its enormous workload, for some 2 billion beats over a lifetime, requires critical regulation of the ATP and O2 supply to the heart, and the rapid detection of their availability. Is there such an O2 sensing mechanism in the heart? If so, what is the sensor and how does it detect O2? In this report we probed the possible molecular site(s) for sensing of O2 in the heart. Our experiments suggest that a motif closely associated with CaM-binding domains of the C-terminal tail of L-type Ca2+ channel may serve as the oxygen sensor of the heart.
Methods
All experiments were carried out on freshly isolated and enzymatically dispersed ventricular myocytes. Anoxic solutions (O2 replaced by N2) were applied rapidly (<50 ms) to an experimental whole-cell voltage-clamped myocyte, and Ica was activated at 0–10 mV from a holding potential of −50 or −60 mV, chosen so as to fully inactivate INa. To prevent secondary effects arising from prolonged hypoxia that include mitochondrial dysfunction and generation of ROS, the experimental cell was exposed to anoxic solution for no longer than 2 min. Po2 values of 5% were measured within 5–10 s of exposure of myocyte to the N2 equilibrated anoxic solutions. All measurements were bracketed such that each cell served as its control and Student’s t-test analysis was used to compare means.
Results
Figure 1 shows the direct effects of anoxic solutions on ICa measured either in ventricular myocytes or in HEK293 cells expressing the recombinant L-type Ca2+ channel. ICa started to decrease within the first 5 s of exposure to anoxic solutions (26 ± 3% in cardiomyocytes and 35 ± 6% in HEK cells within the first 50 s). Ica was equally suppressed at all voltages tested. To exclude the effects of Ca2+ channel run-down, the suppression of ICa only in the first 50 s was quantified for the statistical analysis.
Figure 1.

Acute anoxia suppresses ICa in cardiomyocytes and HEK293 cells expressing recombinant L-type Ca2+ channel. (A) Time course of Ica suppression in response to extracellular O2 deprivation in cardiac and HEK cells. (B) Quantitative representation of Ica suppression in response to acute extracellular anoxia. (C–D) Representative traces of ICa in cardiac (C) and HEK cells (D) before and within 40 s of extracellular O2 deprivation.
PKA phosphorylation prevents anoxic ICa suppression
PKA phosphorylation is well known to increase ICa amplitude and speed its inactivation rate.
Figure 2 compares the suppressive effects of anoxia on ICa in unphosphorylated and phosphorylated channels. Channel phosphorylation significantly relieved the suppressive effects of anoxia on ICa (average suppression decreases to 9 ± 2% from 27 ± 5% in unphosphorylated channels). In order to determine whether channel resistance to the suppressive effects of anoxia is specifically due to protein kinase A (PKA) phosphorylation, we included 10 μM of H-89, a specific PKA inhibitor in the patch pipette, and reexamined the degree of anoxic suppression in the presence of 200 μM cAMP. In a manner similar to that of unphosphorylated myocytes, H-89 exposure reversed the protective effects of PKA phosphorylation and decreased Ica by 26 ± 5% (N = 4, P = 0.002) in response to anoxia. Since it has already been shown that phosphorylation of serine residues by PKA is enhanced under ischemic conditions,4 it is likely that anoxia enhances PKA activity and in the presence of 200 μM cAMP leads to increased conductance of the channel, opposing the suppressive effects of oxygen removal.
Figure 2.

PKA phosphorylation protects against anoxic suppression of ICa. (A–C) Representative I–V traces (−60 to 80 mV) of cardiac Ca2+ current before and after removal of extracellular O2 in myocytes dialyzed with 0 cAMP (A), 10 μM H-89 + 200 μM cAMP (B) and 200 μM cAMP internal solutions (C). (D–F) Cumulative representation of anoxic Ica suppression under conditions shown in A–C.
Role of calcium svignaling in the anoxic response
Phosphorylation by PKA is known to affect Ca2+ channel gating directly by phosphorylating serine 1928 residue on the C-terminus and indirectly by enhancing Ca2+ release pools by phosphorylating phospholamban (PLB) associated with the Ca-ATPase of the SR.5,6 To examine whether PKA-conferred resistance to anoxia was related to the increased Ca2+ release, cAMP-dialyzed cells were incubated with 1 μM thapsigargin to deplete the SR calcium stores. Thapsigargin incubation did not modify the response of phosphorylated ICa to anoxic solutions, but did increase the current amplitude and delayed the fast component of its inactivation by an average of 10 ms (Fig. 3).
