

Abstract
A wide variety of stabilized carbanions have been found to participate as nucleophiles in intramolecular Michael-type conjugate additions to in situ-generated nitrosoalkenes to form bridged carbocyclic systems. The vinylnitroso platforms for these cyclizations have been prepared via two key steps involving ring closing metathesis of vinyl chlorides and regioselective conversion of vinyl chlorides to α-chloroketones with sodium hypochlorite in glacial acetic acid/acetone. An alternative approach to preparation of some cyclization substrates has involved use of more reactive enol ethers as precursors to the requisite α-chloroketones. A sulfonamide anion has also been found to be an effective nucleophile in this type of reaction, leading to formation of a 6-azabicyclo[3.2.1]octane.
Introduction and Background
Although vinylnitroso compounds 3 were first postulated as transient, highly reactive intermediates over 100 years ago,1,2 the first isolable member of this class of unstable electron deficient alkenes was not reported until 1960.3 Two primary modes of reactivity have been described for nitrosoalkenes over the years. It has been observed that species such as 3 can act as a heterodiene in [4+2]-cycloadditions with electron rich alkenes to form 5,6-dihydro-4H-1,2-oxazines. Both inter- and intramolecular versions of this process have been effected.2b Vinylnitroso compounds 3 are also known to act as Michael acceptors in conjugate addition reactions with a variety of carbon and heteroatom nucleophiles to afford adducts 4, but little in the way of systematic study of this type of transformation currently exists (Scheme 1).2,4
SCHEME 1.

Formation and Conjugate Additions of Nitrosoalkenes
The two most commonly used methods for producing nitrosoalkenes utilize oximes of α-chloroaldehydes and -ketones. The large preponderance of examples in the literature of generation of vinylnitroso compounds has involved base-promoted dehydrohalogenation of α-halooximes 1.2 However, for effecting conjugate additions, this process usually involves the use of two equivalents of a nucleophile, one of which acts as a base. In the 1980’s, Denmark and coworkers developed a useful and more efficient alternative procedure based on the fluoride-induced cleavage of easily prepared α-chloro-O-silyloximes 2 to give a nitrosoalkene 3.5
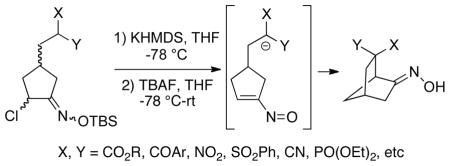
We have recently been engaged in expanding the utility of nitrosoalkenes as enolonium ion equivalents, and in applying this chemistry to total syntheses of some natural product targets.4,6 During the course of these studies, we decided to explore the feasibility of effecting intramolecular conjugate additions of nitrosoalkenes to produce bridged and fused ring systems since such methodology had not previously been reported.7 In a brief preliminary communication,8 we have demonstrated that such cyclizations are indeed easily effected. This initial work was done primarily with a series of substrates using malonate anions as the nucleophile. For example, malonate 5 was first deprotonated, followed by exposure of the α-chloro-O-silyloxime moiety to tetrabutylammonium fluoride in THF to generate the vinylnitroso intermediate 6, which then cyclized to afford bicyclo[2.2.2]octane 7 as a single (E)-oxime isomer (Scheme 2). Cyclization substrates such as 5 were easily prepared via two reactions developed in these laboratories: ring closing metathesis of vinyl chlorides9 and regioselective conversion of vinyl chlorides to α-chloroketones with sodium hypochlorite/glacial acetic acid (vide infra).10 In view of our success in carrying out these cyclizations, we became interested in exploring the possibility of effecting related transformations using a range of soft nucleophiles other than malonates, and these recent studies are the subject of this paper.
SCHEME 2.

Intramolecular Nitrosoalkene Conjugate Addition
Results and Discussion
Our initial experiments were directed towards synthesis of the requisite cyclization substrates for these expanded studies. It was decided to first explore a platform leading to various bicyclo[2.2.1]heptane ring systems, and thus readily prepared iodide 8 was chosen as a precursor (see Supporting Information). Alkylation of malononitrile anion in DMSO with this iodide led to vinyl chloride 9, which upon treatment with sodium hypochlorite in acetone/glacial acetic acid10 afforded α-chloroketone 10 as a mixture of diastereomers (Scheme 3). The O-silyloxime 11 was then prepared (complex mixture of diastereomers and geometrical isomers) from ketone 10 using commercially available O-TBS-hydroxylamine. Using a similar protocol as for the malonate systems,8 malononitrile 11 was deprotonated with potassium hexamethyldisilazide in THF at −78 °C, followed by addition of TBAF and warming slowly to room temperature to generate the transient nitrosoalkene intermediate 12, which then cyclized to bicyclic dinitrile 13 in good yield. Compound 13 is a single oxime isomer we have tentatively assigned the (E)-geometry as shown. It might also be noted that with several of the substrates described in this paper, it was found that yields of cyclization products were slightly improved by using potassium hexamethyldisilazide rather than the corresponding sodium base (Cf Scheme 2).
SCHEME 3.

Formation of a Dicyano Bicyclo[2.2.1]heptane
Since iodide 8 proved rather difficult to manipulate due to its volatility, we therefore opted to prepare other substrates via a modified strategy. Thus, the sequence shown in Scheme 4 was used to synthesize a diverse array of cyclization precursors bearing various groups to be tested as nucleophiles (yields unoptimized; see Supporting Information for detailed experimental procedures). Diene iodide 14, which we have previously described,8 is more easily handled than 8, and could be combined with a number of active methylene compounds to produce alkylated products 15a-g. These dienes all underwent successful ring closing metathesis using the second generation Grubbs catalyst to afford cyclic vinyl chlorides 16a-g.9 Using our sodium hypochlorite methodology,10 vinyl chlorides 16a-g were then converted to the corresponding α-chloroketones 17a-g, respectively, in moderate yields. Finally, these α-chloroketones were transformed into the requisite α-chloro-O-TBS-oxime cyclization substrates 18a-g.
SCHEME 4.

Synthesis of Various Cyclization Substrates
Cyclization studies were then conducted on the 1,3-diketone 18a and the bis-phenylsulfone 18b (Scheme 5). Using our standard protocol, O-silyloxime substrates 18a and 18b were first deprotonated using potassium hexamethyldisilazide, followed by treatment with TBAF to generate the vinylnitroso intermediate, leading to bicyclo[2.2.1]heptanes 19 and 20, respectively. Both of these compounds were found to be single oxime geometric isomers, once again presumed to have the (E)-stereochemistry.
SCHEME 5.

