

Abstract
Aims
To investigate the adaptations of left ventricular function and calcium handling to chronic heart rate reduction with ivabradine in the reperfused heart.
Methods and Results
Rabbits underwent 20-min coronary artery occlusion followed by 3 weeks of reperfusion. Throughout reperfusion, rabbits received ivabradine (10 mg/kg/day) or vehicle (Control). Ivabradine reduced heart rate by about 20% and improved both ejection fraction (+35%) and systolic displacement (+26%) after 3 weeks of treatment. Interestingly, this was associated with a 2-fold increase expression of FKBP12/12.6. There was no difference in the expressions of phospholamban, SERCA2a, calsequestrin, ryanodine, phospho-ryanodine and Na2+/Ca2+ exchanger in the two groups. Infarct scar and vascular density were similar in both groups. Administration of a single intravenous bolus of ivabradine (1 mg/kg) in control rabbits at 3 weeks of reperfusion also significantly improved acutely ejection fraction and systolic displacement.
Conclusion
Chronic heart rate reduction protects the myocardium against ventricular dysfunction induced by myocardial ischaemia followed by 3 weeks of reperfusion. Beyond pure heart rate reduction, ivabradine improves global and regional systolic function of the reperfused heart through a dual mechanism involving a direct mechanical effect and a long-term adaptation in calcium handling, as supported by the increase in FKBP12/12.6 expression.
Keywords: Left ventricular dysfunction, infarction, calcium handling, chronic heart rate reduction, If- channel, ivabradine
INTRODUCTION
Inhibition of the cardiac pacemaker If current by ivabradine induces pure heart rate reduction, exerts anti-ischaemic properties and protects against myocardial stunning and infarction 1–5. Several experimental studies have also demonstrated that chronic treatment with ivabradine improves global left ventricular (LV) function and reduces cardiac collagen accumulation in rats with congestive heart failure induced by permanent coronary artery ligation 6, 7, 8, 9. One possible mechanism is a direct Frank-Starling mechanical effect secondary to an increase in end-diastolic volume with heart rate reduction, as suggested by previous results observed during myocardial stunning 3, 10. An improvement in both calcium transient amplitude and SERCA function could also explain the beneficial effects of ivabradine on left ventricular dysfunction after permanent coronary artery ligation 11. However, the mechanism for this improvement in left ventricular function remains to be determined. Indeed, there is a paucity of results concerning the effects of chronic pure heart rate reduction on calcium handling and especially FKBP12/12.6 expression. This is of importance as FKBP12/12.6 modulates cardiac excitation-contraction coupling through its binding to the ryanodine receptor RyR2 12, 13 and mediates coupled gating observed in RyR2 channel clusters 14. This prevents calcium leak from the sarcoplasmic reticulum, thereby ultimately increasing contractility 12, 13. Finally, it is important to emphasize that these long-term beneficial effects of ivabradine on post-infarction LV dysfunction have been investigated only in models of permanent coronary ligation, i.e., in the absence of coronary artery reperfusion 6–9, 11. Such models do not properly reflect the clinical situation as to date, most of patients with acute myocardial infarction undergo coronary revascularization with angioplasty, thrombolysis surgery or spontaneous recanalization, i.e., they are exposed to LV dysfunction and remodelling resulting from both a loss of contractile tissue and altered salvaged territory.
The aim of the present study was thus to investigate for the first time the effects of chronic heart rate reduction in rabbits undergoing coronary artery occlusion followed by 3 weeks of reperfusion, i.e., a clinically relevant experimental model of post-infarction. We evaluated global and regional LV systolic functions and remodelling in the post-infarction period in the presence or absence of continuous administration of ivabradine. Changes in the proteins involved in calcium handling, including the ryanodine channel (RyR2), SERCA2a, Na+/Ca2+ exchanger (NCX1), calsequestrin as well as modulatory proteins, such as FKBP12/12.6 and phospholamban, were also investigated. Among these, FKBP12/12.6 is of particular importance as its deficiency participates in heart failure 15 whereas its overexpression increases calcium transient amplitude and cell shortening 13, 16.
METHODS
The animal instrumentation and the ensuing experiments were conducted in accordance with French official regulations. All experiments were performed in male New Zealand rabbits (3.0–3.5 kg).
Protocol
As illustrated in figure 1, rabbits were randomly divided into Control and Ivabradine groups receiving saline and ivabradine (10 mg/kg/day during 21 days), respectively.
Figure 1.
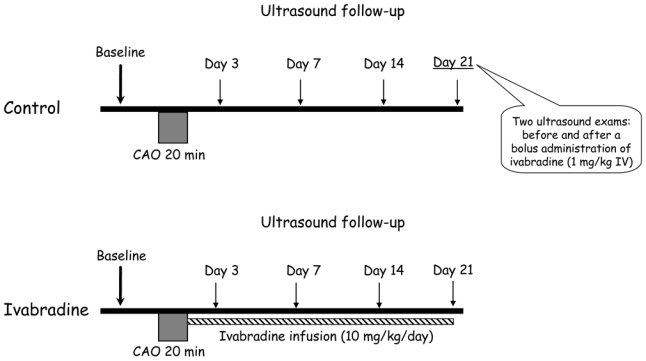
Experimental protocol (CAO, coronary artery occlusion).
Rabbits were anesthetized using zolazepam and tiletamine (20–30 mg/kg each, IV) and then intubated and mechanically ventilated. Anesthesia was maintained by pentobarbital (20–30 mg/kg iv). An external electrocardiogram was recorded. A left thoracotomy was performed under sterile conditions and the pericardium was opened. A 3/0 polypropylene suture was passed beneath a major branch of the left coronary artery. The ends of the ligature were passed through a short segment of propylene tubing to form a snare and coronary artery occlusion (CAO) was induced by pulling the snare through the tubing for 20 min. Reperfusion was subsequently induced by releasing the snare and the chest was then closed in layers. Osmotic pumps (Alzet 2ML4, Durect, Cupertino, CA, USA) were then subcutaneously implanted in order to deliver either saline or ivabradine. Antibiotic (spiramycin, 30 mg/kg/day IM) and analgesics (flunixin meglumine, 1 mg/kg/12h IM; buprenorphine, 0.02 mg/kg/12h SC) were given for 5 and 2 consecutive days following surgery, respectively. Rabbits were submitted to regular echocardiographic evaluations at baseline (1 week before surgery) as well as at days 3, 7, 14 and 21 of reperfusion.
