

Abstract
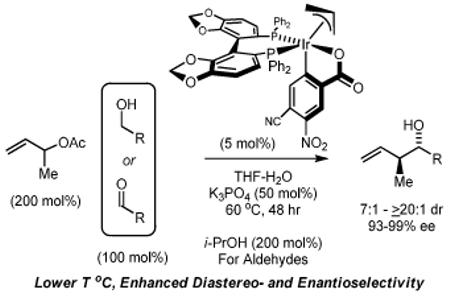 The cyclometallated iridium complex (S)-I derived from [Ir(cod)Cl]2, 4-cyano-3-nitrobenzoic acid, allyl acetate and (S)-SEGPHOS is conveniently isolated by precipitation or through conventional silica gel flash chromatography. This single component precatalyst allows alcohol mediated carbonyl crotylations to be performed at significantly lower temperature, resulting in enhanced levels of anti-diastereo- and enantioselectivity. Most significantly, the chromatographically isolated precatalyst (S)-I enables carbonyl crotylations that are not possible under previously reported conditions involving in situ generation of (S)-I.
The cyclometallated iridium complex (S)-I derived from [Ir(cod)Cl]2, 4-cyano-3-nitrobenzoic acid, allyl acetate and (S)-SEGPHOS is conveniently isolated by precipitation or through conventional silica gel flash chromatography. This single component precatalyst allows alcohol mediated carbonyl crotylations to be performed at significantly lower temperature, resulting in enhanced levels of anti-diastereo- and enantioselectivity. Most significantly, the chromatographically isolated precatalyst (S)-I enables carbonyl crotylations that are not possible under previously reported conditions involving in situ generation of (S)-I.
In the course of developing C-C bond forming hydrogenations and transfer hydrogenations,1 it was found that ortho-cyclometallated iridium C,O-benzoates serve as catalysts for diverse carbonyl allylation processes wherein primary alcohol dehydrogenation triggers reductive generation of allyliridium nucleophiles from allylic carboxylates, thus enabling asymmetric carbonyl allylation directly from the alcohol oxidation level. Under nearly identical conditions, carbonyl allylation is achieved from the aldehyde oxidation level employing isopropanol as terminal reductant.2 Notably, by harnessing the reductive capability of alcohol mediated transfer hydrogenation, asymmetric carbonyl allylation is achieved in the absence of stoichiometric allylmetal reagents or metallic reductants, representing a significant departure from conventional carbonyl allylation protocols.3,4,5
Our initial investigations into stereoselective carbonyl crotylation employing α-methyl allyl acetate as the crotyl donor were achieved using the ortho-cyclometallated iridium C,O-benzoate prepared in situ from [Ir(cod)Cl]2, allyl acetate, 4-cyano-3-nitrobenzoic acid and the chiral phosphine ligand (S)-SEGPHOS.2c,6,7 Although in situ assembly of the catalyst proved convenient and exceptional enantioselectivities typically were observed (>95% ee), only moderate levels of anti-diastereoselectivity were evident (5:1 – 11:1 dr). In subsequent work, conditions for preparation of the discrete ortho-cyclometallated iridium C,O-benzoate precatalyst and its isolation via precipitation were identified.2d Notably, using such single component precatalysts, alcohol mediated carbonyl allylation processes were found to proceed at considerably lower temperatures. This fact prompted the present reinvestigation of alcohol mediated carbonyl crotylation. Here, we report that the ortho-cyclometallated iridium C,O-benzoate derived from [Ir(cod)Cl]2, allyl acetate, 4-cyano-3-nitrobenzoic acid and (S)SEGPHOS, termed (S)-I, is subject to conventional silica gel flash chromatographic purification, and that use of (S)-I purified in this manner enables low temperature (60 °C) alcohol mediated carbonyl crotylation, resulting in enhanced levels of anti-diastereo- and enantioselectivity.
Purification of the ortho-cyclometallated iridium C,O-benzoate precatalyst (S)-I by conventional flash silica gel chromatographically was motivated by the fact that isolation of (S)-I by direct precipitation from the parent reaction mixture failed to exclude small quantities of inorganic byproducts, which contribute to batch dependent variability in catalytic performance. To remove such inorganic impurities, the precatalyst (S)-I was subjected to flash silica gel chromatographically and was found to exhibit excellent chromatographic stability. Thus, using (S)-SEGPHOS as the limiting reagent, the iridium precatalyst (S)-I is obtained in 85% isolated as a yellow powder after purification by silica gel chromatography and subsequent precipitation, as described in the Experimental Section (Scheme 1).
Scheme 1.

Synthesis and isolation of the cyclometallated iridium complex (S)-I.