Formation of Bicyclo[2.2.1]heptanes
Another system to be examined was the phenylsulfone methyl ester 18d, which upon cyclization under the usual reaction conditions led to a single stereoisomeric product 22a in high yield (Scheme 6). Since we could not establish the configuration of 22a directly by NMR methods due to peak overlap, we attempted to effect a Beckmann rearrangement to afford the azabicyclo[3.2.1]octane 23 which we hoped might be more easily analyzed. However, treatment of 22a with tosyl chloride/triethylamine/DMAP11 instead led to the stable crystalline oxime tosylate 22b. 2D NMR studies on tosylate 22b established its configuration to be as shown, primarily due to an nOe enhancement between the methyl group of the ester and an aromatic proton ortho to the sulfonyl group of the tosyl moiety. In addition, we observe an upfield shift for the 1H NMR resonances of the O-methyl group of 22a and 22b (0.17 and 0.49 ppm, respectively) relative to the corresponding uncyclized precursors 15d-18d. Further confirmation of this structure, along with the (E)-geometry of the oxime, came from an X-ray analysis of 22b (see Supporting Information). In the crystal, the methyl ester moiety of 22b is disposed above the face of the aromatic ring of the tosyl group, thus lending support to the NMR upfield shift argument. Thus, the cyclization apparently occurs via an ester enolate conformation like 21. At this point, it is not clear if the preference for formation of the product having the ester group syn to the oxime is a result of steric factors, or if there is some type of dipole effect controlling the stereochemical outcome.
SCHEME 6.

Stereoselective Formation of Oxime 22a
Several other cyclizations involving unsymmetrical substrates are shown in Scheme 7. The nitro ester 18e also was stereoselective and provided a single cyclization product 24. Surprisingly, treatment of oxime 24 with tosyl choride, as was done with oxime 22a, gave no reaction even upon heating. However, based upon the fact that the -OCH2- group of the ethyl ester of 24 is not shifted upfield relative to precursors 15e-18e, we have tentatively assigned the configuration of this compound as having the oxime and ester anti as shown.
SCHEME 7.

Cyclizations of Additional Unsymmetrical Substrates
The β-ketoester substrate 18c also could be successfully cyclized, but in this case an inseparable 2.2:1 mixture of epimeric products 25 was produced. Similarly, phosphonate ester 18f also cyclized in good yield, but with this system an inseparable 1:1 mixture of epimers 26 was obtained. Finally, with phenylsulfone nitrile substrate 18g a 10:1 epimeric mixture of bicycloheptanes 27 was generated, which again was inseparable by chromatography. In each of the above three examples, the products all appeared to be single oxime geometric isomers assumed to be (E), although the configuration of the products at the quaternary carbon was not determined due to our inability to obtain pure compounds. At present, it is unclear why there is so much variation in the stereochemical outcomes of these cyclizations with different functional groups.
One difficulty encountered during the course of these studies is that the hypochlorite-induced halogenation of vinyl chlorides often leads to a variety of undesired side products, including competitive chlorination of the active methine portion of the substrate, thereby lowering the yield of the desired α-chloroketones (Cf Scheme 4).10b It is believed that some of these problems are probably due in part to the electron deficient nature of the olefinic double bond of vinyl chlorides, thus leading to relatively slow conversion to the respective α-chloroketones. We therefore considered the possibility of using more electron rich enol ethers as alternative precursors to α-chloroketones in order to accelerate the halogenation and possibly obviate some of these problems. Moreover, we anticipated that such a strategy should provide greater flexibility in preparation of various precursors for the nitrosoalkene conjugate addition methodology.
To test this idea, the known enol ether alcohol 28, prepared via the Diels-Alder reaction of 2-methoxybutadiene12 with ethyl acrylate followed by ester reduction with lithium aluminum hydride,13 was converted to tosylate 29 (Scheme 8). Alkylation of methyl α-phenylsulfonylacetate with this tosylate then led to 30 as a mixture of diastereomers in 53% yield based on alcohol 28. Using the halogenation conditions described by Cha et al.,14 enol ether 30 was transformed to α-chloroketone 31 with N-chlorosuccinimide in 64% yield. Conversion of this α-chloroketone to O-TBS-oxime 32, followed by cyclization under the usual conditions afforded the bicyclo[3.2.1]octane 33a as a single diastereomer. The configuration and (E)-oxime geometry of this product was established by conversion to the oxime tosylate 33b, which was analyzed by X-ray crystallography. Therefore, the outcome of this cyclization is the same as that observed in the bicyclo[2.2.1]heptane series 22 where the ester group is syn to the oxime in the product (Cf. Scheme 6).
SCHEME 8.

Substrate Preparation via an Enol Ether and Stereoselective Cyclization to a Bicyclo[3.2.1]octane
We have also explored the possibility of effecting intramolecular nitrosoalkene conjugate additions using nitrogen nucleophiles to form bridged azaheterocycles. Thus, Diels-Alder cycloaddition of 2-methoxybutadiene12 with acrylonitrile as reported by Katritzky and coworkers15 gave an 8:1 mixture of the desired adduct 34 along with its regioisomer which was not easily separated (Scheme 9). Therefore, the mixture was reduced to the corresponding amine 35,16 followed by treatment of the crude material with tosyl chloride to afford sulfonamide 36 which could be purified by chromatography. We were pleased to find that under the Cha reaction conditions,14 enol ether 36 was cleanly transformed to α-chloroketone 37 in high yield. It should be added that we have observed that halogenation of some vinyl chloride systems related to enol ether 36 using our sodium hypochlorite method led to significantly lower yields of the respective α-chloroketones, and also produced varying amounts of products resulting from N-chlorination of the sulfonamide.17
SCHEME 9.

Cyclization with a Sulfonamide Nucleophile
Subsequent reaction of compound 37 with O-TBS-hydroxylamine produced the required α-chloro-O-silyloxime 38. Interestingly, it was discovered that TBAF is a sufficiently strong base to deprotonate the sulfonamide for the cyclization onto the derived nitrosoalkene. Thus, exposure of substrate 38 to two equivalents of TBAF in acetonitrile at 0 °C led directly to formation of the desired 6-azabicyclo[3.2.1]octane in good total yield as a 5:1 oxime geometric isomer mixture. The major isomer of 39 was determined by X-ray crystallography to have the (E)-geometry as shown. For further characterization, the oxime mixture could be oxidatively cleaved to afford the bridged ketone 40 in 76% yield.18
Conclusion
In summary, we have found that intramolecular conjugate additions of nitrosoalkenes, generated in situ via the Denmark α-chloro-O-silyloxime methodology,5 can be effected using a wide variety of soft carbon nucleophiles to produce bridged ring systems.19 Some of these cyclizations were found to proceed with high stereoselectivity, although the factors controlling the product configurations are not yet well understood. Similarly, use of a sulfonamide nucleophile leads to the formation of a bridged aza-heterocyclic ring system.20 We are currently investigating further extensions and variations of this methodology, along with applications to the synthesis of some natural products.
