

Abstract
 A nonribosomal peptide synthetase-like enzyme (NRPS325) from Aspergillus terreus was reconstituted in vitro and was shown to synthesize thiopyrazines using an unprecedented mechanism. Substrate promiscuity of NRPS325 towards different amino acids and free thiols was explored to produce > 60 different thiopyrazine compounds.
A nonribosomal peptide synthetase-like enzyme (NRPS325) from Aspergillus terreus was reconstituted in vitro and was shown to synthesize thiopyrazines using an unprecedented mechanism. Substrate promiscuity of NRPS325 towards different amino acids and free thiols was explored to produce > 60 different thiopyrazine compounds.
Filamentous fungi are important microorganisms that biosynthesize structurally diverse and pharmaceutically important natural products.1 Among them, polyketides (such as lovastatin) and nonribosomal peptides (NRPs, such as penicillin) are of particular interest because of their important biological activities.2 The biosynthetic enzymes from fungi that assemble these molecules, such as polyketide synthases (PKSs) and NRP synthetases (NRPSs) are giant megasynthases with a multidomain architecture. Fungal PKSs iteratively utilize a single set of domains to assemble complex metabolites,3 while a typical module of fungal NRPS only catalyzes one round of i) adenylation of an amino acid by the Adenylation (A) domain and transfer to the phosphopantetheinyl (pPant) arm of Thiolation (T) domain; and ii) condensation between the nucleophilic amino group and an electrophilic carbonyl by the Condensation (C) domain.4 Using this biosynthetic logic, fungal NRPS modules can synthesize tetramic acids when fused to a PKS;5 diketopiperizines when paired in tandem;6 and other oligopeptides7 and indole alkaloids8 when multiple modules are connected in an assembly-line like fashion.
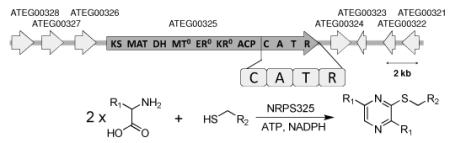
We previously reconstituted the activities of ApdA, an Aspergillus nidulans PKS-NRPS megasynthetase that synthesizes the tetramic acid preaspyridone (Figure 1).9 We demonstrated that the ApdA PKS and NRPS modules of the megasynthetase can be dissected and can interact functionally in trans. When ApdA PKS module and cyclopiazonic acid synthetase10 (CpaS) NRPS module from Aspergillus flavus were combined, a tryptophan-containing preaspyridone analog was obtained. Inspired by this result of combinatorial biosynthesis, we set out to funcationally identify other heterologous NRPS modules from different sequenced fungal species. In this study, we show that serendipitously, the NRPS module (NRPS325) of the only PKS-NRPS megasynthetase (ATEG00325) in Aspergillus terreus can synthesize thiol-substituted pyrazines. The thiopyrazine synthetase activities were independent of any upstream PKS activities. During the preparation of this manuscript, the natural role of ATEG00325 was genetically identified to be involved in the biosynthesis of isoflavipucine and dihydroisoflavipucine (Figure 1),11 highlighting the unexpected biosynthetic potential of this NRPS module unlocked during our genome mining studies.
Figure 1.

Selected tetramic acid natural products.
Bioinformatic analysis predicts that the NRPS325 is capped with a C-terminus NADPH-binding reductase (R) domain that is homologous to those found in equisetin,5b tenellin,5c and CpaS synthetases.10 To analyze the activities of NRPS325 in vitro, we expressed and purified the 158 kDa holo-NRPS module consisting of C-A-T-R from the engineered E. coli BAP1/pKJ75 (Figure S1).12 The relative specificity of the A domain was assessed using a pyrophosphate release assay in the presence of different amino acids (Figure S2). NRPS325 displayed substrate promiscuity towards nearly all the natural aliphatic and aromatic amino acids, including L-Ile, LMet, L-Leu, L-Val, L-Phe, L-Tyr and L-Trp. Towards L-Leu, which is the amino acid that is confirmed by isotopic-labeling experiment to be incorporated into isoflavipucine,11 the A domain displayed kcat of 17.2 ± 2.4 min−1 and Km of 310.0 ± 15.5 μM (Figure S3).
