

Click chemistry[1] has become one of the most important reactions in the field of glycoscience enabling the rapid assembly under very mild conditions of a vast array of glycoconjugates.[2] We were struck, however, by the absence of azidomethyl glycosides which necessarily excludes the whole class of N-azidomethyl triazoles from the arsenal of glycoconjugates accessible by Click chemistry.[3] Zhu and Schmidt addressed the problem indirectly through the synthesis of a series of azidomethyl thioglycosides,[4] but to date the more native O-glycosides have not been described.
Seeking to remedy this deficiency we prepared phenylthiomethanol 1[5] and investigated its use as glycosyl acceptor. Activation of tetrabenzoyl mannopyranosyl bromide 2 with silver triflate in the presence of 1 afforded the anticipated α-glycoside 3 in 61% yield (Scheme 1). With the corresponding trichloroacetimidate donor 4[6,7] activation with catalytic silver triflate in the presence of 1 gave 65% of 3. Interestingly even thioglycoside donors could be applied provided that a preactivation protocol was employed. Thus, preactivation of a 4,6-O-benzylidene protected β-mannopyranosyl donor 5 under our standard benzenesulfinyl piperidine (BSP)/trifluoromethanesulfonic anhydride (Tf2O) conditions[8] resulted in the formation of a β-glycoside 6 (Scheme 1).[9]
Scheme 1.

Synthesis of phenylthiomethyl glycosides
Bn = benzyl, Bz = benzoyl, Tf = trifluoromethanesulfonyl, TTBP = 2,4,6-tri(tert-butyl)pyrimidine
Activation of 3 and 6 with N-iodosuccinimide (NIS) and trifluoromethansulfonic acid in the presence of azidotrimethylsilane resulted in conversion to the corresponding azidomethyl glycosides 7 and 8 in good yield (Scheme 2) thereby affording the first examples of this class of compound.
Scheme 2.
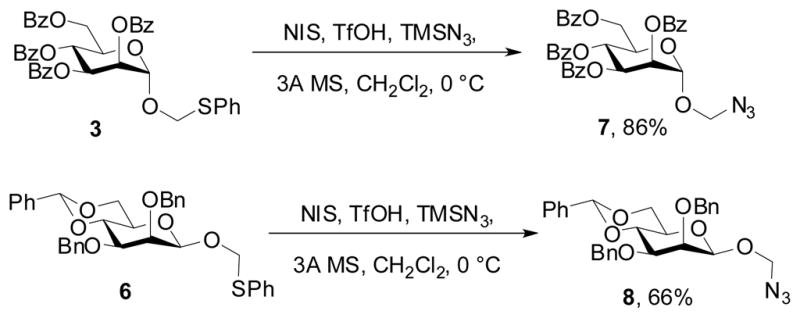
Synthesis of azidomethyl glycosides
Employing the propargyl α-mannoside 9 as reaction partner, Click chemistry was investigated with the azidomethyl glycoside 7 under “classical” copper(I) catalyzed conditions,[1,2] leading preferentially to the 1,4-disubstituted triazoles, and with a more recent ruthenium-based system[10] that afforded the 1,5-isomer (Scheme 3). The application of ruthenium catalysis in this manner, which, to our knowledge, has yet to be reported in glycoconjugate synthesis, provides a closer structural analogue to a branched trisaccharide motif than the more extended array obtained under the copper catalyzed conditions.[11,12]
Scheme 3.