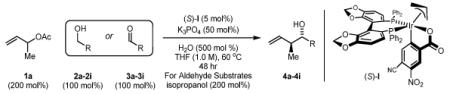
The performance of the chromatographically isolated precatalyst (S)-I was evaluated in carbonyl crotylations from the alcohol or aldehyde oxidation level via transfer hydrogenation, and a comparison with results obtained in reactions involving generation of (S)-I in situ was made.2c Whereas reaction temperatures of 90 °C were necessary using the catalyst generated in situ, the chromatographically isolated precatalyst (S)-I functioned at 60 °C without extending reaction time. A considerable improvement in the level of anti-diastereoselectivity (7:1 – >20:1 dr) was accompanied by a modest but consistent improvement in enantioselectivity (92-99% ee). As demonstrated by the conversion of alcohols 2a-2i to adducts 4a-4i and the conversion of aldehydes 3a-3i to adducts 4a-4i, these favorable trends in selectivity were evident in carbonyl crotylations from the alcohol or aldehyde oxidation level. Most significantly, as illustrated by the formation of adducts 4d and 4h, the chromatographically isolated precatalyst (S)-I enables carbonyl crotylations that are not possible under previously reported conditions involving in situ generation of (S)-I, perhaps due to thermal instability of these particular aldehydes or cross reactivity of these aldehydes with the catalyst at higher temperatures. As established in prior mechanistic studies,2c diastereomeric ratios are generally higher in crotylations conducted from the aldehyde oxidation level, as capture of the kinetically formed (E)-σ-allyliridium via carbonyl addition is more rapid due to higher concentrations of aldehyde (Table 1).
Table 1.
Enhanced levels of anti-diastereo- and enantioselectivity in alcohol mediated carbonyl crotylations using the chromatographically isolated single component iridium catalyst (S)-I.a
|
In Situ Method (ref. 2c) [Ir(cod)Cl]2 (2.5 mol%) (S)-SEGPHOS (5 mol%) 4-CN-3-NO2BzOH (10 mol%) Cs2CO3 (20 mol%) THF (2.0 M), 90 °C, 48 hrs α-Methyl Allyl Acetate (200 mol%) For Aldehyde Substrates isopropanol (200 mol%) |
 |
||
|---|---|---|---|
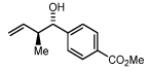
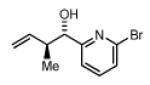
| 2a,3a: R = p-Br-Ph 2d,3d: R = 6-Br-2-Pyr 2g,3g: R = (CH2)2Ph |
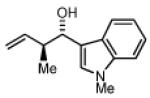
2b,3b: R = p-MeO-Ph 2e,3e: R = 3-Indolyl 2h,3h: R = (CH2)2NHBoc |
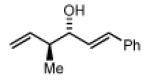
2c,3c: R = p-(CO2Me)-Ph 2f,3f: R = HC=CHPh 2i,3i: R = (CH2)2OPMB |
|
| Oxidation Level |
|
|
|
| Alcohol Aldehyde |
Preformed (S)-I 78% Yield 4a, 16:1 dr, 97% ee 82% Yield 4a, 17:1 dr, 98% ee |
Preformed (S)-I 91% Yield 4b, 10:1 dr, 95% ee 89% Yield 4b, 12:1 dr, 98% ee) |
Preformed (S)-I 78% Yield 4c, 11:1 dr, 98% ee 81% Yield 4c, 13:1 dr, 98% ee |
| Alcohol Aldehyde |
In Situ (S)-I 73% Yield 4a, 8:1 dr, 95% ee 78% Yield 4a, 11:1 dr, 97% ee |
In Situ (S)-I 67% Yield 4b, 5:1 dr, 90% ee 75% Yield 4b, 7:1 dr, 97% ee |
In Situ (S)-I 70% Yield 4c, 7:1 dr, 95% ee 80% Yield 4c, 11:1 dr, 96% ee |
|
|
|
|
| Alcohol Aldehyde |
Preformed (S)-I 50% Yield 4d, 14:1 dr, 98% ee 75% Yield 4d, >20:1 dr, 97% ee |
Preformed (S)-I 75% Yield 4e, 7:1 dr, 98% ee 74% Yield 4e, 10:1 dr, 98% ee |
Preformed (S)-Ib 72% Yield 4f, 10:1 dr, 93% ee 77% Yield 4f, 10:1 dr, 98% ee |
| Alcohol Aldehyde |
In Situ (S)-I no product observed |
In Situ (S)-I 73% Yield 4e, 5:1 dr, 95% ee 78% Yield 4e, 6:1 dr, 97% ee |
In Situ (S)-I 61% Yield 4f, 7:1 dr, 86% ee 66% Yield 4f, 8:1 dr, 98% ee |
|
|
|
|
| Alcohol Aldehyde |
Preformed (S)-I 71% Yield 4g, >20:1 dr, 99% ee 71% Yield 4g, >20:1 dr, 98% ee |
Preformed (S)-Ib 71% Yield 4h, >20:1 dr, 96% ee 66% Yield 4h, >20:1 dr, 99% ee |
Preformed (S)-I 76% Yield 4i, 15:1 dr, 97% ee 76% Yield 4i, >20:1 dr, 99% ee |
| Alcohol Aldehyde |
In Situ (S)-I 69% Yield 4g, 7:1 dr, 98% ee 71% Yield 4g, 11:1 dr, 98% ee |
In Situ (S)-I no product observed |
In Situ (S)-I 73% Yield 4i, 7:1 dr, 95% ee 88% Yield 4i, 7:1dr, 95% ee |
Cited yields are of material isolated by silica gel chromatography. Enantiomeric excess was determined by chiral stationary phase HPLC analysis through comparison of reaction products to racemic diastereomeric mixtures. For assignment of relative and absolute stereochemistry, see reference 2c. See experimental section for further details.