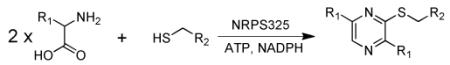
We then attempted to functionally characterize NRPS325 in vitro in the presence of equal molar amounts of ApdAPKS, along with enoyl reductase ApdC, malonyl-CoA, L-Leu, SAM, NADPH and ATP (Figure 2B). The same mixture with the natural AdpA NRPS partner afforded preaspyridone as a single product. In sharp contrast, the reaction extract analyzed by LC-MS revealed an unexpected, conjugated product 1a (Figure 2B, i) (Molecular weight (MWT) = 344) with λmax at 319 nm (Figure S16). Removal of ApdA-PKS had no effect on the product profile, suggesting the turnover of 1a is entirely from the standalone functions of NRPS325 (Figure 2B, ii). Excluding either L-Leu or ATP from the above reaction mixture resulted in no product formation. Formation of 1a was absolutely dependent on NADPH (Figure 2B, iii), which points to a likely role of the R domain in reductive product release. Substitution of L-Leu with L-Val (Figure 2B, iv) in the reaction mixture led to the formation of 1b (MWT = 316) with identical UV absorption. The decrease of 28 mu is consistent with the loss of two methylene groups and is therefore indicative of the product originating from two molecules of the provided amino acid.
Figure 2.

Identification and in vitro reconstitution of NRPS325. (A) Organization of ATEG00325 gene cluster from A. terreus. Highlighting in grey is the 12.4 kb hybrid PKS-NRPS; (B) HPLC analysis (320 nm) of ethyl acetate extracts from the in vitro assays. Each assay contains 10 μM NRPS325 in the presence of different reagents partly shown on the right of the traces. The final concentrations of the different components, when added were: 10 μM ApdA-PKS; 10 μM ApdC; 5 mM thiol; 10 mM amino acid; 2 mM NADPH; 1 mM SAM; and 20 mM ATP. All reactions were performed at room temperature for 12 hours in phosphate buffer (pH = 7.4).
Interestingly, removal of the ~0.5 mM DTT that was present in the buffer used for enzyme storage totally abolished the production of 1a (Figure 2B, v). The role of the thiol in the formation of 1a was investigated by substituting DTT with N-acetylcysteamine (NAC). A different product 2a (MWT = 309) with identical UV spectrum to 1a was formed (Figure 2B, vi). The decrease of 35 mu between 1a and 2a is consistent with the molecular weight difference between DTT and NAC. Taking together, these observations led us to predict that 1a is derived from two molecules of L-Leu and one molecule of DTT.
To obtain a sufficient amount of 1a for structural elucidation, E. coli BAP1/pKJ75 induced with IPTG was supplemented with 5 mM DTT and 10 mM L-Leu. However, the presence of DTT at millimolar concentrations inhibited growth of E. coli. Instead, NAC was supplemented to BAP1/pKJ75 to give 2a at a final titer of 30 mg/L three days after induction. The molecular formula of 2a was determined to be C16H27N3OS by high resolution mass spectrometry ([M+H]+ m/z: observed 310.1951; calculated: 310.1948) (Figure S11). The structure of 2a as shown in Scheme 1 was established by extensive 1D and 2D NMR analysis (Table S3 and Figures S12-15). A comparison of the 1H and 13C NMR spectra of 2a with those of pure NAC and L-Leu revealed that 2a contains an intact NAC molecule and two isobutyl chains. The remaining 1D signals (H-5 at δH 8.25; C-2 at δC 153.9; C-3 at δC 153.6; C-5 at δC 139.0; C-6 at δC 151.7) suggested the presence of a 6-member aromatic heterocycle containing two nitrogen atoms. The structure of 2a was finalized through key 1H-13C and 1H-15N HMBC correlations (shown in Table S3) to be 2-(S-NAC)-3,6-diisobutylpyrazine. NRPS325 was thus confirmed to synthesize a thiopyrazine instead of a diketopiperazine from two molecules of l-Leu and one molecule of NAC, resulting in the NAC being attached to C-2 of the pyrazine via an aryl sulfide linkage.
Scheme 1.

Proposed biosynthetic pathway for 2a.