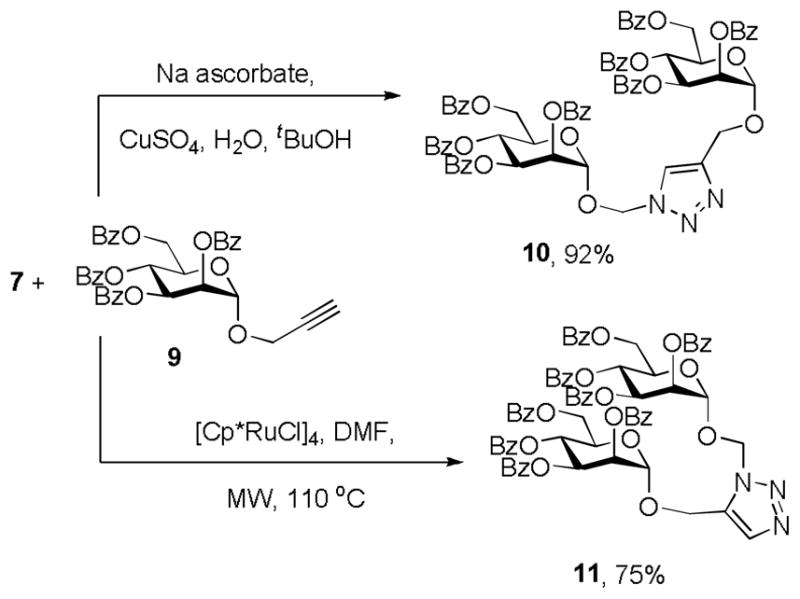
Cu and Ru-Catalyzed cycloadditions with 7
Cp* = pentamethylcyclopentadienyl, MW = microwave irradiation
We next investigated the reaction of these novel azidomethyl glycosides with thioacids, with a view to the formation of amidomethyl glycosides. Perhaps not too surprisingly in view of the relatively electron-rich nature of the azide,[13] the reaction of 7 with thioacetic acid required prolonged microwave heating to afford the unusual amidomethyl glycoside 12 in 51% yield with some 25% of 7 recovered unchanged (Scheme 4).
Scheme 4.
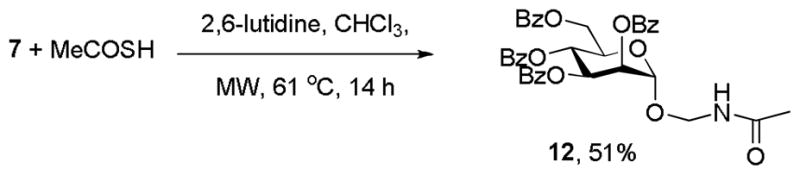
Reaction of 7 with thioacetic acid
Better success in the formation of amidomethyl glycosides was obtained by the Raines variant[14] on the traceless Staudinger reaction.[15] Thus, a series of diphenylphosphinylmethyl thioesters were prepared in the form of their borane adducts. After transfer of the borane to diazabicyclooctane (DABCO) these substituted phosphines were allowed to react with 7 and 8 resulting in the formation of novel amidomethyl glycosides in high yield (Scheme 5).
Scheme 5.
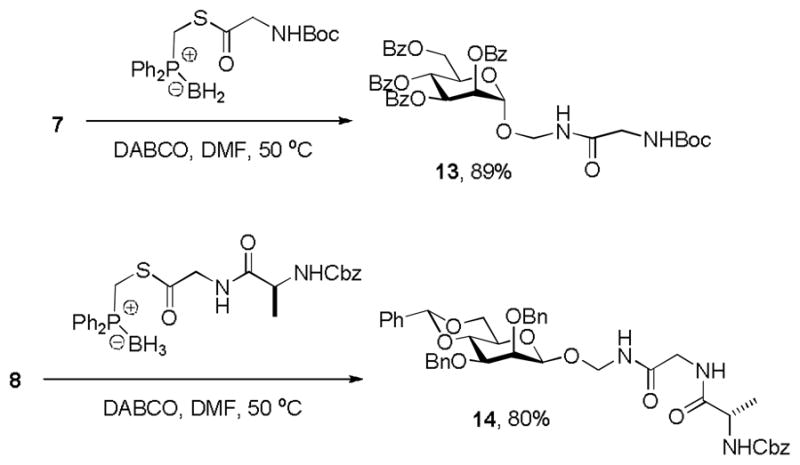
Synthesis of azidomethyl glycosides by traceless Staudinger reaction
Finally, in view of the successful conversion of 3 and 6 to the azidomethyl glycosides 7 and 8 we turned our attention to glycosidic bond formation. Simple “acetal glycosides” of this type have been previously prepared by the reaction of trimethylsilyl glycosides with formaldehyde acetals under catalysis by trimethylsilyl triflate, and by the reaction of anomeric hemiacetals with methylthiomethyl ethers in the presence of N-iodosuccinimide, when the products were obtained as mixtures of stereoisomers.[16] Pleasingly, reaction of both 3 and 6 with NIS and TfOH in the presence of suitable acceptor alcohols provided the corresponding “acetal glycosides” 15, 16 and 17 in excellent yield and as single anomers (Scheme 6). Zemplen deacetylation of 16 gave the free “acetal glycoside” 18 in 91% yield (Scheme 7). The phenylthiomethyl glycosides therefore provide a new convenient and stereoselective means of entry into the “acetal glycosides”, an unusual and somewhat limited class of compounds previously investigated for their potential as enzyme inhibitors.[17,18] We note that the syntheses of both the azidomethyl glycosides (Scheme 2) and the acetal glycosides (Scheme 6) likely proceed through a transient glycosyloxymethyl cation and that this intermediate is trapped by the incoming nucleophile substantially more rapidly than it undergoes decomposition to the glycosyl cation and formaldehyde. As has been previously recorded[17a] the acetal glycosides are considerably less stable to aqueous acid than simple glycosides, nevertheless we observed no difficulties in their purification by chromatography over silica gel, either before or after removal of the protecting groups.