70 °C.
In summary, we report that the iridium precatalyst (S)-I is subject to chromatographic purification, and that the single component precatalyst (S)-I promotes alcohol mediated carbonyl crotylation at significantly lower temperature, resulting in enhanced levels of anti-diastereo- and enantioselectivity.2c Future studies will focus on the development of related C-C bond forming processes that occur in the absence of stoichiometric organometallic reagents, including butadiene mediated carbonyl crotylations from the alcohol oxidation level.7
Experimental Section
Preparation of the Single Component Iridium Precatalyst (S)-I
To a mixture of [Ir(cod)Cl]2 (87.3 mg, 0.13 mmol, 100 mol%), (S)-SEGPHOS (159 mg, 0.26 mmol, 200 mol%), Cs2CO3 (169 mg, 0.52 mmol, 400 mol%), 4-CN-3-NO2BzOH (100 mg, 0.52 mmol, 400 mol%) and allyl acetate (65 mg, 0.65 mmol, 500 mol%) in a sealed tube under an atmosphere of N2 was added THF (2.6 mL, 0.05 M). The reaction mixture was stirred for 30 minutes at ambient temperature and heated for 1.5 hours at 80 °C. Upon cooling to ambient temperature, the reaction mixture was diluted with CH2Cl2 (10 mL), filtered through a celite plug, washed with CH2Cl2 (50 mL) and concentrated in vacuo. The residue was purified by flash chromatography (SiO2, 20% Et2O/CH2Cl2) and concentrated in vacuo. The light yellow gum was dissolved in THF (3 mL). Rapid addition of hexanes (50 mL) to the stirred solution resulted in precipitation of a bright yellow powder, which was collected by gravity filtration. Removal of trace solvents in vacuo delivered (S)-I (228 mg, 0.221 mmol) in 85% yield.
General Procedure for Carbonyl Crotylation from the Alcohol Oxidation Level
An oven-dried sealed tube under an atmosphere of N2 was charged with alcohols 2a-2i, (S)-I (10.3 mg, 0.01 mmol, 5 mol%), K3PO4 (21.5 mg, 0.10 mmol, 50 mol%), THF (0.1 mL, 2.0 M), and H2O (18 μL, 1.0 mmol, 500 mol%). But-3-en-2-yl acetate 1 (45.6 mg, 0.40 mmol, 200 mol%) was added and the mixture was allowed to stir at ambient temperature for 0.5 hr, at which point the reaction vessel was placed in an oil bath at 60 °C and was allowed to stir for 48 hr. The reaction mixture was concentrated in vacuo. Purification of the residue by column chromatography (SiO2; ethyl acetate: hexanes) provided 4a-4i.
General Procedure for Carbonyl Crotylation from the Aldehyde Oxidation Level
An oven-dried sealed tube under an atmosphere of N2 was charged with aldehydes 3a-3i, (S)-I (10.3 mg, 0.01 mmol, 5 mol%), K3PO4 (21.5 mg, 0.10 mmol, 50 mol%), THF (0.1 mL, 2.0 M), isopropanol (31 μL, 0.4 mmol, 200 mol%), and H2O (18 μL, 1.0 mmol, 500 mol%). But-3-en-2-yl acetate 1 (45.6 mg, 0.40 mmol, 200 mol%) was added and the mixture was allowed to stir at ambient temperature for 0.5 hr, at which point the reaction vessel was placed in an oil bath at 60 °C and was allowed to stir for 48 hr. The reaction mixture was concentrated in vacuo. Purification of the residue by column chromatography (SiO2; ethyl acetate: hexanes, 1:20 with 0.1% TEA) provided 4a-4i.
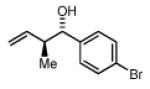
(1S,2S)-1-(4-bromophenyl)-2-methylbut-3-en-1-ol, 4a
The reaction was performed in accordance with the general experimental procedure for carbonyl crotylation from the alcohol oxidation level. Purification of the residue by column chromatography (SiO2; ethyl acetate: hexanes, 1:20 with 0.1% TEA) provided 4a (37.6 mg, 0.156 mmol) as a colorless oil in 78% yield (16:1 dr, 95% ee).