In order to distinguish between the effects of acute heart rate reduction as opposed to those observed during chronic ivabradine administration, all rabbits in the control group received ivabradine at day 21 as a single bolus (1 mg/kg IV). Echocardiographic recordings were performed before and 30 min after this acute administration of ivabradine.
Echocardiography
During each echocardiographic evaluation, rabbits were sedated with diazepam (2–4 mg/kg IP) and a standard transthoracic echocardiography was performed using an ultrasonographic unit instrumented with a 6–12 MHz phased-array transducer Vivid 7 (General Electric Medical System, Waukesha, WI, USA). Ventricular measurements were obtained in the right parasternal location (short-axis view) using a 2D–guided M-mode. Systolic and diastolic LV areas were evaluated and LV volume was estimated using the Simpson approximation. Ejection fraction was calculated and expressed as a percentage. End-systole and end-diastole were determined using ECG as the end of the T wave and the onset of the QRS, respectively.
Using the same ultrasonographic unit, 2D colour tissue Doppler imaging was performed in radial motion with right parasternal ventricular short-axis views. Receiving gain of the grey scale was set to optimize clarity of the endocardial and epicardial boundaries of the left ventricular free wall during each examination. Real-time colour Doppler images were superimposed on the grey scale at frame rate ≥ 200 frames/s. Doppler receiving gain was adjusted to maintain optimal colour of the myocardium. The systolic displacement of the LV free wall was calculated using tissue tracking imaging, i.e., by integration of the velocity signal according to the time. It should be acknowledged that our preliminary experiments demonstrated that the signal-to-noise ratio of strain and strain rate imaging was poor and it did not allow accurate assessment of the regional function compared with tissue tracking.
Measurement of risk area and infarct size
The hearts were collected after 3 weeks of reperfusion. The ascending aorta was cannulated and perfused retrogradely with Alcyan blue (1%) after ligation of the previously occluded artery. The left ventricle was cut into 6 to 8 slices which were weighed and incubated with 1% triphenyltetrazolium chloride (TTC, Sigma, Poole, U.K.) in a pH 7.4 buffer at 37°C. Slices were fixed in 10% formaldehyde and then photographed with a digital camera mounted on a stereomicroscope. The area at risk and the infarcted zones were quantified using a computerized planimetric program (Image J-1.37, National Institute of Health, Bethesda, ML, USA). The area at risk was identified as the non-blue region and was expressed as a percentage of the LV weight. Infarcted area was identified as the TTC-negative tissue and was expressed as a percentage of the area at risk.
Histology and immunohistochemistry
Slices from the middle part of the heart were fixed in formalin and embedded in paraffin for histology and immunohistochemistry. Histological abnormalities were investigated using hematoxylin-eosin-saffron (HES) as previously described 17, 18. The size of infarct scar was also assessed on scanned HES sections using a scanner (Tribvn, Chatillon, France) and Image J program. Anti-CD31 monoclonal antibody (Dako, Trappes, France) was used as an endothelial cell marker in order to identify microcirculatory network. Capillary density was evaluated in the border zone surrounding infarct scar as well as in the myocardium that did not undergo the initial coronary artery occlusion. In each territory, 10 fields were analyzed at X20 magnification: the number of CD-31 labeled capillary sections was counted in each field. Sirius red staining was also performed and used to quantify the interstitial fibrosis that was quantified in the border zone surrounding infarct scar and expressed as a percentage of the field area (10 fields analyzed at X20 magnification): the interstitial fibrosis was measured with image analysis using NIS Elements program (Nikon, Champigny, France).
Western Blots
A second set of rabbits, i.e., 5 control and 4 ivabradine rabbits underwent 20-min coronary artery occlusion followed by 21 days of reperfusion in order to investigate calcium handling. Finally, a third set of animals was investigated, i.e., 4 SHAM-operated and 4 other control rabbits. Myocardial samples were homogenized in ice-cold buffer containing 50mM Tris-HCl (pH 7.4 at 4°C), 150mM NaCl, 1mM EDTA, 5μL/mL protease inhibitor cocktail, 1 mM sodium orthovanadate, 5 mM sodium fluoride and 1 mM sodium Na2β-glycerol phosphate (all from Sigma, L’Isle d’Abeau Chesnes, France) and centrifuged (40,000g for 30 min at 4°C). The protein concentration was determined using a BCA Protein Assay Kit (Pierce, Rockford, IL, USA). Proteins were separated on SDS-polyacrylamide gels and transferred to PVDF membrane (Millipore, Bedford, MA, USA). After blocking with 5% nonfat dry milk in Tris-buffered saline containing 0.1% Tween-20 (TBS-T), membranes were incubated for 1h at room temperature with primary antibodies against cardiac ryanodine receptor (RyR2), FKBP 12/12.6, calsequestrin, SERCA 2a, phospholamban (all from Affinity Bioreagents, Golden, CO, USA), phosphorylated cardiac ryanodine receptor (P-RyR2) (Badrilla, Leeds, UK) or Na2+/Ca2+ exchanger (NCX1) (1/1000, Abcam, Cambridge, UK). The membranes were then washed three times with TBS-T for 10 min and subsequently incubated for 1h with the appropriate secondary antibody conjugated to horseradish peroxidase (Santa Cruz Biotechnology, CA, USA). Bands were visualized with chemiluminescence (ECL Western Blotting Substrate, Pierce, IL, USA), scanned and quantified in a blinded manner using gel analysis software (Image J-1.37, National Institute of Health, Bethesda, MD, USA). Calsequestrin was used as standard for controlling equal loading.