Experimental Section
2-(3-Chlorocyclopent-3-enylmethyl)-malononitrile (9)
A suspension of NaH (60% dispersion in mineral oil, 180 mg, 4.5 mmol) in DMSO (2 mL) was heated at 70 °C for 1 h and cooled to rt. To the resulting mixture was added a solution of malononitrile (298 mg, 4.5 mmol) in DMSO (2 mL) at rt. After stirring the mixture for 5 min, a solution of iodide 8 (545 mg, 2.25 mmol) in DMSO (1 mL) was added, and the mixture was stirred for 12 h at 65 °C. Saturated aqueous NH4Cl and ether were added. The organic layer was separated and the aqueous layer was extracted with ether. The combined ether layer was washed with water and brine. The organic layer was dried over Na2SO4 and the solvent was removed under reduced pressure. The residue was purified by flash chromatography (30% ether/pentane) to afford the vinyl chloride 6 as a colorless oil (203 mg, 50%). 1H NMR (300 MHz, CDCl3) δ 5.47-5.46 (m, 1H), 3.59 (t, J = 7.4 Hz, 1H), 2.67-2.46 (m, 3H), 2.17-2.09 (m, 1H), 2.03-1.91 (m, 3H); 13C NMR (75 MHz, CDCl3) δ 130.2, 124.5, 112.1, 42.1, 36.5, 35.8, 34.4, 20.9; no molecular ion was detected by HRMS.
2-(3-Chloro-4-oxocyclopentylmethyl)-malononitrile (10)
To a solution of vinyl chloride 9 (39 mg, 0.22 mmol), acetone (2.1 mL) and glacial acetic acid (0.9 mL) at 0 °C was added dropwise sodium hypochlorite (0.18 mL of 10% solution, 0.22 mmol) via syringe. The reaction mixture was stirred at 0 °C for 30 min and quenched by addition of saturated aqueous NaHCO3 solution. The mixture was then extracted with dichloromethane. The combined organic layers were washed with brine and dried over Na2SO4. The solvent was removed under reduced pressure and the residue was purified by flash chromatography (80% ether/pentane) affording the α-chloroketone 10 as a clear oil (20 mg, 47%). 1H NMR (300 MHz, CDCl3, diastereomer mixture) δ 4.21-4.11 (m, 1H), 3.79-3.72 (m, 1H), 2.88-2.66 (m, 2H), 2.48-2.40 (m, 1H), 2.23-2.15 (m, 2H), 2.12-1.89 (m, 2H); 13C NMR (75 MHz, CDCl3) δ 207.3, 207.1, 112.3, 112.2, 58.3, 56.6, 41.4, 41.3, 39.5, 39.2, 36.3, 36.1, 31.9, 31.0, 21.8, 21.6; HRMS-AP [M + NH4]+ calcd for C9H13ClN3O, 214.0747; found, 214.0748.
2-(3-Chloro-4-tert-butyldimethylsilyloxyiminocyclopentylmethyl)-malononitrile (11)
To a solution of α-chloroketone 10 (17 mg, 0.086 mmol) in dichloromethane (3 mL) were added O-(tert-butyldimethylsilyl)-hydroxylamine (16.5 mg, 0.11 mmol), 4Å molecular sieves (crushed), and a catalytic amount of PPTS. The mixture was stirred at rt for 24 h and then filtered through a pad of Celite. The solvent was removed under reduced pressure and the residue was purified by flash chromatography (30% ether/hexanes) to afford the α-chloro-O-silylketoxime 10 as a colorless oil (18 mg, 64%). 1H NMR (300 MHz, CDCl3, complex mixture of diastereomers including oxime geometric isomers) δ 4.88-4.54 (m, 1H), 3.63-3.57 (m, 1H), 2.86-2.42 (m, 2H), 2.24-1.89 (m, 4H), 1.76-1.61 (m, 1H), 0.77-0.70 (m, 9H), 0.01-0.00 (m, 6H); 13C NMR (75 MHz, CDCl3) δ 165.5, 165.0, 164.5, 164.1, 112.6, 112.5, 58.4, 56.0, 50.7, 42.3, 41.2, 36.7, 36.1, 35.8, 33.4, 32.1, 31.9, 26.4, 26.3, 26.3, 22.0, 21.9, 18.5, 18.5, −4.8, −4.8; HRMS-AP [M + H]+ calcd for C15H25ClN3OSi, 326.1455; found, 326.1459.
Typical Intramolecular Nitrosoalkene Cyclization: 6-Hydroxyiminobicyclo[2.2.1]heptane-2,2-dicarbonitrile (13)
To a stirred solution of O-silyloxime 11 (16.3 mg, 0.05 mmol) in THF (3 mL) at −78 °C was added dropwise KHMDS (0.5 M in toluene, 0.12 mL, 0.06 mmol) via syringe, and the reaction mixture was stirred at −78 °C for 1 h. TBAF (1 M in THF, 0.05 mL, 0.05 mmol) was added dropwise via syringe, and the mixture was warmed to rt over 3 h. Saturated aqueous NH4Cl was then added. The mixture was extracted with ether and the combined extracts were dried over Na2SO4. The solvent was removed under reduced pressure and the residue was purified by flash chromatography (75% ether/pentane) to afford the bridged bicyclic oxime 13 as a single geometric isomer (7.1 mg, 82%). 1H NMR (300 MHz, CDCl3) δ 7.94 (br s, 1H), 3.37 (d, J = 1.2 Hz, 1H), 2.70-2.66 (m, 1H), 2.47-2.31 (m, 2H), 2.14 (dd, J = 3.3, 17.9 Hz, 1H), 2.02 (dd, J = 2.5, 13.4 Hz, 1H), 1.90-1.84 (m, 1H), 1.79-1.67 (m, 1H); 13C NMR (75 MHz, CDCl3) δ 156.7, 114.5, 113.5, 50.9, 42.2, 36.9, 34.4, 32.8, 32.5; HRMS-EI [M]+ calcd for C9H9N3O, 175.0746; found, 175.0759.
6,6-Dibenzoylbicyclo[2.2.1]heptan-2-one Oxime (19)
78% (8 mg, prepared from 15 mg (0.031 mmol) of 18a); 1H NMR (300 MHz, CDCl3) δ 7.80-7.76 (m, 2H), 7.71-7.68 (m, 2H), 7.35-7.28 (m, 2H), 7.23-7.17 (m, 4H), 6.60 (br s, 1H), 3.89 (d, J = 0.9 Hz, 1H), 3.08 (dd, J =2.6, 12.8 Hz, 1H), 2.53 (s, 1H), 2.33-2.16 (m, 2H), 1.93-1.85 (m, 2H), 1.64-1.60 (m, 1H); 13C NMR (75 MHz, CDCl3) δ 197.9, 197.2, 163.4, 137.2, 135.3, 133.7, 133.5, 129.7, 129.5, 129.0, 128.9, 70.1, 51.2, 40.5, 38.6, 35.8, 33.6; HRMS-ES [M+H]+ calcd for C21H20NO3, 334.1443; found, 334.1447.
6,6-Bis-benzenesulfonylbicyclo[2.2.1]heptan-2-one Oxime (20)
61% (5.7 mg, prepared from 13 mg (0.023 mmol) of 18b); 1H NMR (300 MHz, CDCl3) δ 8.03-8.00 (m, 2H), 7.64-7.61 (m, 2H), 7.56-7.49 (m, 2H), 7.44-7.34 (m, 4H), 6.98 (br s, 1H), 3.25 (s, 1H), 2.66-2.60 (m, 2H), 2.45-2.34 (m, 3H), 2.24 (dd, J = 3.5, 17.3 Hz, 1H), 1.47-1.34 (m, 1H); 13C NMR (75 MHz, CDCl3) δ 162.3, 139.6, 137.1, 135.0, 134.6, 131.0, 131.0, 129.3, 129.0, 95.5, 50.8, 40.8, 35.9, 35.1, 33.5; HRMS-ES [M+H]+ calcd for C19H20NO5S2, 406.0783; found, 406.0774.