Figure 3.
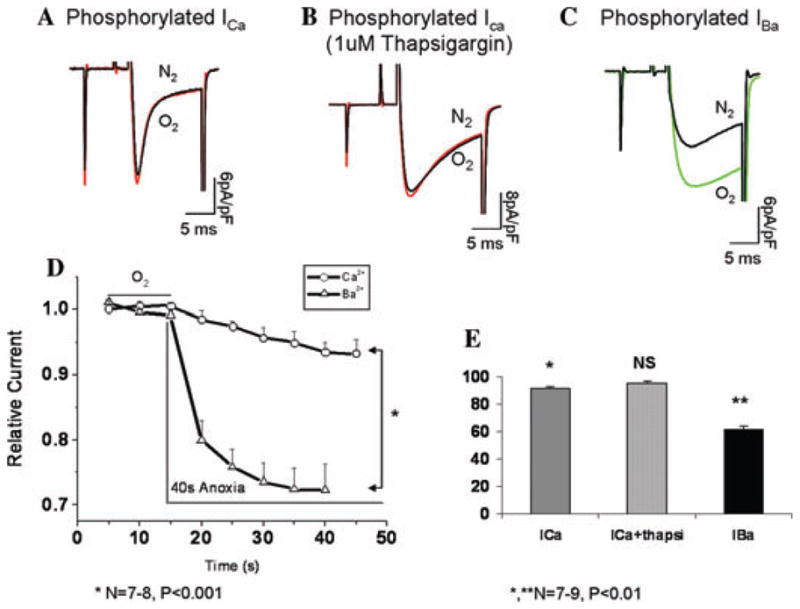
Channel phosphorylation does not protect against the effects of O2 deprivation when Ba2+ is the charge carrier through the channel. (A–C) Representative traces of phosphorylated ICa (A), phosphorylated ICa in presence of 1 uM thapsigargin, and phosphorylated IBa before and after oxygen deprivation in left ventricular myocytes. (D) Time course of current suppression in response to anoxia with Ca2+ and Ba2+ as charge carriers. (E) Quantitative representation of A–C (N = 7–9).
Thus, it appears that direct effects on the channel and not CICR are the critical steps in relieving the suppressive effect of anoxia on the phosphorylated channel. Furthermore, the protective effect of PKA phosphorylation against anoxia appears to be strongly mediated by Ca2+ influx through the channel. This assertion is supported by the finding that IBa is not only more sensitive to anoxic suppression, but is also insensitive to PKA-mediated protection, suggesting that both phosphorylation and the nature of permeating cation through the channel are critical in the O2-sensing mechanism. In phosphorylated channels, the anoxia-induced suppression of IBa occurred within the first 5 s of exposure to oxygen removal, reaching a maximum value of 38% within 40 s. Under the same conditions and time frame, phosphorylated Ca2+ currents were suppressed by only 6% (Fig. 3). Given that the influx of Ba2+ through the channel fails to gate Ca2+ release from the ryanodine receptors, these results suggest that the observed effects of PKA are directly linked to molecular interactions between the channel and the permeating Ca2+.
Where on the channel is oxygen sensed?
The rapid (~5 s) suppressive effects of O2 removal on Ica, the enhancement of this inhibitory effect when Ba2+ is transported through the channel, and the relief of this suppressive effect only when the phosphorylated channel is transporting Ca2+ suggests that a domain of the channel closely associated with permeating Ca2+ may contain the O2 sensor motif. To further identify the site of the sensor, we probed the role of the CaM-binding domain of the C-terminus in O2 sensing. Taking into account that CaM is unable to fully associate with the channel in presence of Ba2+, we hypothesized that sensitivity of IBa to anoxia may result from loss of channel regulation by CaM, which results in exposure of possible O2-sensing residues within the CaM-binding region. Thus, mutation of this region should eliminate the anoxic response of the channel even with Ba2+ as the charge carrier. Because the exact C-terminal site involved in CaM-mediated regulation is a matter of debate and the proposed binding region ranges from 75 residues (1572–1647) down to the 11 residue of the putative IQ motif,7–10 we examined the effect of anoxia on the α1c86 channel, which has an alternative splice variance in exon 40–41 (residues 1572–1651) within the C-tail. The 80 amino acid mutation of α1c86 channel incorporates all the Ca2+/CaM-binding sites (LA, IQ, and K motifs) and lacks Ca2+-dependent inactivation.10,11 When we measured IBa under anoxia in control (α1c77) and the mutant (α1c86) channels, we found that IBa through the α1c86 channel was totally insensitive to deprivation of O2 in sharp contrast to 38% suppression of IBa through the α1c77 channel (Fig. 4). This finding supports the idea that the differential anoxic responses seen between Ca2+ and Ba2+ are due to association of Ca2+/CaM with the C-terminus and that oxygen-sensing ability of the channel is masked by local Ca2+/CaM-dependent regulation.