Statistical Analysis
All data were expressed as mean ± SEM. Statistical analysis was performed using animals that successfully underwent the whole protocol. Heart rate and echocardiographic parameters were compared at baseline using an unpaired Student t test. Data collected during reperfusion (day 3 to day 21) were analysed using two-way ANOVA for repeated measures (repeated times nested in treatments) with treatment effect, time effect and time*treatment interaction. Individual comparisons were performed using an unpaired Student t test. Values at day 21 in the control group were also compared before and after the IV bolus of ivabradine using a paired Student t test. Western blot results were analysed using a Mann-Whitney test. Differences were considered significant for P≤0.05 and the tests were two-sided. Statistical analyses were performed using Statview 5.0 (Abacus Concepts, Berkeley, CA, USA).
RESULTS
Out of the 26 rabbits that underwent to the initial surgery, 22 survived coronary artery occlusion and the first hours of reperfusion. Another 5 rabbits (4 Control and 1 Ivabradine) died at days 2, 7, 14, 15 and 20 following surgery. There were therefore 9 and 8 rabbits in the control and ivabradine groups, respectively.
Heart rate
Heart rate was similar between Control and Ivabradine at baseline but was significantly decreased throughout reperfusion in Ivabradine as compared to Control (−20%, −12%, −18% and −20%, at days 3, 7, 14 and 21, respectively) (Table 1).
Table I.
Hemodynamic parameters
| Baseline | Reperfusion |
P value (Reperfusion period) |
||||||
|---|---|---|---|---|---|---|---|---|
| Day 3 | Day 7 | Day 14 | Day 21 | Group effect | Time effect | Group x time | ||
| Heart rate | ||||||||
| Control | 263±11 | 292±10 | 261±10 | 264±8 | 266±14 | P<0.0001 | P=0.0742 | P=0.5930 |
| Ivabradine | 255±8 | 234±10 | 229±13 | 216±9 | 214±9 | |||
| Left ventricular diastolic volume (ml/kg) | ||||||||
| Control | 0.74±0.07 | 0.92±0.07 | 1.10±0.14 | 1.13±0.10 | 1.37±0.11 | P=0.0452 | P=0.0019 | P=0.7138 |
| Ivabradine | 0.82±0.06 | 1.16±0.06 | 1.17±0.06 | 1.29±0.07 | 1.42±0.10 | |||
| Left ventricular systolic volume (ml/kg) | ||||||||
| Control | 0.27±0.02 | 0.42±0.03 | 0.59±0.09 | 0.63±0.08 | 0.79±0.10 | P=0.0276 | P=0.0022 | P=0.2768 |
| Ivabradine | 0.27±0.02 | 0.46±0.05 | 0.45±0.04 | 0.52±0.03 | 0.59±0.05 | |||
Values are mean ± SEM.
In Control, bolus administration of ivabradine at day 21 also induced a significant decrease in heart rate (−24±2%).
Left ventricular function
As shown in Table 1, the reperfusion period was associated with a LV dilatation as shown by the progressive increases in LV diastolic volumes in Control. Heart rate reduction with ivabradine induced significant increases in LV diastolic volumes and also improved LV systolic volumes. As a result (Figure 2), ejection fraction was preserved in Ivabradine with significantly greater values as compared to Control (e.g., a 35% increase at day 21). Similarly, regional function assessed by systolic displacement was increased in ivabradine as compared to Control (e.g., by 26% at Day 21) (Figure 2).
Figure 2.
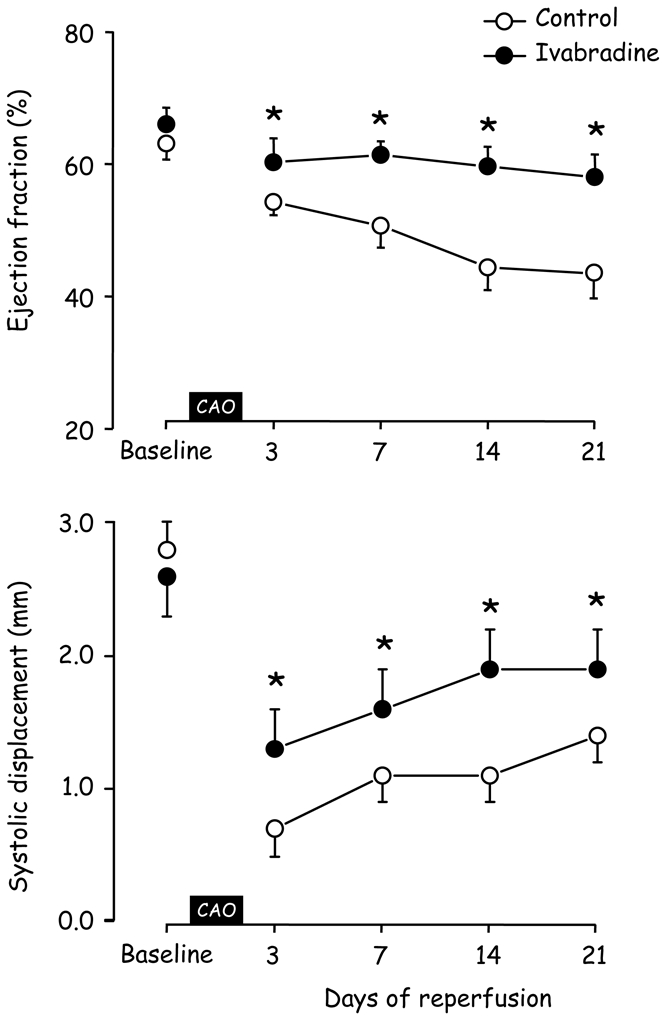
Evolution of ejection fraction and systolic displacement measured at baseline and during the 3 weeks of reperfusion in Control and Ivabradine (CAO, coronary artery occlusion). *, P<0.05 vs Control.