2-Benzenesulfonyl-6-hydroxyiminobicyclo[2.2.1]heptane-2-carboxylic Acid Methyl Ester (22a)
96% (17 mg, prepared from 26 mg (0.055 mmol) of 18d); 1H NMR (300 MHz, CDCl3) δ 7.71 (d, J = 7.4 Hz, 2H), 7.56 (t, J = 7.4 Hz, 1H), 7.43 (t, J = 7.6 Hz, 2H), 3.41 (s, 3H), 3.26 (s, 1H), 2.76-2.65 (m, 2H), 2.46-2.41 (m, 1H), 2.33-2.15 (m, 2H), 1.98 (dd, J = 3.4, 17.8 Hz, 1H), 1.44 (d, J = 10.7 Hz, 1H); 13C NMR (75 MHz, CDCl3) δ 167.7, 161.9, 137.7, 134.7, 130.1, 129.3, 79.2, 53.3, 50.4, 38.2, 36.0, 34.0, 34.0; HRMS-ES [M + H]+ calcd for C15H18NO5S, 324.0906; found, 324.0901.
6-Hydroxyimino-2-nitrobicyclo[2.2.1]heptane-2-carboxylic Acid Ethyl Ester (24)
In this case, the reaction mixture was warmed to 0 °C after the TBAF addition. 70% (6.4 mg, prepared from 15 mg (0.038 mmol) of 18e); 1H NMR (300 MHz, CDCl3) δ 7.22 (br s, 1H), 4.38-4.22 (m, 2H), 3.74 (d, J = 1.0 Hz, 1H), 2.80-2.71 (m, 2H), 2.55-2.47 (m, 1H), 2.37 (dd, J = 3.3, 17.8 Hz, 1H), 2.27-2.17 (m, 1H), 2.00-1.95 (m, 1H), 1.86-1.81 (m, 1H), 1.31 (t, J = 7.1 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ 166.2, 159.8, 97.3, 63.1, 51.4, 40.0, 38.3, 34.8, 33.2, 13.8; HRMS-ES [M+H]+ calcd for C10H15N2O5, 243.0981; found, 243.0979.
6-Hydroxyimino-2-(4-methoxybenzoyl)-bicyclo[2.2.1]heptane-2-carboxylic Acid Ethyl Ester (25)
66% (16.2 mg, prepared from 39 mg (0.081 mmol) of 18c); 1H NMR (300 MHz, CDCl3, inseparable ~2.2:1 mixture of isomers) δ 7.96-7.92 (m, 2H, major), 7.88-7.84 (m, 2H, minor), 7.04 (br s, 1H, minor), 6.91 (d, J = 6.6 Hz, 2H, major and minor), 6.71 (br s, 1H, major), 4.12-4.04 (m, 2H, major and minor), 3.87 (s, 3H, major and minor), 3.70 (s, 1H, major), 3.66 (s, 1H, minor), 2.88 (dd, J = 2.1, 9.8 Hz, 1H, major), 2.71 (dd, J = 1.8, 9.7 Hz, 1H, minor), 2.59 (s, 1H, major and minor), 2.43-2.30 (m, 1H, major and minor), 2.19-2.11 (m, 1H, major and minor), 1.92-1.86 (m, 1H, major and minor), 1.75-1.64 (m, 2H, major and minor), 1.07-1.01 (m, 3H, major and minor); 13C NMR (75 MHz, CDCl3)δ 193.4, 192.5, 173.6, 172.3, 163.8, 163.8, 163.7, 162.8, 131.6, 131.3, 129.2, 127.4, 114.2, 64.1, 63.5, 62.4, 61.9, 55.8, 55.8, 50.5, 50.1, 40.5, 40.2, 38.7, 37.8, 36.1, 34.9, 34.0, 33.3, 14.2, 14.1; HRMS-ES [M + H]+ calcd for C18H22NO5, 332.1498; found, 332.1512.
2-(Diethoxyphosphoryl)-6-hydroxyiminobicyclo[2.2.1]heptane-2-carboxylic Acid Ethyl Ester (26)
75% (5.4 mg, prepared from 10.5 mg (0.022 mmol) of 18f); 1H NMR (300 MHz, CDCl3, inseparable ~1:1 mixture of isomers) δ 7.83 (br s, 1H), 4.26-4.00 (m, 6H), 3.40-3.34 (m, 1H), 2.60 (s, 1H), 2.46-1.95 (m, 6H), 1.39-1.23 (m, 9H); 13C NMR (75 MHz, CDCl3) δ 169.9, 169.9, 163.1, 162.9, 63.1, 63.0, 63.0, 62.9, 61.8, 55.3, 53.5, 48.8, 38.4, 35.5, 35.4, 33.7, 33.6, 33.5, 16.4, 16.4, 16.3, 16.3, 13.9; HRMS-ES [M+H]+ calcd for C14H25NO6P, 334.1420; found, 334.1426.
2-Benzenesulfonyl-6-hydroxyiminobicyclo[2.2.1]heptane-2-carbonitrile (27)
85% (7 mg, prepared from 12.5 mg (0.028 mmol) of 18g); 1H NMR (300 MHz, CDCl3, inseparable ~10:1 mixture of isomers) δ 7.98 (d, J = 7.5 Hz, 2H, minor), 7.89 (d, J = 7.5 Hz, 2H, major), 7.62 (t, J = 7.3 Hz, 1H, major and minor), 7.49 (t, J = 7.7 Hz, 2H, major and minor), 7.33 (br s, 1H, minor), 7.22 (br s, 1H, major), 3.21 (s, 1H, major), 2.99 (s, 1H, minor), 2.67-2.60 (m, 2H, major and minor), 2.40-2.26 (m, 2H, major and minor), 2.06 (dd, J = 3.5, 18.1 Hz, 1H, major and minor), 1.90 (dd, J = 2.6, 13.1 Hz, 1H, major and minor), 1.51 (d, J = 10.4 Hz, 1H, major and minor); 13C NMR (75 MHz, CDCl3) δ 160.3, 159.8, 135.8, 135.6, 135.0, 130.9, 129.9, 129.7, 118.5, 117.7, 67.1, 65.7, 50.1, 40.6, 39.3, 37.6, 37.2, 36.0, 35.6, 33.6, 33.5; HRMS-ES [M + H]+ calcd for C14H15N2O3S, 291.0803; found, 291.0800.