Scheme 6.
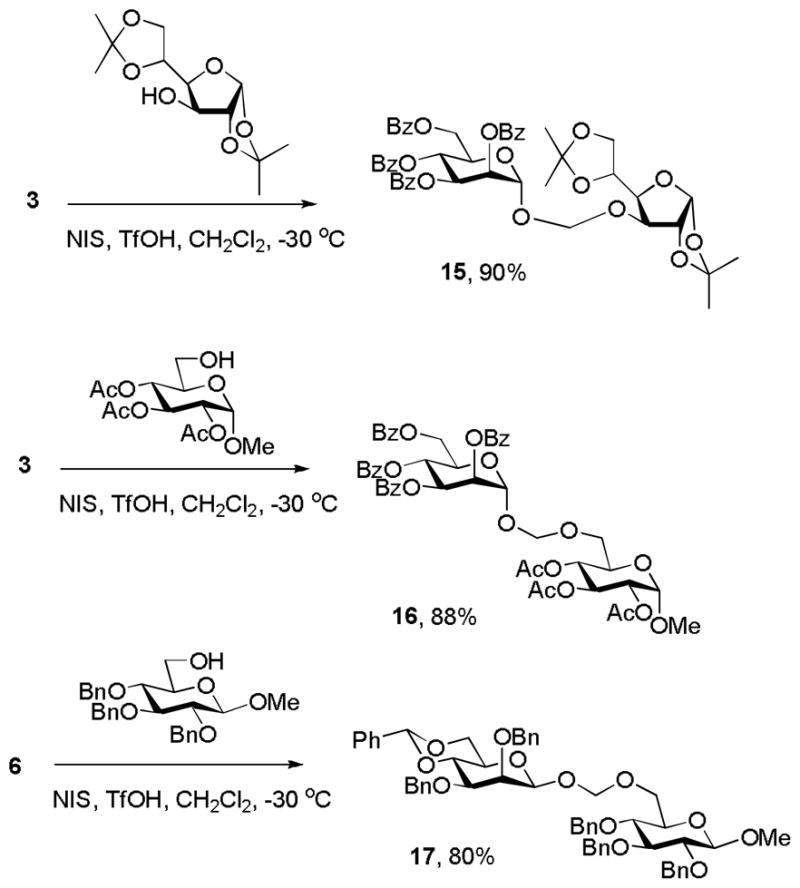
Synthesis of alkoxymethyl glycosides
Scheme 7.
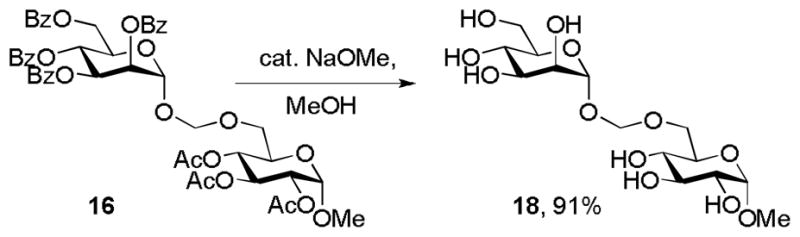
Deprotection of a glycosyloxymethyl glycoside
Overall, the phenylthiomethyl glycosides may be obtained from thioglycosides and or trichloroacetimidates by reaction with phenylthiomethanol under typical glycosylation conditions. They are stable compounds that on activation of the phenylthiomethyl moiety provide direct access to the azidomethyl methyl glycosides and the “acetal glycosides”. As the anomeric carbon is not implicated in these transformations the anomeric stereochemistry of the phenylthiomethyl glycosides is completely retained. The azidomethyl glycosides take part in “Click” reactions with alkynes under copper or ruthenium-catalyzed conditions providing access to new classes of 1,4- and 1,5-triazoles. Finally, the azidomethyl glycosides take part readily in traceless Staudinger reactions enabling the formation of amidomethyl glycosides.