As shown in Figure 3, single bolus administration of ivabradine in Control at day 21 also induced a significant increase in ejection fraction (+26±7%) along with an improved systolic displacement.
Figure 3.
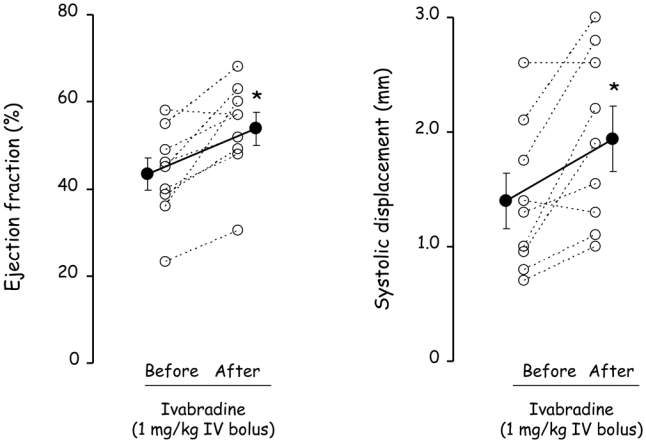
Ejection fraction and systolic displacement measured before and after a single administration of ivabradine (1 mg/kg, IV bolus) in Control at day 21. Open circles represent individual data and closed circles are mean ± SEM. *, P<0.05 vs “Before”.
Western Blots
There was no significant difference in calsequestrin, NCX1, RyR2, P-RyR2, SERCA2a and phospholamban between Ivabradine (n=4) and Control (n=5) (Figure 4). Interestingly as illustrated in Figure 5, FKBP12/12.6 was increased by 96% in Ivabradine as compared to Control.
Figure 4.
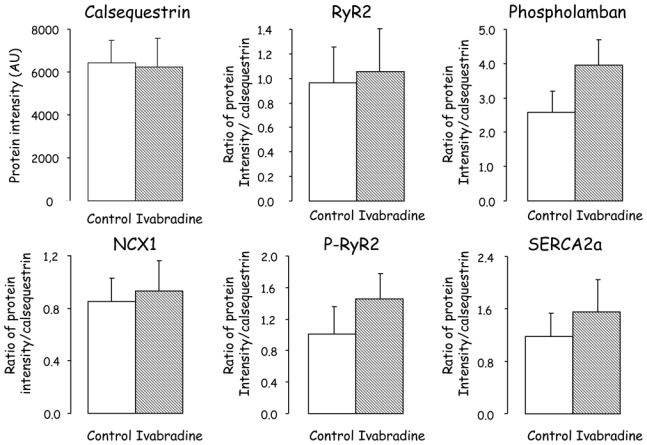
Western blots of calsequestrin, Na2+/Ca2+ exchanger (NCX1), ryanodine (RyR2), phosphorylated ryanodine (P-RyR2), SERCA2a, and phospholamban obtained after 3 weeks of reperfusion in Control and Ivabradine.
Figure 5.
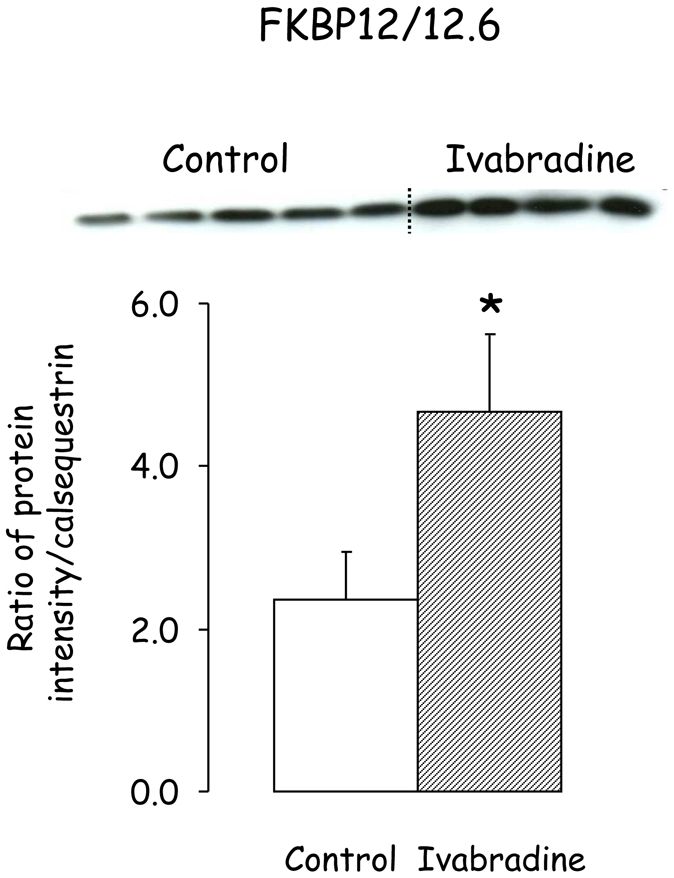
Western blot of FKBP12/12.6 obtained after 3 weeks of reperfusion in Control and Ivabradine. *, P<0.05 vs Control.
In the third set of investigated animals, the comparison between SHAM (n=4) and Control (n=4) revealed significant decreases in RyR2 (1.1±0.1 vs 0.4±0.1, respectively) and SERCA2a (0.8±0.1 vs 0.5±0.1) and increase in NCX1 (1.4±0.1 vs 1.6±0.1). Calsequestrin, phospholamban and FKBP12/12.6 remained similar between the 2 groups (15760±555 vs 17058±1321 Arbitrary Units, 1.2±0.2 vs 1.0±0.1 and 1.0±0.1 vs 0.9±0.1, respectively).