2-Benzenesulfonyl-6-(p-toluenesulfonyloxyimino)bicyclo[2.2.1]heptane-2-carboxylic Acid Methyl Ester (22b)
To a stirred mixture of p-toluenesulfonyl chloride (23.4 mg, 0.123 mmol), triethylamine (0.017 mL, 0.123 mmol) and a catalytic amount of 4-dimethylaminopyridine in dichloromethane (1 mL) was added a solution of oxime 22a (15.7 mg, 0.049 mmol) in dichloromethane (2 mL) at rt. The resulting mixture was stirred for 2 h at rt. Dichloromethane was then added and the organic layer was washed with water and brine. The organic layer was dried over MgSO4 and the solvent was removed under reduced pressure. The residue was purified by flash chromatography (30% ethyl acetate/hexanes) to afford the bicyclic O-tosyl oxime 22b as a white solid (19.0 mg, 82%). A sample of 22b was recrystallized for X-ray analysis from CH2Cl2:EtOAc. mp 121-125 °C; 1H NMR (300 MHz, CDCl3) δ 7.77-7.69 (m, 4H), 7.65-7.59 (m, 1H), 7.48 (t, J = 7.6 Hz, 2H), 7.23 (d, J = 8.2 Hz, 2H), 3.46 (s, 1H), 3.14 (s, 3H), 2.78-2.69 (m, 2H), 2.54-2.49 (m, 1H), 2.42-2.30 (m, 5H), 2.15 (dd, J = 3.8, 18.4 Hz, 1H), 1.50 (d, J = 9.6 Hz, 1H); 13C NMR (75 MHz, CDCl3) δ 170.7, 167.0, 145.4, 137.4, 134.9, 133.0, 130.1, 130.0, 129.4, 129.2, 78.5, 53.1, 50.8, 38.3, 35.9, 35.8, 33.9, 22.0; HRMS- ES [M + H]+ calcd for C22H24NO7S2, 478.0994; found, 478.0989.
Methyl 3-(4-Methoxycyclohex-3-enyl)-2-(phenylsulfonyl)propanoate (30)
To a solution of alcohol 28 (230 mg, 1.62 mmol) in pyridine (10 mL) was added tosyl chloride (617 mg, 3.24 mmol). The mixture was stirred at rt for 18 h and washed with saturated NaHCO3. The organic layer was removed and the aqueous layer was extracted with EtOAc. The combined organic extracts were dried over Na2SO4, and the solvent was removed under reduced pressure to give the tosylate 29 as a yellow oil. The compound was used for the next step without purification. 1H NMR (300 MHz, CDCl3) δ 7.72 (d, J = 8.3 Hz, 2H), 7.28 (d, J = 8.3 Hz, 2H), 4.434 (br s, 1H), 3.84 (d, J = 6.8 Hz, 2H), 3.40 (s, 3H), 2.34 (s, 3H), 2.11-1.66 (m, 7H).
Methyl phenylsulfonylacetate (0.29 mL, 1.62 mmol) was added to a stirred solution of sodium hydride (77 mg, 1.94 mmol, 60% dispersion in mineral oil) in DMF (8 mL) at 0 °C. The reaction mixture was warmed to rt and stirred for 1 h. A solution of crude tosylate 29 in DMF (2 mL) and NaI (98 mg, 0.65 mmol) were added at rt. The resulting mixture was heated at 65 °C for 18 h and then cooled to rt. The mixture was washed with brine, the organic layer was separated and the aqueous layer was extracted with EtOAc. The combined organic extracts were dried over Na2SO4. The solvent was removed under reduced pressure to give a residue purified by flash column chromatography on silica gel (1:3:0.01 EtOAc:hexanes:triethylamine) to afford the title compound 30 as a clear oil containing a mixture of diastereomers in an 1:1 ratio (290 mg, 53% yield for 2 steps). 1H NMR (300 MHz, CDCl3) δ 7.89-7.81 (m, 2H), 7.68-7.62 (m, 1H), 7.60-7.51 (m, 2H), 4.47 (br s, 1H), 4.08-3.98 (m, 1H), 3.61-3.61 (m, 3H), 3.46-3.42 (m, 3H), 2.12-1.95 (m, 6H), 1.94-1.66 (m, 4H), 1.661.21 (m, 3H); 13C NMR (75 MHz, CDCl3) δ 165.9, 165.9, 154.4, 154.2, 136.3. 136.2, 133.6, 128.6, 128.4, 90.9, 90.5, 68.6, 68.4. 53.3, 52.3, 52.3, 32.0, 31.2, 30.8, 30.2, 29.3, 28.6, 28.2, 27.0, 26.4, 26.2; HRMS-ES [M+H]+ calcd for C17H23O5S, 339.1266; found, 339.1259.
Methyl 3-(3-Chloro-4-oxocyclohexyl)-2-(phenylsulfonyl)propanoate (31)
To a solution of enol ether 30 (110 mg, 0.33 mmol) in THF:H2O (4:3, 3 mL) was added NaOAc (29 mg, 0.36 mmol) and N-chlorosuccinimide (48 mg, 0.36 mmol). The reaction mixture was stirred at 0 °C for 5 h and then extracted with EtOAc. The combined organic extracts were dried over Na2SO4. The solvent was removed under reduced pressure to give a residue purified by flash column chromatography on silica gel (1:3 EtOAc:hexanes) to afford the title compound 31 as clear oil (74 mg, 64%). 1H NMR (300 MHz, CDCl3) (complex diastereomer mixture) δ 7.89-7.82 (m. 2H), 7.71-7.66 (m, 1H), 7.58-7.54 (m, 2H), 4.16 (br s, 1H), 3.98 (dd, J = 11.0, 4.2 Hz, 1H), 3.65-5.60 (m, 3H), 3.13-2.84 (m, 1H), 2.29-2.12 (m, 3H), 2.07-1.97 (m, 3H), 1.89-1.75 (m, 1H), 1.48-1.30 (m, 1H); 13C NMR (75 MHz, CDCl3) δ 207.4, 204.0, 203.9, 166.7, 166.6, 137.2, 137.1, 135.0, 129.7, 129.7, 129.6, 69.3, 69.2, 59.6, 59.3, 53.9, 53.7, 53.6, 41.3, 40.1, 35.7, 35.5, 32.9, 31.8, 31.7, 31.7, 31.3, 29.2, 29.1.
O-Silyloximes 32
To a solution of α-chloroketones 31 (19 mg, 0.05 mmol) in CH2Cl2 (5 mL) were added O-(t-butyldimethylsilyl)-hydroxylamine (16 mg, 0.11 mmol), 4Å molecular sieves (crushed), and a catalytic amount of PPTS. The mixture was stirred at rt for 84 h and then filtered through a pad of Celite which was washed with EtOAc. The filtrate was evaporated under reduced pressure to give a residue purified by flash column chromatography on silica gel (1:3 EtOAc:hexanes) to afford compound 32 as a clear oil containing a complex mixture of diastereomers including oxime geometric isomers (17 mg, 66%). 1H NMR (300 MHz, CDCl3) δ 7.89-7.82 (m, 2H), 7.73-7.64 (m, 1H), 7.62-7.52 (m, 2H), 5.60-4.70 (m, 1H), 4.07-3.97 (m, 1H), 3.63-3.60 (m, 3H), 3.28-1.83 (m, 7H), 1.84-1.03 (m, 2H), 0.92-0.87 (m, 9H), 0.13-0.03 (m, 6H); 13C NMR (75 MHz, CDCl3) δ166.9, 166.8, 161.0, 159.7, 137.2, 134.8, 129.8, 129.7, 129.5, 69.3, 69.1, 59.0, 58.9, 53.6, 53.5, 47.0, 46.8, 41.9, 40.7, 32.7, 32.7, 32.6, 31.8, 30.8, 30.0, 29.7, 27.1, 26.4, 20.0, 19.8, 18.6, −4.9; HRMS-ES [M+H]+ calcd for C22H35ClNO5SSi, 488.1694; found, 488.1691.