Experimental Section
Preparation of phenylthiomethyl tetra-O-benzoyl-α-D-mannopyranoside (3)
2,3,4,6-Tetra-O-benzoyl-α-D-mannopyranosyl bromide (2.24 g, 3.41 mmol), phenythiomethanol (1.91 g, 13.6 mmol) and activated 4A powdered molecular sieves (900 mg) were mixed in dichloromethane (17 mL) and stirred at room temperature for 10 min before AgOTf (964 mg, 3.75 mmol) was added at 0 °C. The reaction mixture was allowed to warm to room temperature until TLC showed that the donor had been consumed (2–4 h). Saturated aqueous NaHCO3 then was added at 0°C, and the reaction mixture was filtered, and the filtrate was washed with brine. The organic layer was dried and concentrated under reduced pressure and the product was isolated by silica gel column chromatography (eluent: hexane/ethyl acetate from 20/1 to 10/1) to give 3 (1.58 g, 61%) as a white foam. [α]D23 +42.0° (c, 2.6, CHCl3); 1H NMR (500 MHz, CDCl3) δ: 8.15-8.13 (m, 2H), 8.11-8.10 (m, 2H), 7.99-7.97 (m, 2H), 7.88-7.86 (m, 2H), 7.64-7.59 (m, 4H), 7.54-7.51 (m, 1H), 7.47-7.43 (m, 5H), 7.41-7.37 (m, 4H), 7.34-7.29 (m, 3H), 6.17 (t, J = 10.0 Hz, 1H), 5.96 (dd, J = 3.5, J = 10.5 Hz, 1H), 5.75 (m, 1H), 5.62 (d, J = 1.5 Hz, 1H), 5.27 (d, J = 12.0 Hz, 1H), 5.18 (d, J = 12.0 Hz, 1H), 4.72 (dd, J = 2.5, J = 12.5 Hz, 1H), 4.50 (dd, J = 4.5, J = 12.0 Hz, 1H), 4.41 (m, 1H); 13CNMR (125 MHz, CDCl3) δ: 166.4, 165.74, 165.68, 165.6, 134.8, 133.81, 133.75, 133.5, 133.4, 131.3, 130.2, 130.1, 130.0, 129.5, 129.3, 129.2, 128.9, 128.8, 128.7, 128.6, 127.8, 94.9, 72.6, 70.6, 70.2, 69.9, 67.1, 63.0; ESIHRMS Calcd. for C41H34O10S [M+Na]+ 741.1770, found 741.1738.
Supplementary Material
Acknowledgments
We thank the NIH (GM 62160) for partial support of this work.
Footnotes
Dedicated to Andrea Vasella
Supporting information for this article is available on the WWW under http://www.angewandte.org or from the author.
Contributor Information
Prof. Dr. David Crich, Department of Chemistry, Wayne State University, 5101 Cass Avenue, Detroit, MI 48202 (USA). Centre de Recherche de Gif, Institut de Chimie des Substances Naturelles, CNRS, Avenue de la Terrasse, 91198 Gif-sur-Yvette CEDEX (France), Fax: (+33) 1-6907-7752.
Fan Yang, Department of Chemistry, Wayne State University, 5101 Cass Avenue, Detroit, MI 48202 (USA).