Macroscopic pathology and histology
Analyses were performed in all animals that completed the whole protocol (9 Control and 8 ivabradine). LV weights, area at risks and infarct sizes were similar in Control and Ivabradine (6.6±0.4 vs 6.7±0.4g, 29±2 vs 29±3% and 25±3 vs 28±5%, respectively). Histology also revealed similar infarct scar sizes in Control and Ivabradine.
Concerning fibrosis (Figure 6), Sirius red staining of histological left ventricular slices did not reveal interstitial fibrosis in the remote non ischaemic zone in either group. However, a huge interstitial fibrosis was observed in the peri-infarction border zone with a similar extent in both groups (12.9±2.3 and 9.5±2.6% of the microscopic fields’ area in Ivabradine and Control).
Figure 6.
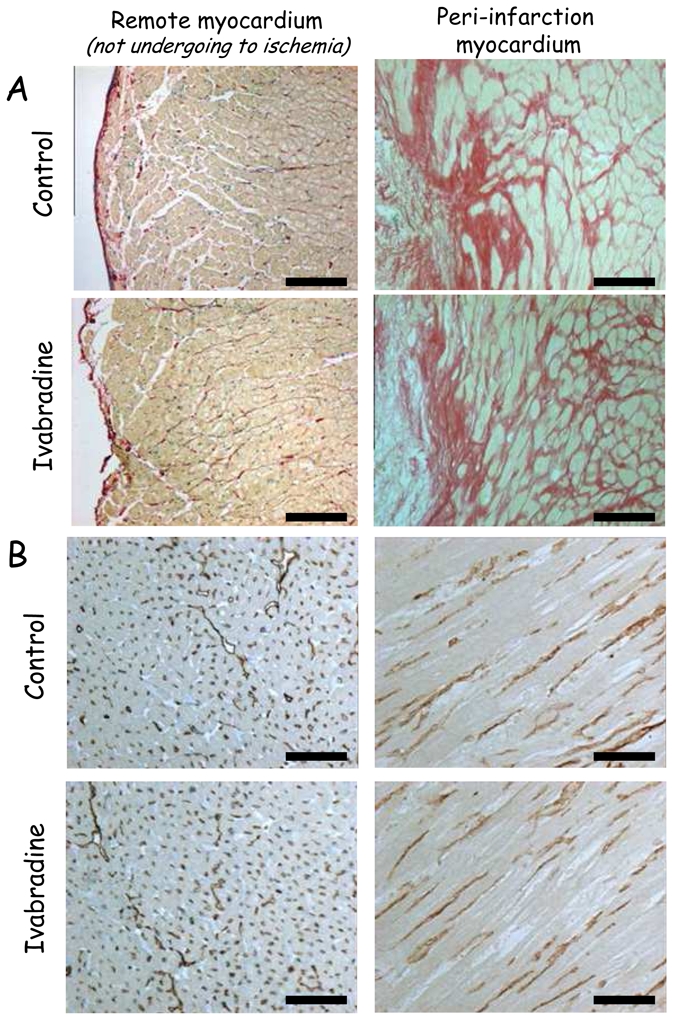
Sirius red staining (Panel A, bar=100μm) and immunohistochemical labelling of the endothelial marker CD31 (Panel B, bar=200μm) of the left ventricle in the remote and peri-infarction territories in Control and Ivabradine.
Microvascular labelling using CD31 immunohistochemistry revealed a dramatic decrease in the microvessel density in the peri-infarction zone vs remote non ischaemic myocardium in both groups. Microvessel densities were similar in the two groups. They averaged 366±33 and 302±30 vessels/μm2 in the peri-infarction territory vs 1021±36 and 944±41 vessels/mm2 in the remote non ischaemic territory in Control and Ivabradine, respectively.
DISCUSSION
The present study provides new informations on the beneficial effects of chronic pure heart rate reduction in the post-infarcted and reperfused myocardium. Our results show that ivabradine-induced heart rate reduction protects against LV dysfunction following myocardial infarction and reperfusion through two potential mechanisms: 1) a pure mechanical effect as demonstrated by the functional consequences of acute administration of ivabradine in the control group and 2) a chronic adaptation of calcium handling in the myocardium as supported by an increase in FKBP12/12.6 expression, which is known to be deficient in failing hearts 15.
The functional results obtained in this study cannot be superimposed on those of previous investigations of ivabradine in a post-infarction situation 6–9, 11. Indeed, these reports involve experimental models of permanent coronary artery ligation which induces profound ventricular remodelling associated with the development of congestive heart failure. In the context of LV dysfunction that occurs after myocardial infarction, we believe that our experimental setting is more clinically relevant because the ischaemic area was reperfused. In our rabbit model of ischaemia-reperfusion, systolic dysfunction and ventricular dilation were related to an infarct scar size averaging 7% of the left ventricle, i.e., dramatically smaller than that observed in models of permanent coronary artery ligation (~40% of the left ventricle in rats) 6, 7. We did not observe any histological evidence of myocardial remodelling in the remote territory not submitted to ischaemia, which means that our model can not be considered as a classical global deleterious remodelling and that our results cannot be superimposed on previous results obtained using ivabradine in congestive heart failure following permanent coronary artery ligation 6, 7. Finally our model leads to a mixed region containing both infarcted and salvaged tissues. Importantly, it also allows investigation of the regional function of the reperfused area.
The observed LV dysfunction could result from myocardial stunning of the salvaged areas with reperfusion, the loss of contractile tissue resulting from infarction and ventricular remodeling. We did not specifically investigate this issue but several aspects could be discussed. Firstly, infarction affected only a small part of the left ventricle (7%) and therefore the direct loss of contractile tissue secondary to necrosis did not induce per se a dramatic decrease in ejection fraction. Secondly, the contribution of LV remodeling could be suggested from the progressive deterioration in ejection fraction of control rabbits that was prevented by chronic administration of ivabradine. Finally, the analysis of systolic displacement within the area at risk clearly revealed myocardial stunning as a progressive return to baseline occurred in control animals. In agreement with our previous results 3, it is reasonable to conclude that ivabradine reduced myocardial stunning in the present study.