The proposed mechanism of thiopyrazine synthesis is shown in Scheme 1. Since two molecules of L-Leu must be activated sequentially by a single A domain, we propose NRPS325 must transfer the first leucyl moiety from the T domain to the free thiol of NAC to form leucyl-S-NAC 3 by transthioesterification. In the mixture containing L-Leu, ATP, NAC and NRPS325, a compound with [M+H]+ m/z = 233 can be identified using selected ion monitoring, and its retention time matched precisely to a chemically synthesized 3 (Figure S5). This transthioesterification reaction frees up the T domain to be loaded with the second leucyl group, and is consistent with the thiol-dependent formation of 2a. 3 as an intermediate in the reaction can be further supported through the synthesis of 2a by NRPS325 in the presence of only 3 and NADPH (Figure S6). In this case, the T domain can be loaded with leucyl group through transthioesterification between the pPant arm of holo-NRPS325 and 3, hence no ATP or free l-Leu is required for activation. Attack of the α-amino group of the second leucyl moiety on the carbonyl of 3 then yields a tetrahedral intermediate, which is dehydrated to afford the ethanimidothioate 4. Notably, the formation of the proposed intermediate 4 requires an unusual dehydration step occurred on the tetrahedral intermediate instead of the expected thiol elimination to form the amide bond. This step may be reminiscent of the cyclodehydration reactions catalyzed by the cyclization (Cy) domain in some bacterial NRPSs to afford oxazole and thiazole rings.13 Subsequently, reductive release of aldehyde 5 by the R domain (Scheme 1), followed by imine formation and air oxidation result in the formation of 2a.
To gain further insights into the unusual mechanism of thiopyrazine synthesis as shown in Scheme 1, the roles of the individual domains were probed. First, apo-NRPS325 lost the ability to produce 2a, confirming the dependence on the pPant arm of the loaded T domain. The C domain of NRPSs catalyzes the canonical C-N bond formation, and therefore should play a role in the formation of the tetrahedral intermediate in the proposed pathway.4 The two histidine residues located within the signature motif of C domain HHxxxDG were mutated to Ala separately.14 Both H193A and H194A mutants of NRPS325 were impaired in the synthesis of thiopyrazine compounds, as the apparent rate of 2a formation was <10% of the wild type NRPS325 (Figure S8). A comparable ~13-fold reduction in the rate of 2a synthesis was also observed when we truncated the C domain and used the ATR tridomain (Figure S1) for the synthesis of 2a. Therefore, the proposed nucleophilic attack of the free amine leucyl-S-T on the carbonyl of 3 can take place spontaenously, but its rate can be significantly enhanced in the presence of the C domain. The C domain may achieve rate enhancement through favored binding of the two substrates. Similar observations in reduction in C-N bond formation rates upon C domain inactivation were also observed in the vibriobactin biosynthetic enzymes VibF (C2) and the free-standing VibH C domain.15
Clearly, the proposed reductive release of aldehyde 5 by the R domain is a critical requirement for thiopyrazine formation. The R domain contains the intact catalytic triad Ser-Tyr-Lys and the well-conserved NADPH binding site GxxGxxG found in short chain dehydrogenase/reductase.5b, 10, 11 Mutation of the GxxGxxG motif to GxxAxxA completely abolished the production of 2a (Figure S7). The requirement of NADPH by NRPS325 was also monitored spectrometrically at 340 nm (Figure S9). Consumption of NADPH was only observed in the presence of all the required building blocks, including l-Leu, ATP and NAC. Only background change in absorbance at 340 nm were observed in the absence of the free thiol, thereby excluding the possibility of direct reduction of aminoacyl-S-T by the R domain. The reductive release of 5 observed here is consistent with the proposed role of the R domain in the synthesis of isoflavipucine.11 A number of R domains in fungal PKS-NRPSs were previously identified to lack reductive function and instead catalyze Dieckmann condensation to form tetramic acids.5a, 5b, 5c, 10 As expected, no trace of thiopyrazines were detected when the NRPS modules from ApdA and CpaS were assayed as standalone enzymes. Lastly, formation of 2a by the truncated TR didomain in the presence of 3 and NADPH (Figure S7) suggests that the R domain may also be involved in the dehydration of the tetrahedral intermediate to form the ethanimidothioate, instead of the thermodynamically favored peptide bond. However, this putative new function of the R domain remains unverified.