References
- 1.a) Kolb HC, Finn MG, Sharpless KB. Angew Chem Int Ed. 2001;40:2004–2021. doi: 10.1002/1521-3773(20010601)40:11<2004::AID-ANIE2004>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]; b) Tornoe CW, Christensen C, Meldal M. J Org Chem. 2002;67:3057–3064. doi: 10.1021/jo011148j. [DOI] [PubMed] [Google Scholar]; c) Rostovtsev VV, Green LG, Fokin VV, Sharpless KB. Angew Chem Int Ed. 2002;41:2596–2599. doi: 10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]; d) Meldal M, Tornoe CW. Chem Rev. 2008;108:2952–3015. doi: 10.1021/cr0783479. [DOI] [PubMed] [Google Scholar]
- 2.a) Dondoni A. Chem Asian J. 2007;2:700–708. doi: 10.1002/asia.200700015. [DOI] [PubMed] [Google Scholar]; b) Dedola S, Nepogodiev SA, Field RA. Org Biomol Chem. 2007:1006–1017. doi: 10.1039/b618048p. [DOI] [PubMed] [Google Scholar]; c) Nicotra F, Cipolla L, Peri F, La Ferta B, Redaelli C. Adv Carbohydr Chem Biochem. 2007;61:353–398. doi: 10.1016/S0065-2318(07)61007-5. [DOI] [PubMed] [Google Scholar]; d) Gyorgydeak Z, Thiem J. Adv Carbohydr Chem Biochem. 2006;60:103–182. doi: 10.1016/S0065-2318(06)60004-8. [DOI] [PubMed] [Google Scholar]; e) Santoyo-Gonzalez F, Hernandez-Mateo F. Top Heterocycl Chem. 2007;7:133–177. [Google Scholar]; f) Pieters RJ, Rijkers DTS, Liskamp RMS. QSAR Comb Sci. 2007;26:1181–1190. [Google Scholar]; g) Hanson SR, Greenberg WA, Wong CH. QSAR Comb Sci. 2007;26:1243–1252. [Google Scholar]; h) Wilkinson BL, Bornaghi LF, Houston TA, Poulsen S-A. In: Drug Des Res Perspectives. Kaplan SP, editor. Nova Science Publishers; 2007. pp. 57–102. [Google Scholar]
- 3.Simple azidomethyl ethers have been prepared from methylthio ethers and chloromethyl ethers in the presence of azide anion, and have been used as protecting groups in nucleoside chemistry but the azidomethyl glycosides appear to be unknown. Zavgorodny S, Polyanski M, Besidsky E, Kriukov E, Sanin A, Pokrovskaya M, Gurskaya G, Lonnberg H, Azhayev A. Tetrahedron Lett. 1991;32:7593–7596.Zavgorodny SG, Pechenov AE, Shvets VI, Miroshnikov AI. Nucleosides, Nucleotides Nucleic Acids. 2000;19:1977–1991. doi: 10.1080/15257770008045472.Guo J, Xu N, Li Z, Zhang S, Wu J, Kim DH, Marma MS, Meng Q, Cao H, Li X, Shi S, Yu L, Kalachikov S, Russo JJ, Turro NJ, Ju J. Proc Natl Acad Sci U S A. 2008;105:9145–9150. doi: 10.1073/pnas.0804023105.Loubinoux B, Tabbache S, Gerardin P, Mlazimbakana J. Tetrahedron. 1988;44:6055–6064.Young T, Kiessling LL. Angew Chem Int Ed. 2002;41:3449–3451. doi: 10.1002/1521-3773(20020916)41:18<3449::AID-ANIE3449>3.0.CO;2-U.
- 4.Zhu X, Schmidt RR. J Org Chem. 2004;69:1081–1085. doi: 10.1021/jo035300o. [DOI] [PubMed] [Google Scholar]
- 5.Gundersen LL, Benneche T. Acta Chem Scand. 1991;45:975–977. [Google Scholar]
- 6.a) Schmidt RR, Kinzy W. Adv Carbohydr Chem Biochem. 1994;50:21–123. doi: 10.1016/s0065-2318(08)60150-x. [DOI] [PubMed] [Google Scholar]; b) Bien F, Ziegler T. Tetrahedron: Asymmetry. 1998;9:781–790. [Google Scholar]
- 7.We note that the use of more than 1.1 equiv of silver triflate resulted in lower yields of product 3 and over activation leading, inter alia, to the formation of phenylthiomethoxymethyl glycosides. The moderate yields for the formation of 3 and 6 are to some extent the result of the relative instability of phenylthiomethanol under the coupling conditions.
- 8.a) Crich D, Smith M. J Am Chem Soc. 2001;123:9015–9020. doi: 10.1021/ja0111481. [DOI] [PubMed] [Google Scholar]; b) Crich D, Lim LBL. Org React. 2004;64:115–251. [Google Scholar]
- 9.The moderate yield is most likely due to the persistence of thiophilic species in the reaction mixture after the pre-activation step.
- 10.Boren BC, Narayan S, Rasmussen LK, Zhang L, Zhao H, Lin Z, Jia G, Fokin VV. J Am Chem Soc. 2008;130:8923–8930. doi: 10.1021/ja0749993. [DOI] [PubMed] [Google Scholar]
- 11.Zemplen de-O-benzoylation of 10 and 11 gave mimics of methyl 3,6-di-O-(α-D-mannopyranosyl)-α-D-mannopyranoside, the binding motif for the mannose-binding lectin concanavalin A. These compounds were found by isothermal titration calorimetry to have binding constants of 80.0 and 13.7 M−1 × 103, respectively, for binding to concanavalin A as opposed to the 390 M−1 × 103 exhibited by methyl 3,6-di-O-(α-D-mannopyranosyl)-α-D-mannopyranoside itself. The significantly reduced binding affinity exhibited by the two triazoles is attributed in part to the absence of the 2- and 4- hydroxyl groups of the “reducing sugar” in methyl 3,6-di-O-(α-D-mannopyranosyl)-α-D-mannopyranoside, which are known to contribute substantially to binding. Veeneman GH, van der Marel GA, Vandenelst H, van Boom JH. Recl Trav Chim Pays-Bas. 1990;109:449–451.