In the present study, chronic administration of ivabradine significantly decreased heart rate by an average of 18 %, i.e., similar to that reported in previous studies performed in rats 7, dogs 3, 19 and pigs 4. This effect was constant over the 3 weeks of reperfusion. In these conditions, heart rate reduction preserved ejection fraction as opposed to its progressive alteration observed in Control. These beneficial effects were not related to differences in infarct sizes as a similar amount of myocardial damage was observed in the two groups. This is in apparent contradiction with a previous investigation 4 demonstrating a significant reduction in infarct size with ivabradine. However it must be emphasized that those authors administered ivabradine before ischaemia whereas its administration was started only after reperfusion in the present study, thus avoiding the infarct-limiting effect of ivabradine 4. Furthermore, no significant difference in the size of the area at risk was observed between the two groups of rabbits. It should also be stressed that the improvement in LV function cannot be the consequence of a reduction in fibrosis or an increase in capillary density with ivabradine, as opposed to previous results described in models of heart failure with permanent coronary artery ligation 7, 9. This apparent discrepancy is most likely the consequence of differences in experimental models, i.e., a reperfused vs a ligated territory. A longer period of follow-up over several months as opposed to the 3 weeks of reperfusion in our study might be necessary to reveal such differences.
The beneficial effects of ivabradine on post-infarction systolic function can be explained by two mechanisms. The single bolus administration performed at day 21 of reperfusion in control animals induced a significant increase in systolic displacement and ejection fraction. This suggests that heart rate reduction also improves global and regional systolic function through a direct and acute mechanical effect. This observation is in accordance with previous reports demonstrating that acute heart rate reduction with ivabradine improves systolic wall thickening and reduces post-systolic motion in the stunned myocardium 3, 10. Improved left ventricular filling, a Frank-Starling mechanism associated with a greater diastolic coronary perfusion, is certainly involved in this direct effect.
The second and novel mechanism is related to a chronic change in calcium handling. In our experimental conditions, expressions of SERCA2a and RyR2 were significantly reduced in control as compared to SHAM rabbits and that of NCX1 was increased, without any change in FKBP12/12.6, calsequestrin or phospholamban. Administration of ivabradine did not change the expression of NCX1, RyR2 and its phosphorylated form, SERCA2a, phospholamban or calsequestrin as compared to Control at 3 weeks of reperfusion. However, the important observation is that the expression of FKBP12/12.6 was doubled by chronic administration of ivabradine. Such increase was not only a return to baseline values but rather a significant increase in expression which went beyond normal levels. Although it was not directly demonstrated, it is reasonable to speculate that the increase in FKBP12/12.6 expression observed in the present study participates to the improvement in global and regional cardiac function induced by chronic administration of ivabradine. Indeed in the heart, FKBP12/12.6 modulates cardiac excitation-contraction coupling through its binding to RyR2 12, 13. It mediates coupled gating of RyR2 channel clusters 14 and prevents calcium leak from the sarcoplasmic reticulum, thereby ultimately increasing contractility 12, 13. Interestingly, FKBP12/12.6 deficiency has been shown to participate in heart failure 15 and arrhythmias 20. Conversely, overexpression of FKBP12 in rabbit cardiomyocytes using adenoviral-mediated gene transfer increases systolic calcium transient amplitude secondary to a higher degree of excitation-contraction coupling synchrony 12. A similar increase in FKBP12/12.6 expression has also been previously demonstrated with late preconditioning against myocardial stunning 21. Interestingly and in particular relevance to our results, its overexpression increases calcium transient amplitude and cell shortening 13, 16, and can improve cardiac function in postmyocardial infarction in rats 22. To our knowledge, the present study is the first to describe an increase in FKBP12/12.6 with chronic heart rate reduction. This observation is particularly interesting in the context of the development of pharmacological agents that stabilize the RyR2-FKBP12/12.6 interaction and that are known to improve myocardial function, to inhibit calcium leak from the sarcoplasmic reticulum and to prevent arrhythmias 20, 23.
Regarding the effects of ivabradine in the present study, one can conclude that part of its protective effects are directly related to heart rate reduction per se, as suggested by the improvement in LV function observed after the single bolus administration of ivabradine in the control group at 21 days of reperfusion. Similar acute changes have been previously reported in the stunned myocardium 3. However, as we did not perform experimental studies in which heart rate was corrected with pacing, one cannot rule out the possibility of pleiotropic properties of ivabradine beyond pure heart rate reduction 24. Indeed, a previous report has demonstrated in an established pig model of regional myocardial ischemia-reperfusion that the protective effects of ivabradine on myocardial infarction were only partially reversed by atrial pacing 4. Interestingly during heart failure, If expression is increased in the ventricular myocytes during heart failure 25. Such channel can also contribute to calcium currents 26 and possibly to calcium overload. Therefore part of the observed beneficial effects of ivabradine could be exerted directly on the left ventricular myocardium.
These results may have general implications regarding heart rate reduction after myocardial infarction. It is reasonable to speculate that the effects observed in the present study could be generalized to other bradycardic agents, i.e., mostly β-blockers. However, it should be noted that the potential pleiotropic effects would be restricted to If channel blockers such as ivabradine or UL-FS 49 27. However additional negative inotropic properties of β-blockers might alter the consequences of heart rate reduction. As previously demonstrated during acute stunning, the negative inotropic of β-blockers effect might have deleterious effects on LV function 3. In addition, β-blockers slow the rate of isovolumic relaxation and therefore, impede coronary blood flow whereas this effect is not observed with ivabradine 28, 29. Importantly, β-blockade unmasks α-adrenergic mediated coronary vasocontriction 29. In contrast, ivabradine has no direct effect on coronary vasomotion: it does not unmask α-adrenergic coronary vasoconstriction and also preserves endothelium-mediated vasodilation that is typically observed in large coronary arteries in response to increased shear stress 1.