Given the broad substrate specificities of the A domain in activating different amino acids and NRPS325 in perfoming transthioesterification with different free thiols, we tested the biocatalytic prowess of NRPS325 in the synthesis of a library of trisubstituted pyrazines. Using different combinations of amino acids and free thiol substrates, we showed that 63 different compounds can be synthesized by this single NRPS in good yields (Table S4 and Figure S16-46). A subset of this is shown in Table 1, in which six different amino acids and four different thiols were combinatorially mixed to produce 24 compounds in vitro. Notably, the unnatural amino acids trifluoroleucine (Tfl) and azidohomoalanine (Aha) were each incorporated into pyrazine scaffolds efficiently. The only previously reported microbial source of thiopyrazine is the marine bacterium Sulfitobacter pontiacus (BIO-007).16 Our work here uncovers the hidden capabilities of a fungal NRPS module in the synthesis of thiopyrazines, which is vastly different from the recently confirmed natural role of the parent enzyme. Such unexpected findings further underscore the untapped biocatalytic potential of megasynthetases from natural product biosynthetic pathways.
Table 1.
A subset of selected products that are synthesized by NRPS325.
 | ||||
|---|---|---|---|---|
| R1 | R2 = CH2NHCOCH3 | R2 = CH(OH)CH(OH)CH2SH | R2 = CH2OH | R2 = CH2CH2CH2OH |
| CH2CH(CH3)2 | 2a, 100%a | 1a, 52% | 6a, 60% | 7a, 36% |
| CH(CH3)2 | 2b, 18% | 1b, 14% | 6b, 9% | 7b, 8% |
| CH2CH2SCH3 | 2c, 51% | 1c, 50% | 6c, 39% | 7c, 25% |
| CH2Ph | 2e, 71% | 1e, 29% | 6e, 29% | 7e, 19% |
| CH2CH(CH3)CF3 | 2g, 19% | 1g, 23% | 6g, 29% | 7g, 30% |
| CH2CH2N3 | 2h, 25% | 1h, 36% | 6h, 17% | 7h, 26% |
The percentages in parenthesis indicate the relative yields of thiopyrazines normalized to the yield of 2a.
Supplementary Material
Acknowledgment
We thank Dr. Liansuo Zu, Dr. Xinkai Xie and Dr. Yit-Heng Chooi (UCLA) for insightful discussions; Prof. David Tirrell (Caltech) for a gift of Tfl and Aha; and Prof. Christopher Walsh (HMS) for advice. This research is supported by NIH 1R01GM092217 and by the Packard Foundation
Footnotes
Supporting Information Available: Experimental details, NMR spectra of 2a and biochemical data. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Hoffmeister D, Keller NP. Nat. Prod. Rep. 2007;24:393. doi: 10.1039/b603084j. [DOI] [PubMed] [Google Scholar]
- 2.Tobert JA. Nat. Rev. Drug. Discov. 2003;2:517. doi: 10.1038/nrd1112. [DOI] [PubMed] [Google Scholar]
- 3.Cox RJ. Org. Biomol. Chem. 2007;5:2010. doi: 10.1039/b704420h. [DOI] [PubMed] [Google Scholar]
- 4.Finking R, Marahiel MA. Annu. Rev. Microbiol. 2004;58:453. doi: 10.1146/annurev.micro.58.030603.123615. [DOI] [PubMed] [Google Scholar]