- 12.a) Simanek EE, McGarvey GJ, Jablonowski JA, Wong CH. Chem Rev. 1998;98:833–862. doi: 10.1021/cr940226i. [DOI] [PubMed] [Google Scholar]; b) Bernardi A, Cheshev P. Chem Eur J. 2008;14:7434–7441. doi: 10.1002/chem.200800597. [DOI] [PubMed] [Google Scholar]
- 13.a) Shangguan N, Katukojvala S, Greenberg R, Williams LJ. J Am Chem Soc. 2003;125:7754–7755. doi: 10.1021/ja0294919. [DOI] [PubMed] [Google Scholar]; b) Merkx R, Brouwer AR, Rijkers DTS, Liskamp RMJ. Org Lett. 2005;7:1125–1128. doi: 10.1021/ol0501119. [DOI] [PubMed] [Google Scholar]
- 14.a) Soellner MB, Nilsson BL, Raines RT. J Am Chem Soc. 2006;128:8820–8828. doi: 10.1021/ja060484k. [DOI] [PubMed] [Google Scholar]; b) Grandjean C, Boutonnier A, Guerreiro C, Fournier JM, Mulard LA. J Org Chem. 2005;70:7123–7132. doi: 10.1021/jo0505472. [DOI] [PubMed] [Google Scholar]
- 15.a) Nilsson BL, Kiessling LL, Raines RT. Org Lett. 2000;2:1939–1941. doi: 10.1021/ol0060174. [DOI] [PubMed] [Google Scholar]; b) Saxon E, Armstrong JI, Bertozzi CR. Org Lett. 2000;2:2141–2143. doi: 10.1021/ol006054v. [DOI] [PubMed] [Google Scholar]; c) Hackenberger CPR, Schwarzer D. Angew Chem Int Ed. 2008;47:10030–10074. doi: 10.1002/anie.200801313. [DOI] [PubMed] [Google Scholar]; d) Best MD. Biochemistry. 2009;48:6571–6584. doi: 10.1021/bi9007726. [DOI] [PubMed] [Google Scholar]; e) Dirksen A, Dawson PE. Curr Opin Chem Bio. 2008;12:760–766. doi: 10.1016/j.cbpa.2008.10.009. [DOI] [PubMed] [Google Scholar]
- 16.a) Tietze LF, Fischer R, Guder HJ, Neumann M. Liebigs Ann Chem. 1987:847–856. [Google Scholar]; b) Tietze LF, Fischer R. Angew Chem Int Ed Engl. 1981;20:969–970. [Google Scholar]; c) Tietze LF, Fischer R. Tetrahedron Lett. 1981;22:3239–3242. [Google Scholar]
- 17.a) Jegge S, Lehmann J. Carbohydr Res. 1985;142:47–59. doi: 10.1016/s0008-6215(00)90732-8. [DOI] [PubMed] [Google Scholar]; b) Tietze LF, Seele R, Leiting B, Krach T. Carbohydr Res. 1988;180:253–262. doi: 10.1016/0008-6215(88)80082-x. [DOI] [PubMed] [Google Scholar]
- 18.The basic structural unit of the “acetal glycosides” occurs naturally in the 1,1′-disaccharides, in sucrose, in the iridoid glycosides and in the seco-iridoid glycosides, albeit in a somewhat more complex form. Tietze LF, Niemeyer U, Marx P, Glusenkamp KH, Schwenen L. Tetrahedron. 1980;36:735–739.Tietze LF, Niemeyer U, Marx P, Glusenkamp KH. Tetrahedron. 1980;36:1231–1236.Tietze LF. Angew Chem Int Ed Engl. 1983;22:828–841.Tietze LF, Fischer R. Angew Chem Int Ed Engl. 1983;22:888.Oscarson S, Sehgelmeble FW. J Am Chem Soc. 2000;122:8869–8872.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.