(1S,2S)-1-(4-bromophenyl)-2-methylbut-3-en-1-ol, 4a
The reaction was performed in accordance with the general experimental procedure for carbonyl crotylation from the aldehyde oxidation level. Purification of the residue by column chromatography (SiO2; ethyl acetate: hexanes, 1:20 with 0.1% TEA) provided 4a (39.5 mg, 0.164 mmol) as a colorless oil in 82% yield (17:1 dr, 98% ee).
TLC (SiO2): Rf = 0.4 (ethyl acetate: hexanes, 1:5)
1H NMR (400 MHz, CDCl3): δ 7.46 (d, J = 8.0 Hz, 2H), 7.18 (d, J = 8.0 Hz, 2H), 5.81-5.71 (m, 1H), 5.22-5.16 (m, 2H), 4.32 (d, J = 7.6 Hz, 1H), 2.45-2.37 (m, 1H), 2.20 (br s, 1H), 0.87 (d, J = 6.8 Hz, 3H).
13 C NMR (100 MHz, CDCl3): δ141.4, 140.1, 131.3, 128.6, 121.4, 117.3, 77.1, 46.4, 16.4.
HPLC (Chiralpak AS-H/AS-H column, hexanes:i-PrOH = 98:2, 0.5 mL/min, 230 nm), tminor = 27.9 min, tmajor = 31.8 min.
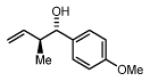
(1S,2S)-1-(4-methoxyphenyl)-2-methylbut-3-en-1-ol, 4b
The reaction was performed in accordance with the general experimental procedure for carbonyl crotylation from the alcohol oxidation level. Purification of the residue by column chromatography (SiO2; ethyl acetate: hexanes, 1:20 with 0.1% TEA) provided 4b (35.0 mg, 0.182 mmol) as a colorless oil in 91% yield (10:1 dr, 94% ee).
(1S,2S)-1-(4-methoxyphenyl)-2-methylbut-3-en-1-ol, 4b
The reaction was performed in accordance with the general experimental procedure for carbonyl crotylation from the aldehyde oxidation level. Purification of the residue by column chromatography (SiO2; ethyl acetate: hexanes, 1:20 with 0.1% TEA) provided 4b (34.2 mg, 0.178 mmol) as a colorless oil in 89% yield (12:1 dr, 98% ee).
TLC (SiO2): Rf = 0.4 (ethyl acetate: hexanes, 1:5).
1H NMR (400 MHz, CDCl3): δ 7.25 (d, J = 8.0 Hz, 2H), 6.87 (d, J = 8.0 Hz, 2H), 5.86-5.76 (m, 1H), 5.23-5.16 (m, 2H), 4.29 (d, J = 8.4 Hz, 1H), 3.80 (s, 3H), 2.48-2.42 (m, 1H), 2.15 (br s, 1H), 0.83 (d, J = 6.8 Hz, 3H).
13C NMR (100 MHz, CDCl3): δ159.3, 141.2, 134.8, 128.2, 117.0, 113.9, 77.7, 55.5, 46.7, 16.8.
HPLC (Chiralpak AD-H/AD-H column, hexanes:i-PrOH = 95:5, 0.5 mL/min, 230 nm), tminor = 41.2 min, tmajor = 48.9 min.
Methyl 4-((1S,2S)-1-hydroxy-2-methylbut-3-enyl)benzoate, 4c
The reaction was performed in accordance with the general experimental procedure for carbonyl crotylation from the alcohol oxidation level. Purification of the residue by column chromatography (SiO2; ethyl acetate: hexanes, 1:20 with 0.1% TEA) provided 4c (34.4 mg, 0.156 mmol) as a colorless oil in 78% yield (11:1 dr, 97% ee).
Methyl 4-((1S,2S)-1-hydroxy-2-methylbut-3-enyl)benzoate, 4c
The reaction was performed in accordance with the general experimental procedure for carbonyl crotylation from the aldehyde oxidation level. Purification of the residue by column chromatography (SiO2; ethyl acetate: hexanes, 1:20 with 0.1% TEA) provided 4c (35.7 mg, 0.162 mmol) as a colorless oil in 81% yield (13:1 dr, 98% ee).
TLC (SiO2): Rf = 0.4 (ethyl acetate: hexanes, 1:5).
1H NMR (400 MHz, CDCl3): δ 7.97 (d, J = 8.0 Hz, 2H), 7.36 (d, J = 8.0 Hz, 2H), 5.79-5.69 (m, 1H), 5.17-5.12 (m, 2H), 4.40 (d, J = 7.2 Hz, 1H), 3.88 (s, 3H), 2.49-2.36 (m, 2H), 0.86 (d, J = 6.8 Hz, 3H).