In conclusion, chronic heart rate reduction with ivabradine protects the myocardium against ventricular dysfunction in a clinically relevant model of myocardial infarction followed by reperfusion. Beyond pure heart rate reduction, chronic administration of ivabradine improves global and regional systolic function of the reperfused heart through a dual mechanism involving a direct mechanical effect and a long-term adaptation in calcium handling, as supported by an increase in FKBP12/12.6 expression. As FKBP12/12.6 deficiency has been reported as one possible trigger of LV dysfunction and heart failure, the present study opens new insights regarding the long-term benefits of chronic heart rate reduction with ivabradine.
Acknowledgments
The present study was supported by Servier. The authors are greatly indebted to Fanny Lidouren and Alain Bizé for their excellent technical assistance. They also wish to thank Drs J. Roussel, P. Gluais, M. Bouly for fruitful discussions.
Footnotes
CONFLICT OF INTEREST
Alain Berdeaux has received honoraria for lectures and has served as consultant to Servier. The other authors have no conflict of interest to declare.
References
- 1.Simon L, Ghaleh B, Puybasset L, Giudicelli JF, Berdeaux A. Coronary and hemodynamic effects of S 16257, a new bradycardic agent, in resting and exercising conscious dogs. J Pharmacol Exp Ther. 1995;275:659–66. [PubMed] [Google Scholar]
- 2.Colin P, Ghaleh B, Monnet X, Su J, Hittinger L, Giudicelli JF, Berdeaux A. Contributions of heart rate and contractility to myocardial oxygen balance during exercise. Am J Physiol. 2003;284:H676–82. doi: 10.1152/ajpheart.00564.2002. [DOI] [PubMed] [Google Scholar]
- 3.Monnet X, Colin P, Ghaleh B, Hittinger L, Giudicelli JF, Berdeaux A. Heart rate reduction during exercise-induced myocardial ischaemia and stunning. Eur Heart J. 2004;25:579–86. doi: 10.1016/j.ehj.2003.10.010. [DOI] [PubMed] [Google Scholar]
- 4.Heusch G, Skyschally A, Gres P, van Caster P, Schilawa D, Schulz R. Improvement of regional myocardial blood flow and function and reduction of infarct size with ivabradine: protection beyond heart rate reduction. Eur Heart J. 2008;29:2265–75. doi: 10.1093/eurheartj/ehn337. [DOI] [PubMed] [Google Scholar]
- 5.Thollon C, Cambarrat C, Vian J, Prost JF, Peglion JL, Vilaine JP. Electrophysiological effects of S 16257, a novel sino-atrial node modulator, on rabbit and guinea-pig cardiac preparations: comparison with UL-FS 49. Br J Pharmacol. 1994;112:37–42. doi: 10.1111/j.1476-5381.1994.tb13025.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dedkov EI, Zheng W, Christensen LP, Weiss RM, Mahlberg-Gaudin F, Tomanek RJ. Preservation of coronary reserve by ivabradine-induced reduction in heart rate in infarcted rats is associated with decrease in perivascular collagen. Am J Physiol Heart Circ Physiol. 2007;293:H590–8. doi: 10.1152/ajpheart.00047.2007. [DOI] [PubMed] [Google Scholar]
- 7.Mulder P, Barbier S, Chagraoui A, Richard V, Henry JP, Lallemand F, Renet S, Lerebours G, Mahlberg-Gaudin F, Thuillez C. Long-term heart rate reduction induced by the selective I(f) current inhibitor ivabradine improves left ventricular function and intrinsic myocardial structure in congestive heart failure. Circulation. 2004;109:1674–9. doi: 10.1161/01.CIR.0000118464.48959.1C. [DOI] [PubMed] [Google Scholar]
- 8.Christensen LP, Zhang RL, Zheng W, Campanelli JJ, Dedkov EI, Weiss RM, Tomanek RJ. Postmyocardial infarction remodeling and coronary reserve: effects of ivabradine and beta blockade therapy. Am J Physiol Heart Circ Physiol. 2009;297:H322–30. doi: 10.1152/ajpheart.01337.2008. [DOI] [PubMed] [Google Scholar]
- 9.Milliez P, Messaoudi S, Nehme J, Rodriguez C, Samuel JL, Delcayre C. Beneficial effects of delayed ivabradine treatment on cardiac anatomical and electrical remodeling in rat severe chronic heart failure. Am J Physiol Heart Circ Physiol. 2009;296:H435–41. doi: 10.1152/ajpheart.00591.2008. [DOI] [PubMed] [Google Scholar]
- 10.Lucats L, Ghaleh B, Monnet X, Colin P, Bize A, Berdeaux A. Conversion of post-systolic wall thickening into ejectional thickening by selective heart rate reduction during myocardial stunning. Eur Heart J. 2007;28:872–9. doi: 10.1093/eurheartj/ehm030. [DOI] [PubMed] [Google Scholar]
- 11.Maczewski M, Mackiewicz U. Effect of metoprolol and ivabradine on left ventricular remodelling and Ca2+ handling in the post-infarction rat heart. Cardiovasc Res. 2008;79:42–51. doi: 10.1093/cvr/cvn057. [DOI] [PubMed] [Google Scholar]
- 12.Loughrey CM, Seidler T, Miller SL, Prestle J, MacEachern KE, Reynolds DF, Hasenfuss G, Smith GL. Over-expression of FK506-binding protein FKBP12.6 alters excitation-contraction coupling in adult rabbit cardiomyocytes. J Physiol. 2004;556:919–34. doi: 10.1113/jphysiol.2003.057166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Prestle J, Janssen PM, Janssen AP, Zeitz O, Lehnart SE, Bruce L, Smith GL, Hasenfuss G. Overexpression of FK506-binding protein FKBP12.6 in cardiomyocytes reduces ryanodine receptor-mediated Ca(2+) leak from the sarcoplasmic reticulum and increases contractility. Circ Res. 2001;88:188–94. doi: 10.1161/01.res.88.2.188. [DOI] [PubMed] [Google Scholar]