Methyl 4-(Hydroxyimino)-6-(phenylsulfonyl)bicyclo[3.2.1]octane-6-carboxylate (33a)
To a stirred solution of oxime 32 (40 mg, 0.08 mmol) in THF (2 mL) at −78 °C was added KHMDS (0.5 M in toluene, 0.24 mL, 0.12 mmol) dropwise. After stirring the mixture for 1 h at −78 °C, TBAF (1 M in THF, 0.13 mL, 0.12 mmol) was added and reaction mixture was stirred at 0 °C for 3 h. Saturated NH4Cl was added and the mixture was extracted with EtOAc. The combined organic layers were dried over Na2SO4 and the solvent was removed under reduced pressure. The residue was purified by flash column chromatography on silica gel (2:1 EtOAc:hexanes) to afford the title compound 33a as a white solid (23 mg, 83% yield). 1H NMR (300 MHz, CDCl3) δ 7.71-7.67 (m, 2H), 7.56-7.50 (m, 1H), 7.43-7.38 (m, 2H), 3.40 (s, 3H), 3.38-3.29 (m, 1H), 2.97-2.87 (m, 2H), 2.58-2.39 (m, 3H), 1.63-1.42 (m, 4H), 1.21-1.04 (m, 1H); 13C NMR (75 MHz, CDCl3) δ168.0, 158.8, 137.0, 134.7, 130.4, 129.3, 83.0, 53.3, 49.6, 37.5, 35.0, 33.4, 30.5, 18.5; HRMS-ES [M+H]+ calcd for C16H20NO5S, 338.1062; found, 338.1061.
Synthesis of Oxime Tosylate 33b
To a stirred mixture of triethylamine (0.02 mL, 0.11 mmol), tosyl chloride (20 mg, 0.11 mmol), and a catalytic amount of 4-dimethylaminopyridine in CH2Cl2 (1 mL) was added a solution of oxime 33a (12 mg, 0.036 mmol) in CH2Cl2 (2 mL) at rt. The resulting mixture was stirred at rt for 2 h and washed with saturated NaHCO3. The organic layer was removed and the aqueous layer was extracted with EtOAc. The combined organic extracts were dried over Na2SO4 and the solvent was removed under reduced pressure to give a residue purified by flash column chromatography on silica gel (1:2 EtOAc:hexanes) to afford compound 33b as a white solid (11 mg, 63%). A sample was crystallized from MeOH/CH2Cl2 for X-ray analysis. mp 120-124 °C; 1H NMR (300 MHz, CDCl3) δ 7.77-7.71 (m, 4H), 7.71-7.65 (m, 1H), 7.60-7.47 (m, 2H), 7.28-2.25 (m, 2H), 3.61 (d, J = 4.8 Hz, 1H), 3.19 (s, 3H), 3.06-2.92 (m, 2H), 2.70-2.58 (m, 2H), 2.42 (s, 3H), 1.76-1.51 (m, 4H), 1.55-1.15 (m, 1H); 13C NMR (100 MHz, CDCl3) δ 166.3, 166.2, 143.8, 135.5, 133.4, 131.6, 128.8, 128.4, 128.0, 81.1, 51.8, 47.2, 36.3, 33.0, 32.6, 29.2, 20.66, 19.3; HRMS-ES [M+H]+ calcd for C23H26NO7S2, 492.1151; found, 492.1134.
4-Methoxycyclohex-3-ene Carbonitrile (34) and 3-Methoxycyclohex-3-ene Carbonitrile.15
To a pressure flask containing 2-methoxy-1,3-butadiene12 (3.21 g, 38.2 mmol) in toluene (12 mL) was added acrylonitrile (2.75 mL, 42.0 mmol). The flask was sealed tightly and the mixture was stirred for 16 h at 145 °C. The solvent was removed under reduced pressure, and the residue was purified by flash column chromatography on silica gel (1:1 EtOAc:hexanes) to afford the adducts as a clear oil containing a mixture of regioisomers in an 8:1 ratio (2.93 g, 56% yield). 1H NMR (300 MHz, CDCl3) δ 4.60 (t, J = 3.9 Hz, 0.11H), 4.49 (t, J = 12.9 Hz, 0.88H), 3.44 (s, 3H), 2.72-2.69 (m, 0.88H), 2.38-2.35 (m, 0.11H), 2.35-2.18 (m, 2H), 2.18-2.11 (m, 1H), 2.11-1.96 (m, 1H), 1.96-1.74 (m, 2H); 13C NMR (75 MHz, CDCl3) δ 155.1, 152.0, 122.7, 93.2, 90.2, 54.5, 30.9, 27.3, 26.0, 25.9, 25.7, 25.3, 21.6; HRMS-EI [M+H]+ calcd for C8H12NO, 138.0919; found, 138.0921.
N-(4-Methoxycyclohex-3-enylmethyl)-4-methylbenzenesulfonamide (36)
To a stirred suspension of LiAlH4 (276 mg, 7.29 mmol) in THF (10 mL) at 0 °C was added the mixture of the above nitriles (500 mg, 3.64 mmol) in THF (2 mL) dropwise and the mixture was stirred at rt for 2 h. The reaction was quenched by slow addition of a 5:1 mixture of THF:H2O at 0 °C, followed by addition of 1 M NaOH with stirring. The solids were filtered off and washed with EtOAc. The organic layer was separated, and the aqueous layer was extracted with EtOAc. The combined organics were dried over Na2SO4, and the solvent was removed under reduced pressure to afford a yellow oil. The mixture of amines was used for the next step without purification. Data for major regioisomer 35: 1H NMR (300 MHz, CDCl3) δ 4.51 (br s, 1H), 3.44 (s, 3H), 2.56 (d, J = 6.5 Hz, 2H), 2.15-1.92 (m, 5H), 1.76-1.64 (m, 2H), 1.64-1.36 (m, 1H), 1.36-1.06 (m, 1H); 13C NMR (75 MHz, CDCl3) δ 155.7, 92.3, 54.4, 47.8, 37.6, 28.1, 27.7, 26.9; HRMS-EI [M+H]+ calcd for C8H16NO, 142.1232; found, 142.1240.