13C NMR (100 MHz, CDCl3): δ167.2, 147.9, 140.1, 129.7, 129.6, 127.0, 117.5, 77.3, 52.3, 46.5, 16.6.
HPLC (Chiralpak AD-H column, hexanes:i-PrOH = 95:5, 0.5 mL/min, 254 nm), tminor = 27.1 min, tmajor = 32.3 min.
(1S,2S)-1-(6-bromopyridin-2-yl)-2-methylbut-3-en-1-ol, 4d
The reaction was performed in accordance with the general experimental procedure for carbonyl crotylation from the alcohol oxidation level. Purification of the residue by column chromatography (SiO2; ethyl acetate: hexanes, 1:20 with 0.1% TEA) provided 4d (24.2 mg, 0.100 mmol) as a colorless oil in 50% yield (14:1 dr, 98% ee).
(1S,2S)-1-(6-bromopyridin-2-yl)-2-methylbut-3-en-1-ol, 4d
The reaction was performed in accordance with the general experimental procedure for carbonyl crotylation from the aldehyde oxidation level. Purification of the residue by column chromatography (SiO2; ethyl acetate: hexanes, 1:20 with 0.1% TEA) provided 4d (36.3 mg, 0.150 mmol) as a colorless oil in 75% yield (>20:1 dr, 97% ee).
TLC (SiO2): Rf = 0.3 (ethyl acetate: hexanes, 1:5).
1H NMR (400 MHz, CDCl3): δ 7.53 (t, J = 7.6 Hz, 1H), 7.38 (d, J = 7.6 Hz, 1H), 7.25 (d, J = 8.0 Hz, 1H), 5.73 (dt, J = 17.2, 10.4, 1H), 5.10-4.99 (m, 2H), 4.58 (t, J = 5.2 Hz, 1H), 3.28 (d, J = 6.0 Hz, 1H), 2.72-2.64 (m, 1H), 1.06 (d, J = 6.8 Hz, 3H).
13C NMR (100 MHz, CDCl3): δ 162.8, 141.0, 138.7, 138.6, 126.7, 120.0, 116.5, 44.6, 16.1.
HPLC (Chiralcel OD-H column, hexanes:i-PrOH = 95:5, 0.5 mL/min, 210 nm), tmajor = 12.1 min, tminor = 16.8 min.
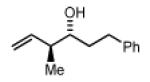
(1S,2S)-2-methyl-1-(1-methyl-1H-indol-3-yl)but-3-en-1-ol, 4e
The reaction was performed in accordance with the general experimental procedure for carbonyl crotylation from the aldehyde oxidation level. Purification of the residue by column chromatography (SiO2; ethyl acetate: hexanes, 1:20 with 0.1% TEA) provided 4e (32.3 mg, 0.150 mmol) as a colorless oil in 75% yield (7:1 dr, 98% ee).
(1S,2S)-2-methyl-1-(1-methyl-1H-indol-3-yl)but-3-en-1-ol, 4e
The reaction was performed in accordance with the general experimental procedure for carbonyl crotylation from the aldehyde oxidation level. Purification of the residue by column chromatography (SiO2; ethyl acetate: hexanes, 1:20 with 0.1% TEA) provided 4e (31.9 mg, 0.148 mmol) as a colorless oil in 74% yield (10:1 dr, 98% ee).
TLC (SiO2): Rf = 0.4 (ethyl acetate: hexanes, 1:5).
1H NMR (400 MHz, CDCl3): δ 7.62 (d, J = 8.0 Hz, 1H), 7.34 (d, J = 8.4 Hz, 1H), 7.26 (t, J = 8.0 Hz, 1H), 7.14 (t, J = 8.0 Hz, 1H), 6.48 (s, 1H), 5.98-5.88 (m, 1H), 5.33-5.25 (m, 2H), 4.60 (d, J = 8.4 Hz, 1H), 3.81 (s, 3H), 2.86-2.78 (m, 1H), 2.22 (br s, 1H), 1.03 (d, J = 6.8 Hz, 3H).
13C NMR (100 MHz, CDCl3): δ 140.7, 140.3, 138.2, 127.5, 121.9, 120.9, 119.8, 117.4, 109.4, 100.8, 71.4, 44.3, 30.7, 17.5.
HPLC (Chiralcel OJ-H column, hexanes:i-PrOH = 93:7, 0.5 mL/min, 254 nm), tmajor = 53.3 min, tminor = 60.0 min
(3R,4S,E)-4-methyl-1-phenylhexa-1,5-dien-3-ol, 4f
The reaction was performed in accordance with the general experimental procedure for carbonyl crotylation from the alcohol oxidation level. Purification of the residue by column chromatography (SiO2; ethyl acetate: hexanes, 1:20 with 0.1% TEA) provided 4f (27.1 mg, 0.144 mmol) as a colorless oil in 72% yield (10:1 dr, 93% ee).