Figure 4.

Mutation of 80 amino acids in the Ca2+/CaM binding domain of the C-terminus inhibits the effect of anoxia on Ba2+-carried currents through the channel. (A) Representative IBa trace in a HEK cell expressing the alpha1C77 subunit of the L-type channel before and after removal of extracellular oxygen. (B) Amino acid sequence 1572–1651 of native L-type channel C-terminus. (C) Representative trace showing IBa response to anoxia in a HEK cell expressing the mutant alpha1c86 subunit. (D) Amino acid sequence 1572–1651 in the mutant 86 channel C-tail.
Discussion
In this report we have demonstrated a novel oxygen-sensing property of the cardiac Ca2+ channel that is regulated by PKA phosphorylation and involves a sequence of amino acids within the Ca2+-binding region of the C-terminus. Similar to previous reports12,13 our findings also indicate that cardiac ICa is sensitive to extracellular O2 levels. The sensitivity of ICa to oxygen deprivation has been attributed to various mechanisms involving redox modification of specific channel residues,14 mitochondrial ROS-mediated regulation,15 regulation by specific isoforms of PKC,13,17 and involvement of 36 amino acids in the C-terminus region of the alpha1c subunit of the channel.16 Aside from the latter study, most proposed mechanisms involve indirect modulation of the channel by intermediary molecules. In this report, however, we have made several new observations that further support direct-channel O2-sensing mechanisms. To directly study channel effects, we studied Ica modulation within 5–50 s of O2 deprivation to avoid activation of secondary pathways. Using this paradigm, we observed that the anoxic response of Ica occurs as early as 5 s of O2 deprivation and maximizes within 40 s. Furthermore, the channel sensitivity to anoxia was highly regulated by PKA phosphorylation such that intracellular addition of 200 μM cAMP completely masked O2 sensing of the channel, while addition of PKA inhibitor H-89 to 200 μM cAMP-dialyzed cells restored the channel sensitivity to anoxia. The observed PKA-induced anoxic resistance was strongly dependent on Ca2+ entry through the channel, such that anoxic suppression of IBa was insensitive to PKA-mediated protection. Treatment of myocytes with thapsigargin further revealed that Ca2+ entry–dependent inactivation and not calcium release from the SR, CICR, mediates the anoxia-produced resistance with PKA activation. This observation revealed a direct link between Ca2+-dependent inactivation and the channel’s O2-sensing ability. Ca2+-dependent inactivation involves 75 amino acids (residues 1572–1647) spanning the CaM association region of the C-terminus, and indeed we have shown that mutation of these residues abolishes IBa sensitivity to anoxia.
This observation along with that of Fearon and colleagues,16 involving different residues 1787–1822, of calcium channel, indicates that the C-tail of the cardiac Ca2+ channel is directly involved in oxygen sensing of the myocardium during the first few seconds of oxygen deprivation. In addition, the criticality of Ca2+-dependent regulation and phosphorylation by PKA in mediating Ca2+ channel O2-sensing capability under acute anoxia may provide some insight into the pathology of ischemic insult. Considering the complexity of the oxidative process and the involvement of numerous signaling pathways, identification of a direct O2-sensing site on the cardiac Ca2+ channel is an important step toward understanding the pathobiology of oxygen deprivation.
Footnotes
Conflicts of interest
The authors declare no conflicts of interest.