- 5.(a) Bergmann S, Schumann J, Scherlach K, Lange C, Brakhage AA, Hertweck C. Nat. Chem. Biol. 2007;3:213. doi: 10.1038/nchembio869. [DOI] [PubMed] [Google Scholar]; (b) Sims JW, Schmidt EW. J. Am. Chem. Soc. 2008;130:11149. doi: 10.1021/ja803078z. [DOI] [PubMed] [Google Scholar]; (c) Halo LM, Marshall JW, Yakasai AA, Song Z, Butts CP, Crump MP, Heneghan M, Bailey AM, Simpson TJ, Lazarus CM, Cox RJ. ChemBioChem. 2008;9:585. doi: 10.1002/cbic.200700390. [DOI] [PubMed] [Google Scholar]; (d) Seshime Y, Juvvadi PR, Tokuoka M, Koyama Y, Kitamoto K, Ebizuka Y, Fujii I. Bioorg. Med. Chem. Lett. 2009;19:3288. doi: 10.1016/j.bmcl.2009.04.073. [DOI] [PubMed] [Google Scholar]; (e) Maiya S, Grundmann A, Li X, Li SM, Turner G. ChemBioChem. 2007;8:1736. doi: 10.1002/cbic.200700202. [DOI] [PubMed] [Google Scholar]
- 6.(a) Maiya S, Grundmann A, Li SM, Turner G. ChemBioChem. 2006;7:1062. doi: 10.1002/cbic.200600003. [DOI] [PubMed] [Google Scholar]; (b) Balibar CJ, Walsh CT. Biochemistry. 2006;45:15029. doi: 10.1021/bi061845b. [DOI] [PubMed] [Google Scholar]
- 7.(a) Zocher R, Nihira T, Paul E, Madry N, Peeters H, Kleinkauf H, Keller U. Biochemistry. 1986;25:550. doi: 10.1021/bi00351a005. [DOI] [PubMed] [Google Scholar]; (b) Daniel JF, Filho ER. Nat. Prod. Rep. 2007;24:1128. doi: 10.1039/b618086h. [DOI] [PubMed] [Google Scholar]
- 8.(a) Yin WB, Grundmann A, Cheng J, Li SM. J. Biol. Chem. 2009;284:100. doi: 10.1074/jbc.M807606200. [DOI] [PubMed] [Google Scholar]; (b) Ames BD, Walsh CT. Biochemistry. 2010;49:3351. doi: 10.1021/bi100198y. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Gao X, Chooi Y, Ames BD, Wang P, Walsh CT, Tang Y. J. Am. Chem. Soc. 2011 doi: 10.1021/ja1101085. DOI: 10.1021/ja1101085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xu W, Cai X, Jung ME, Tang Y. J. Am. Chem. Soc. 2010;132:13604. doi: 10.1021/ja107084d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu X, Walsh CT. Biochemistry. 2009;48:8746. doi: 10.1021/bi901123r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gressler M, Zaehle C, Scherlach K, Hertweck C, Brock M. Chem. Biol. 2011 doi: 10.1016/j.chembiol.2010.12.011. DOI: 10.1016/j.chembiol.2010.12.011. [DOI] [PubMed] [Google Scholar]
- 12.Pfeifer BA, Admiraal SJ, Gramajo H, Cane DE, Khosla C. Science. 2001;291:1790. doi: 10.1126/science.1058092. [DOI] [PubMed] [Google Scholar]
- 13.(a) Duerfahrt T, Eppelmann K, Muller R, Marahiel MA. Chem. Biol. 2004;11:261. doi: 10.1016/j.chembiol.2004.01.013. [DOI] [PubMed] [Google Scholar]; (b) Kelly WL, Hillson NJ, Walsh CT. Biochemistry. 2005;44:13385. doi: 10.1021/bi051124x. [DOI] [PubMed] [Google Scholar]
- 14.(a) Samel SA, Schoenafinger G, Knappe TA, Marahiel MA, Essen LO. Structure. 2007;15:781. doi: 10.1016/j.str.2007.05.008. [DOI] [PubMed] [Google Scholar]; (b) Bergendahl V, Linne U, Marahiel MA. Eur. J. Biochem. 2002;269:620. doi: 10.1046/j.0014-2956.2001.02691.x. [DOI] [PubMed] [Google Scholar]
- 15.(a) Marshall CG, Hillson NJ, Walsh CT. Biochemistry. 2002;41:244. doi: 10.1021/bi011852u. [DOI] [PubMed] [Google Scholar]; (b) Keating TA, Marshall CG, Walsh CT, Keating AE. Nat. Struct. Biol. 2002;9:522. doi: 10.1038/nsb810. [DOI] [PubMed] [Google Scholar]
- 16.Dickschat JS, Reichenbach H, Wagner-Dobler I, Schulz S. Eur. J. Org. Chem. 2005:4141. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.