(3R,4S,E)-4-methyl-1-phenylhexa-1,5-dien-3-ol, 4f
The reaction was performed in accordance with the general experimental procedure for carbonyl crotylation from the aldehyde oxidation level. Purification of the residue by column chromatography (SiO2; ethyl acetate: hexanes, 1:20 with 0.1% TEA) provided 4f (29.0 mg, 0.154 mmol) as a colorless oil in 77% yield (10:1 dr, 98% ee).
TLC (SiO2): Rf = 0.3 (ethyl acetate:hexanes, 1:10).
1H NMR (400 MHz, CDCl3): δ 7.41-7.23 (m, 5H), 6.61 (d, J = 16.0 Hz, 1H), 6.21 (dd, J = 16.0, 7.2 Hz, 1H), 5.88-5.78 (m, 1H), 5.21-5.16 (m, 2H), 4.06 (t, J = 6.8 Hz, 1H), 2.41-2.35 (m, 1H), 1.99 (br s, 1H), 1.06 (d, J = 6.8 Hz, 3H).
13C NMR (100 MHz, CDCl3): δ 140.4, 136.9, 132.0, 130.4, 128.8, 127.9, 126.8, 117.0, 76.4, 44.9, 16.3.
HPLC (Chiralpak AS-H/AS-H column, hexanes:i-PrOH = 98:2, 0.5 mL/min, 254 nm), tminor = 26.8 min, tmajor = 31.5 min.
(3R,4S)-4-methyl-1-phenylhex-5-en-3-ol, 4g
The reaction was performed in accordance with the general experimental procedure for carbonyl crotylation from the alcohol oxidation level. Purification of the residue by column chromatography (SiO2; ethyl acetate: hexanes, 1:20 with 0.1% TEA) provided 4g (27.0 mg, 0.142 mmol) as a colorless oil in 71% yield (>20:1 dr, 99% ee).
(3R,4S)-4-methyl-1-phenylhex-5-en-3-ol, 4g
The reaction was performed in accordance with the general experimental procedure for carbonyl crotylation from the aldehyde oxidation level. Purification of the residue by column chromatography (SiO2; ethyl acetate: hexanes, 1:20 with 0.1% TEA) provided 4g (27.0 mg, 0.142 mmol) as a colorless oil in 71% yield (>20:1 dr, 98% ee).
TLC (SiO2): Rf = 0.4 (ethyl acetate: hexanes, 1:5).
1H NMR (400 MHz, CDCl3): δ 7.31-7.17 (m, 5H), 5.80-5.70 (m, 1H), 5.15-5.10 (m, 2H), 3.43-3.40 (m, 1H), 2.89-2.81 (m, 1H), 2.72-2.64 (m, 1H), 2.26-2.20 (m, 1H), 1.89-1.80 (m, 1H), 1.75-1.62 (m, 2H), 1.03 (d, J = 6.8 Hz, 3H).
13C NMR (100 MHz, CDCl3): δ 142.6, 140.4, 128.7, 128.6, 126.0, 116.8, 74.2, 44.6, 36.4, 32.4, 16.5.
HPLC (Chiralcel OD-H column, hexanes:i-PrOH = 97:3, 0.7 mL/min, 254 nm), tminor = 11.5 min, tmajor = 18.7 min.
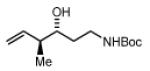
tert-butyl (3R,4S)-3-hydroxy-4-methylhex-5-enylcarbamate, 4h
The reaction was performed in accordance with the general experimental procedure for carbonyl crotylation from the alcohol oxidation level. Purification of the residue by column chromatography (SiO2; ethyl acetate: hexanes, 1:20 with 0.1% TEA) provided 4h (32.6 mg, 0.142 mmol) as a colorless oil in 71% yield (>20:1 dr, 96% ee).
tert-butyl (3R,4S)-3-hydroxy-4-methylhex-5-enylcarbamate, 4h
The reaction was performed in accordance with the general experimental procedure for carbonyl crotylation from the aldehyde oxidation level. Purification of the residue by column chromatography (SiO2; ethyl acetate: hexanes, 1:20 with 0.1% TEA) provided 4h (30.3 mg, 0.142 mmol) as a colorless oil in 66% yield (>20:1 dr, 99% ee).
TLC (SiO2): Rf = 0.5 (ethyl acetate:hexanes, 1:3).
1H NMR (400 MHz, CDCl3): δ 5.76 (dtd, J = 17.2, 10.0, 0.4 Hz, 1H), 5.12-5.05 (m, 2H), 4.91 (br, 1H), 3.48-3.41 (m, 2H), 3.20-3.11 (m, 1H), 2.58 (d, J = 2.8 Hz, 1H), 2.30-2.17 (m, 1H), 1.72-1.64 (m, 1H), 1.54-1.45 (m, 1H), 1.44 (s, 9H), 1.03 (d, J = 6.8 Hz,3H).