- 14.Marx SO, Gaburjakova J, Gaburjakova M, Henrikson C, Ondrias K, Marks AR. Coupled gating between cardiac calcium release channels (ryanodine receptors) Circ Res. 2001;88:1151–8. doi: 10.1161/hh1101.091268. [DOI] [PubMed] [Google Scholar]
- 15.Marx SO, Reiken S, Hisamatsu Y, Jayaraman T, Burkhoff D, Rosemblit N, Marks AR. PKA phosphorylation dissociates FKBP12.6 from the calcium release channel (ryanodine receptor): defective regulation in failing hearts. Cell. 2000;101:365–76. doi: 10.1016/s0092-8674(00)80847-8. [DOI] [PubMed] [Google Scholar]
- 16.Gomez AM, Schwaller B, Porzig H, Vassort G, Niggli E, Egger M. Increased exchange current but normal Ca2+ transport via Na+-Ca2+ exchange during cardiac hypertrophy after myocardial infarction. Circ Res. 2002;91:323–30. doi: 10.1161/01.res.0000031384.55006.db. [DOI] [PubMed] [Google Scholar]
- 17.Tissier R, Souktani R, Bruneval P, Giudicelli JF, Berdeaux A, Ghaleh B. Adenosine A(1)-receptor induced late preconditioning and myocardial infarction: reperfusion duration is critical. Am J Physiol Heart Circ Physiol. 2002;283:H38–43. doi: 10.1152/ajpheart.00866.2001. [DOI] [PubMed] [Google Scholar]
- 18.Tissier R, Waintraub X, Couvreur N, Gervais M, Bruneval P, Mandet C, Zini R, Enriquez B, Berdeaux A, Ghaleh B. Pharmacological postconditioning with the phytoestrogen genistein. J Mol Cell Cardiol. 2007;42:79–87. doi: 10.1016/j.yjmcc.2006.10.007. [DOI] [PubMed] [Google Scholar]
- 19.Lucats L, Ghaleh B, Colin P, Monnet X, Bize A, Berdeaux A. Heart rate reduction by inhibition of If or by beta-blockade has different effects on postsystolic wall thickening. Br J Pharmacol. 2007;150:335–41. doi: 10.1038/sj.bjp.0706996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lehnart SE, Wehrens XH, Laitinen PJ, Reiken SR, Deng SX, Cheng Z, Landry DW, Kontula K, Swan H, Marks AR. Sudden death in familial polymorphic ventricular tachycardia associated with calcium release channel (ryanodine receptor) leak. Circulation. 2004;109:3208–14. doi: 10.1161/01.CIR.0000132472.98675.EC. [DOI] [PubMed] [Google Scholar]
- 21.Lucats L, Vinet L, Bize A, Monnet X, Morin D, Su JB, Rouet-Benzineb P, Cazorla O, Mercadier JJ, Hittinger L, Berdeaux A, Ghaleh B. The inotropic adaptation during late preconditioning against myocardial stunning is associated with an increase in FKBP12.6. Cardiovasc Res. 2007;73:560–7. doi: 10.1016/j.cardiores.2006.11.020. [DOI] [PubMed] [Google Scholar]
- 22.Huang F, Shan J, Reiken S, Wehrens XH, Marks AR. Analysis of calstabin2 (FKBP12.6)-ryanodine receptor interactions: rescue of heart failure by calstabin2 in mice. Proc Natl Acad Sci U S A. 2006;103:3456–61. doi: 10.1073/pnas.0511282103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ito K, Shigematsu S, Sato T, Abe T, Li Y, Arita M. JTV-519, a novel cardioprotective agent, improves the contractile recovery after ischaemia-reperfusion in coronary perfused guinea-pig ventricular muscles. Br J Pharmacol. 2000;130:767–76. doi: 10.1038/sj.bjp.0703373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Heusch G. Pleiotropic action(s) of the bradycardic agent ivabradine: cardiovascular protection beyond heart rate reduction. Br J Pharmacol. 2008;155:970–1. doi: 10.1038/bjp.2008.347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cerbai E, Sartiani L, DePaoli P, Pino R, Maccherini M, Bizzarri F, DiCiolla F, Davoli G, Sani G, Mugelli A. The properties of the pacemaker current I(F)in human ventricular myocytes are modulated by cardiac disease. J Mol Cell Cardiol. 2001;33:441–8. doi: 10.1006/jmcc.2000.1316. [DOI] [PubMed] [Google Scholar]
- 26.Michels G, Brandt MC, Zagidullin N, Khan IF, Larbig R, van Aaken S, Wippermann J, Hoppe UC. Direct evidence for calcium conductance of hyperpolarization-activated cyclic nucleotide-gated channels and human native If at physiological calcium concentrations. Cardiovasc Res. 2008;78:466–75. doi: 10.1093/cvr/cvn032. [DOI] [PubMed] [Google Scholar]
- 27.Guth BD, Heusch G, Seitelberger R, Ross J., Jr Elimination of exercise-induced regional myocardial dysfunction by a bradycardiac agent in dogs with chronic coronary stenosis. Circulation. 1987;75:661–9. doi: 10.1161/01.cir.75.3.661. [DOI] [PubMed] [Google Scholar]
- 28.Colin P, Ghaleh B, Hittinger L, Monnet X, Slama M, Giudicelli JF, Berdeaux A. Differential effects of heart rate reduction and β-blockade on left ventricular relaxation during exercise. Am J Physiol. 2002;282:H672–9. doi: 10.1152/ajpheart.00547.2001. [DOI] [PubMed] [Google Scholar]
- 29.Heusch G. Heart rate in the pathophysiology of coronary blood flow and myocardial ischaemia: benefit from selective bradycardic agents. Br J Pharmacol. 2008;153:1589–601. doi: 10.1038/sj.bjp.0707673. [DOI] [PMC free article] [PubMed] [Google Scholar]