To a solution of the above crude amines (513 mg, 3.64 mmol) in CH2Cl2 (12 mL) was added triethylamine (0.5 mL, 3.59 mmol), tosyl chloride (2 g, 10.5 mmol), and a catalytic amount of 4-dimethylaminopyridine. The mixture was stirred at rt for 18 h and washed with saturated NaHCO3. The organic layer was removed and the aqueous layer was extracted with EtOAc. The combined organic extracts were dried over Na2SO4, and the solvent was removed under reduced pressure to give a residue which was purified by flash column chromatography on silica gel (1:4 EtOAc:hexanes) to afford the title compound 36 as a white solid (617 mg, 57% for 2 steps). 1H NMR (300 MHz, CDCl3) δ 7.68 (d, J = 8.3 Hz, 2H) 7.23 (d, J = 8.0 Hz, 2H), 4.90 (t, J = 10.7 Hz, 1H), 4.42 (br s, 1H), 3.40 (s, 3H), 2.77 (t, J = 6.0 Hz, 2H), 2.35 (s, 3H), 2.07-1.90 (m, 3H), 1.73-1.55 (m, 3 H), 1.25-1.16 (m, 1H); 13C NMR (100 MHz, CDCl3) δ 155.4, 143.8, 137.4, 130.1, 127.5, 112.1, 109.6, 91.8, 54.4, 48.5, 34.4, 27.8, 27.2, 26.6, 22.0; HRMS-EI [M+H]+ calcd for C15H22NO3S, 296.1320; found, 296.1316.
N-(3-Chloro-4-oxocyclohexylmethyl)-4-methylbenzenesulfonamide (37)
To a solution of sulfonamide enol ether 36 (950 mg, 3.22 mmol) in THF:H2O (4:3, 35 mL) was added NaOAc (26 mg, 0.32 mmol) and N-chlorosuccinimide (475 mg, 3.54 mmol). The reaction mixture was stirred at rt for 1 h and then was extracted with EtOAc. The combined organic extracts were dried over Na2SO4. The solvent was removed under reduced pressure and the residue was purified by flash column chromatography on silica gel (1:2 EtOAc:hexanes) to afford the α-chloroketone 37 as a mixture of two diastereomers (935 mg, 92%). A sample of the isomers was separated by chromatography for characterization purposes. Less polar major diastereomer of 37: 1H NMR (300 MHz, CDCl3) δ 7.68 (d, J = 8.3 Hz, 2H) 7.25 (d, J = 7.9 Hz, 2H), 5.35 (t, J = 6.6 Hz, 1H), 4.14-4.11 (m, 1H), 2.86-2.76 (m, 3H), 2.36 (s, 3H), 2.25-2.16 (m, 3H), 1.98-1.93 (m, 1H), 1.82-1.74 (m, 1H), 1.35 -1.13 (m, 1H); 13C NMR (75 MHz, CDCl3) δ 204.6, 144.2, 137.1, 130.3, 127.4, 59.7, 47.5, 38.6, 35.5, 31.5, 30.1, 22.0. More polar minor diastereomer of 37: 1H NMR (300 MHz, CDCl3) δ 7.68 (d, J = 10 Hz, 2H) 7.26 (d, J = 8.2 Hz, 2H), 5.38 (t, J = 6.6 Hz, 1H), 4.44-4.39 (m, 1H), 2.82-2.77 (m, 2H), 2.51-2.48 (m, 1H), 2.36 (s, 3H), 2.33-2.00 (m, 3H), 2.02-1.98 (m, 1H), 1.60-1.48 (m, 1H), 1.36-1.28 (m, 1H); 13C NMR (75 MHz, CDCl3) δ 202.3, 144.3, 137.0, 130.3, 127.4, 63.2, 47.6, 42.1, 39.9, 37.8, 30.7, 22.0.
O-Silylketoximes 38
To a solution of α-chloroketones 37 (128 mg, 0.41 mmol) in CH2Cl2 (5 mL) were added O-(t-butyldimethylsilyl)-hydroxylamine (66 mg, 0.45 mmol), 4Å molecular sieves (crushed), and a catalytic amount of PPTS. The mixture was stirred at rt for 48 h, and then filtered through a pad of Celite which was washed with EtOAc. The filtrate was evaporated under reduced pressure to give a residue which was purified by flash column chromatography on silica gel (1:3 EtOAc:hexanes) to afford compound 38 as a clear oil which was an inseparable complex mixture of diastereomers and silyloxime geometric isomers (121 mg, 67% yield). 1H NMR (300 MHz, CDCl3) δ 7.60 (d, J = 8.3 Hz, 2H) 7.16 (d, J = 8 Hz, 2H), 5.55-4.48 (m, 2H), 4.00-3.11, (m, 1H), 2.66-2.29 (m, 2H), 2.21 (s, 3H), 2.08-1.94 (m, 3H), 1.89-1.08 (m, 2H), 0.91-0.78 (m, 1H), 0.78-0.69 (m, 9H), 0.04-0.00 (m, 6H); HRMS-EI [M+H]+ calcd for C20H34N2O3SSiCl, 445.1748; found, 445.1751.
6-(Toluene-4-sulfonyl)-6-azabicyclo[3.2.1]octan-4-one Oxime (39)
To a solution of O-silyloxime 38 (190 mg, 0.43 mmol) in acetonitrile (5 mL) was added TBAF (1 M in THF, 1.07 mL, 1.07 mmol) dropwise at 0 °C and the mixture was stirred at that temperature for 1 h. Saturated NH4Cl was added and the aqueous layer was extracted with EtOAc. The combined organic layers were dried over Na2SO4 and the solvent was removed under reduced pressure to give a residue which was purified by flash column chromatography on silica gel (1:1 EtOAc:hexanes) to afford the title compound 39 as a white solid (106 mg, 84%) containing a mixture of E/Z oxime isomers in a 5:1 ratio. The solid was recrystallized from chloroform to afford colorless crystals of the (E)-isomer suitable for X-ray analysis. Data for mixture: 1H NMR (300 MHz, CDCl3) δ 7.69-7.57 (m, 2H), 7.19 (d, J = 8 Hz, 2H), 5.38 (d, J = 5.8 Hz, 0.17H), 4.42 (d, J = 5.6 Hz, 0.83H), 3.41-3.33 (m, 1H), 3.22 (d, J = 9.5 Hz, 0.17H), 3.06 (d, J = 13.2 Hz, 0.83H), 2.82 (dd, J = 16.5, 6.6 Hz, 1H), 2.45 (br s, 1H), 2.3 (s, 3H), 1.73-1.40 (m, 4H), 1.47-1.13 (m, 2H); 13C NMR (75 MHz, CDCl3) δ157.1, 142.7, 142.6, 134.8, 134.8, 128.9, 128.8, 128.6,126.9, 126.4, 126.2, 59.6, 51.9, 50.8, 50.4, 49.3, 48.5, 36.7, 36.3, 34.2, 33.4, 29.7, 27.8, 25.6, 24.3, 20.7, 17.0; HRMS-EI [M+H]+ calcd for C14H19N2O3S, 295.1116; found, 295.1107.