13C NMR (100 MHz, CDCl3): δ 156.7, 140.4, 116.1, 79.3, 72.6, 44.2, 37.7, 34.2, 28.4, 16.2.
HPLC Enantiomeric excess was determined by HPLC analysis of the 4-nitrobenzoate derivative of the product (Chiralcel OJ-H column, hexanes:i-PrOH = 98:2, 0.75 mL/min, 254 nm), tminor = 24.4 min, tmajor = 28.1 min.
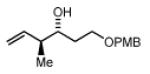
(3R,4S)-1-(4-methoxybenzyloxy)-4-methylhex-5-en-3-ol, 4i
The reaction was performed in accordance with the general experimental procedure for carbonyl crotylation from the alcohol oxidation level. Purification of the residue by column chromatography (SiO2; ethyl acetate: hexanes, 1:20 with 0.1% TEA) provided 4i (38.1 mg, 0.152 mmol) as a colorless oil in 76% yield (15:1 dr, 97% ee).
(3R,4S)-1-(4-methoxybenzyloxy)-4-methylhex-5-en-3-ol, 4i
The reaction was performed in accordance with the general experimental procedure for carbonyl crotylation from the aldehyde oxidation level. Purification of the residue by column chromatography (SiO2; ethyl acetate: hexanes, 1:20 with 0.1% TEA) provided 4i (38.1 mg, 0.152 mmol) as a colorless oil in 76% yield (>20:1 dr, 99% ee).
TLC (SiO2): Rf = 0.5 (ethyl acetate:hexanes, 1:4).
1H NMR (400 MHz, CDCl3): δ 7.26-7.24 (m, 2H), 6.90-6.86 (m, 2H), 5.80 (dt, J = 17.2, 10.0 Hz, 1H), 5.09-5.02 (m, 2H), 4.45 (s, 2H), 3.80 (s, 3H), 3.71-3.61 (m, 3H), 2.79 (d, J = 2.8, 1H), 2.23 (qt, J = 6.8, 0.8 Hz, 1H), 1.74-1.70 (m, 2H), 1.03 (d, J = 6.8 Hz, 3H).
13C NMR (100 MHz, CDCl3): δ 159.2, 140.5, 130.1, 129.3, 115.4, 113.8, 74.3, 73.0, 68.9, 55.3, 44.0, 33.5, 15.8.
HPLC Enantiomeric excess was determined by HPLC analysis of the 4-nitrobenzoate derivative of the product (Chiralcel AD-H column, hexanes:i-PrOH = 98:2, 1.0 mL/min, 210 nm), tminor = 15.4 min, tmajor = 24.8 min.
Supplementary Material
Acknowledgment
Acknowledgment is made to the Robert A. Welch Foundation (F-0038), the NIH-NIGMS (RO1-GM069445), the American Chemical Society Green Chemistry Institute Pharmaceutical Roundtable and the University of Texas at Austin, Center for Green Chemistry and Catalysis. Dr. Yasunori Ino, Dr. Wataru Kuriyama and Dr. Taichiro Touge of Takasago are thanked for the generous donation of SEGPHOS.
Footnotes
Supporting Information Available: Spectral data for adducts 4a-4i, including scanned images of 1H and 13C NMR spectra and chiral stationary phase HPLC traces. This material is available free of charge via the internet at http://pubs.acs.org.
References
- 1.For selected reviews on C-C bond forming hydrogenation and transfer hydrogenation, see: Ngai M-Y, Kong JR, Krische MJ. J. Org. Chem. 2007;72:1063. doi: 10.1021/jo061895m. Skucas E, Ngai M-Y, Komanduri V, Krische MJ. Acc. Chem. Res. 2007;40:1394. doi: 10.1021/ar7001123. Shibahara F, Krische MJ. Chem. Lett. 2008;37:1102. doi: 10.1246/cl.2008.1102. Patman RL, Bower JF, Kim IS, Krische MJ. Aldrichim. Acta. 2008;41:95. Bower JF, Kim IS, Patman RL, Krische MJ. Angew. Chem. Int. Ed. 2009;48:34. doi: 10.1002/anie.200802938. Han SB, Kim IS, Krische MJ. Chem. Commun. 2009:7278. doi: 10.1039/b917243m.