References
- 1.Schneider MF, Chandler WK. Voltage dependent charge movement of skeletal muscle: a possible step in excitation-contraction coupling. Nature. 1973;242:244–246. doi: 10.1038/242244a0. [DOI] [PubMed] [Google Scholar]
- 2.Woo SH, Soldatov NM, Morad M. Modulation of Ca2+ signalling in rat atrial myocytes: possible role of the alpha1C carboxyl terminal. J Physiol. 2003;552:437–447. doi: 10.1113/jphysiol.2003.048330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Morad M, Soldatov N. Calcium channel inactivation: possible role in signal transduction and Ca2+ signaling. Cell Calcium. 2005;38:223–231. doi: 10.1016/j.ceca.2005.06.027. [DOI] [PubMed] [Google Scholar]
- 4.Vittone L, et al. Time course and mechanisms of phosphorylation of phospholamban residues in ischemia-reperfused rat hearts: dissociation of phospholamban phosphorylation pathways. J Mol Cell Cardiol. 2002;34:39–50. doi: 10.1006/jmcc.2001.1488. [DOI] [PubMed] [Google Scholar]
- 5.Yamaoka K, Kameyama M. Regulation of L-type Ca2+ channels in the heart: overview of recent advances. Mol Cell Biochem. 2003;253:3–13. doi: 10.1023/a:1026036931170. [DOI] [PubMed] [Google Scholar]
- 6.Reuter H, Scholz H. The regulation of the calcium conductance of cardiac muscle by adrenaline. J Physiol. 1977;264:49–62. doi: 10.1113/jphysiol.1977.sp011657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Anderson ME. Ca2+-dependent regulation of cardiac L-type Ca2+ channels: is a unifying mechanism at hand? J Mol Cell Cardiol. 2001;33:639–650. doi: 10.1006/jmcc.2000.1354. [DOI] [PubMed] [Google Scholar]
- 8.Fallon JL, et al. Structure of calmodulin bound to the hydrophobic IQ domain of the cardiac Ca(v)1.2 calcium channel. Structure. 2005;13:1881–1886. doi: 10.1016/j.str.2005.09.021. [DOI] [PubMed] [Google Scholar]
- 9.Van Petegem F, Chatelain FC, Minor DL., Jr Insights into voltage-gated calcium channel regulation from the structure of the CaV1.2 IQ domain-Ca2+ calmodulin complex. Nat Struct Mol Biol. 2005;12:1108–1115. doi: 10.1038/nsmb1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Erickson MG, et al. FRET two-hybrid mapping reveals function and location of L-type Ca2+ channel CaM preassociation. Neuron. 2003;39:97–107. doi: 10.1016/s0896-6273(03)00395-7. [DOI] [PubMed] [Google Scholar]
- 11.Soldatov NM, et al. Molecular structures involved in L-type calcium channel inactivation: role of the carboxyl-terminal region encoded by exons 40–42 in alpha1C subunit in the kinetics and Ca2+ dependence of inactivation. J Biol Chem. 1997;272:3560–3566. doi: 10.1074/jbc.272.6.3560. [DOI] [PubMed] [Google Scholar]
- 12.Fearon IM, et al. Hypoxia inhibits the recombinant alpha 1C subunit of the human cardiac L-type Ca2+ channel. J Physiol. 1997;500(Pt 3):551–556. doi: 10.1113/jphysiol.1997.sp022041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hool LC. Hypoxia increases the sensitivity of the L-type Ca(2+) current to beta-adrenergic receptor stimulation via a C2 region-containing protein kinase C isoform. Circ Res. 2000;87:1164–1171. doi: 10.1161/01.res.87.12.1164. [DOI] [PubMed] [Google Scholar]
- 14.Fearon IM, et al. Modulation of recombinant human cardiac L-type Ca2+ channel alpha1C subunits by redox agents and hypoxia. J Physiol. 1999;514(Pt 3):629–637. doi: 10.1111/j.1469-7793.1999.629ad.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hool LC, et al. Role of NAD(P)H oxidase in the regulation of cardiac L-type Ca2+ channel function during acute hypoxia. Cardiovasc Res. 2005;67:624–635. doi: 10.1016/j.cardiores.2005.04.025. [DOI] [PubMed] [Google Scholar]
- 16.Fearon IM, et al. Splice variants reveal the region involved in oxygen sensing by recombinant human L-type Ca(2+) channels. Circ Res. 2000;87:537–539. doi: 10.1161/01.res.87.7.537. [DOI] [PubMed] [Google Scholar]
- 17.Kamp TJ, Hell JW. Regulation of cardiac L-type calcium channels by protein kinase A and protein kinase C. Circ Res. 2000;87:1095–1102. doi: 10.1161/01.res.87.12.1095. [DOI] [PubMed] [Google Scholar]