6-(Toluene-4-sulfonyl)-6-azabicyclo[3.2.1]octan-4-one (40)
To a stirred solution of oximes 39 (40 mg, 0.14 mmol) in 2:1 acetonitrile:H2O (4 mL) was added KMnO4 (43 mg, 0.272 mmol). The mixture was refluxed for 2.5 h and then cooled to rt. The mixture was extracted with CH2Cl2, and the aqueous layer was washed with CH2Cl2. The combined organic extracts were dried over Na2SO4 and the solvent was removed under reduced pressure to give a residue which was purified by flash column chromatography on silica gel (1:1 EtOAc:hexanes) to afford the ketone 40 as a white solid (29 mg, 76% yield). 1H NMR (300 MHz, CDCl3) δ 7.63 (d, J = 8.3 Hz, 2H), 7.25 (d, J = 7.9 Hz, 2H), 4.07 (d, J = 6.0 Hz, 1H), 3.51-3.46 (m, 1H) 3.39 (d, J = 9.7 Hz, 1H), 2.54 (m, 1H), 2.36 (s, 3H), 2.30-2.21 (m, 1H), 2.13-2.05 (m, 1H), 1.91-1.79 (m, 2H), 1.72-1.51 (m, 2H); 13C NMR (75 MHz, CDCl3) δ 207.3, 144.2, 136.0, 130.3, 127.4, 66.2, 52.3, 37.9, 34.8, 34.7, 30.7, 22.0; HRMS-EI [M+H]+ calcd for C14H18NO3S, 280.1007; found, 280.0984.
Supplementary Material
Acknowledgments
We are grateful to the National Institutes of Health (GM-087733) and the National Science Foundation (CHE-0806807) for financial support of this research.
Footnotes
Supporting Information Available: Copies of proton and carbon NMR spectra of new compounds and experimental procedures for some sequences. Included also are X-ray data for compounds 22b, 33b and (E)-39. This material is available free of charge on the Internet at http://pubs.acs.org.
References
- 1.Mathaipoulos G. Chem Ber. 1898;31:2396. [Google Scholar]
- 2.For reviews of vinylnitroso compounds and lead references see: Gilchrist TL. Chem Soc Rev. 1983;12:53.Lyapkalo IM, Ioffe SL. Russ Chem Rev. 1998;67:467.
- 3.Griffin CE, Haszeldine RN. J Chem Soc. 1960:1398. [Google Scholar]
- 4.(a) Li P, Majireck MM, Witek JA, Weinreb SM. Tetrahedron Lett. 2010;51:2032. doi: 10.1016/j.tetlet.2010.02.050. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Witek JA, Weinreb SM. Org Lett. 2011;13:0000. doi: 10.1021/ol2000793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.(a) Denmark SE, Dappen MS. J Org Chem. 1984;49:798. [Google Scholar]; (b) Denmark SE, Dappen MS, Sternberg JA. J Org Chem. 1984;49:4741. [Google Scholar]; (c) Denmark SE, Dappen MS, Sear NL, Jacobs RT. J Am Chem Soc. 1990;112:3466. [Google Scholar]
- 6.For examples of enolonium ion equivalents see: Sacks CE, Fuch PL. J Am Chem Soc. 1975;97:7372.Fuchs PL. J Org Chem. 1976;41:2935.Stork G, Ponaras AA. J Org Chem. 1976;41:2937.Wender PA, Erhardt JM, Letendre LJ. J Am Chem Soc. 1981;103:2114.Hatcher JM, Coltart DM. J Am Chem Soc. 2010;132:4546. doi: 10.1021/ja100932q.Miyoshi T, Miyakawa T, Ueda M, Miyata O. Angew Chem Int Ed. 2011;50:928. doi: 10.1002/anie.201004374.
- 7.For a review of intramolecular Michael reactions see: Little RD, Masjedizadeh MR, Wallquist O, McLoughlin JI. Org React. 1995;47:315.
- 8.Korboukh I, Kumar P, Weinreb SM. J Am Chem Soc. 2007;129:10342. doi: 10.1021/ja074108r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.(a) Chao W, Weinreb SM. Org Lett. 2003;5:2505. doi: 10.1021/ol034775z. [DOI] [PubMed] [Google Scholar]; (b) Chao W, Meketa ML, Weinreb SM. Synthesis. 2004:2058. [Google Scholar]
- 10.(a) VanBrunt MP, Ambenge RO, Weinreb SM. J Org Chem. 2003;68:3323. doi: 10.1021/jo020739m. [DOI] [PubMed] [Google Scholar]; (b) Meketa ML, Mahajan YR, Weinreb SM. Tetrahedron Lett. 2005;46:4749. [Google Scholar]
- 11.White JD, Choi Y. Org Lett. 2000;2:2373. doi: 10.1021/ol0001463. [DOI] [PubMed] [Google Scholar]
- 12.Norris RO, Verbanc JJ, Hennion GF. J Am Chem Soc. 1938;60:1159. [Google Scholar]
- 13.Schill G, Priester CU, Windhovel UF, Fritz H. Tetrahedron. 1987;43:3765. [Google Scholar]
- 14.Lee J, Oh J, Jin SJ, Choi JR, Atwood JL, Cha JK. J Org Chem. 1994;59:6955. [Google Scholar]
- 15.Boxton TJ, Butt G, Liu R, Teo LH, Topsom RD, Katritzky AR. J Chem Soc, Perkin Trans. 1974;2:463. [Google Scholar]
- 16.This amine is known but was not well characterized: Petrov AA, Vladimirova MG. Zh Obsch Khim. 1947;17:1543.
- 17.Korboukh I. PhD Thesis. The Pennsylvania State University; University Park, PA: 2010. [Google Scholar]
- 18.Wali A, Ganeshpure PA, Satish S. Bull Chem Soc Jpn. 1993;66:1847. [Google Scholar]
- 19.For examples of intermolecular conjugate additions of carbon nucleophiles to vinylnitroso compounds via the Denmark procedure see ref. 4 and: Hassner A, Maurya R. Tetrahedron Lett. 1989;30:5803.Kaiser A, Wiegrebe W. Monat Chem. 1998;129:937.
- 20.For some examples of intermolecular conjugate additions of heteronucleophiles to nitrosoalkenes generated by the Denmark methodology see: Hassner A, Murthy K. Tetrahedron Lett. 1987;28:683.Padwa A, Chiacchio U, Dean DC, Schoffsatll AM, Hassner A, Murthy KSK. Tetrahedron Lett. 1988;29:4169.Hassner A, Maurya R, Mesko E. Tetrahedron Lett. 1988;29:5313.Hassner A, Murthy KSK, Padwa A, Bullock WH, Stull PD. J Org Chem. 1988;53:5063.Hassner A, Murthy KSK, Padwa A, Chiacchio U, Dean DC, Schoffstall AM. J Org Chem. 1989;54:5277.Hassner A, Maurya R, Friedman O, Gottlieb HE, Padwa A, Austin D. J Org Chem. 1993;58:4539.Trewartha G, Burrows JN, Barrett AGM. Tetrahedron Lett. 2005;46:3553.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.