- 2.For enantioselective carbonyl allylation via iridium catalyzed C-C bond forming transfer hydrogenation and related processes, see: Kim IS, Ngai M-Y, Krische MJ. J. Am. Chem. Soc. 2008;130:6340. doi: 10.1021/ja802001b. Kim IS, Ngai M-Y, Krische MJ. J. Am. Chem. Soc. 2008;130:14891. doi: 10.1021/ja805722e. Kim IS, Han S-B, Krische MJ. J. Am. Chem. Soc. 2009;131:2514. doi: 10.1021/ja808857w. Han SB, Kim I-S, Han H, Krische MJ. J. Am. Chem. Soc. 2009;131:6916. doi: 10.1021/ja902437k. Lu Y, Kim I-S, Hassan A, Del Valle DJ, Krische MJ. Angew. Chem., Int. Ed. 2009;48:5018. doi: 10.1002/anie.200901648. Itoh J, Han SB, Krische MJ. Angew. Chem., Int. Ed. 2009;48:6313. doi: 10.1002/anie.200902328. Lu Y, Krische MJ. Org. Lett. 2009;11:3108. doi: 10.1021/ol901096d. Hassan A, Lu Y, Krische MJ. Org. Lett. 2009;11:3112. doi: 10.1021/ol901136w. Bechem B, Patman RL, Hashmi S, Krische MJ. J. Org. Chem. 2010;75:1795. doi: 10.1021/jo902697g. Han SB, Han H, Krische MJ. J. Am. Chem. Soc. 2010;132:1760. doi: 10.1021/ja9097675. Zhang YJ, Yang JH, Kim SH, Krische MJ. J. Am. Chem. Soc. 2010;132:4562. doi: 10.1021/ja100949e. Han SB, Gao X, Krische MJ. J. Am. Chem. Soc. 2010;132:9153. doi: 10.1021/ja103299f. Zbieg JR, Fukuzumi T, Krische MJ. Adv. Synth. Catal. 2010;352:2416. doi: 10.1002/adsc.201000599. (n) For recent applications in total synthesis, see: Harsh P, O'Doherty GA. Tetrahedron. 2009;65:5051. doi: 10.1016/j.tet.2009.03.097. Han SB, Hassan A, Kim I-S, Krische MJ. J. Am. Chem. Soc. 2010;132:15559. doi: 10.1021/ja1082798.
- 3.For selected reviews on enantioselective carbonyl allylation, see: Yamamoto Y, Asao N. Chem. Rev. 1993;93:2207. Ramachandran PV. Aldrichim. Acta. 2002;35:23. Kennedy JWJ, Hall DG. Angew. Chem. Int. Ed. 2003;42:4732. doi: 10.1002/anie.200301632. Denmark SE, Fu J. Chem. Rev. 2003;103:2763. doi: 10.1021/cr020050h. Yu C-M, Youn J, Jung H-K. Bull. Korean Chem. Soc. 2006;27:463. Marek I, Sklute G. Chem. Commun. 2007:1683. doi: 10.1039/b615042j. Hall DG. Synlett. 2007:1644.
- 4.For selected reviews of carbonyl allylation based on the reductive coupling of metallo-π-allyls derived from allylic alcohols, ethers or carboxylates, see: Masuyama Y. Palladium-Catalyzed Carbonyl Allylation via π-Allylpalladium Complexes. In: Liebeskind LS, editor. Advances in Metal-Organic Chemistry. Vol. 3. JAI Press; Greenwich: 1994. pp. 255–303. Tamaru Y. Palladium-Catalyzed Reactions of Allyl and Related Derivatives with Organoelectrophiles. In: Negishi E.-i., Meijere A. de., editors. Handbook of Organopalladium Chemistry for Organic Synthesis. Vol. 2. Wiley; New York: 2002. pp. 1917–1943. Tamaru Y. J. Organomet. Chem. 1999;576:215. Kondo T, Mitsudo T.-a. Curr. Org. Chem. 2002;6:1163. Tamaru Y. Eur. J. Org. Chem. 2005;13:2647. Zanoni G, Pontiroli A, Marchetti A, Vidari G. Eur. J. Org. Chem. 2007;22:3599.
- 5.For selected examples of carbonyl allylation via catalytic Nozaki-Hiyama-Kishi coupling of allylic halides, see: Fürstner A, Shi N. J. Am. Chem. Soc. 1996;118:2533. Bandini M, Cozzi PG, Umani-Ronchi A. Polyhedron. 2000;19:537. McManus HA, Cozzi PG, Guiry PJ. Adv. Synth. Catal. 2006;348:551. Hargaden GC, Müller-Bunz H, Guiry PJ. Eur. J. Org. Chem. 2007:4235. Hargaden GC, O'Sullivan TP, Guiry PJ. Org. Biomol. Chem. 2008;6:562. doi: 10.1039/b715834c.
- 6.Saito T, Yokozawa T, Ishizaki T, Moroi T, Sayo N, Miura T, Kumobayashi H. Adv. Synth. Catal. 2001;343:264. For single crystal x-ray diffraction analysis of a closely related ortho-cyclometallated iridium π-allyl complex modified by (S)-SEGPHOS, see reference 2d. [Google Scholar]
- 7.As illustrated in reference 2m, investigation into the use of butadiene as a crotyl donor in iridium catalyzed C-C bond forming transfer hydrogenation is ongoing.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.