


Abstract
Splicing factor 1 (SF1) binds to the branch point sequence (BPS) of mammalian introns and is believed to be important for the splicing of some, but not all, introns. To help identify BPSs, particularly those that depend on SF1, we generated a BPS profile model in which SF1 binding affinity data, validated by branch point mapping, were iteratively incorporated into computational models. We searched a data set of 117 499 human introns for best matches to the SF1 Affinity Model above a threshold, and counted the number of matches at each intronic position. After subtracting a background value, we found that 87.9% of remaining high-scoring matches identified were located in a region upstream of 3′-splice sites where BPSs are typically found. Since U2AF65 recognizes the polypyrimidine tract (PPT) and forms a cooperative RNA complex with SF1, we combined the SF1 model with a PPT model computed from high affinity binding sequences for U2AF65. The combined model, together with binding site location constraints, accurately identified introns bound by SF1 that are candidates for SF1-dependent splicing.
INTRODUCTION
Most metazoan gene expression requires the splicing of introns to generate mature mRNAs. Splicing is mediated by the spliceosome, which consists of U1, U2, U4, U5 and U6 small nuclear ribonucleoproteins (snRNPs) and more than 150 non-snRNP proteins (1). Splicing proceeds through two trans-esterification reactions: the first forms a lariat intermediate with the 5′-end of the intron linked to an adenosine positioned within the branch point sequence (BPS) and the second results in complete intron removal and exon ligation (1).
Mammalian spliceosomes typically assemble on a pre-mRNA substrate by forming an ATP-independent E (early) complex, in which U1 snRNP binds the 5′-splice site and splicing factor 1 (SF1) and U2 auxiliary factor (U2AF) bind cooperatively to elements near the 3′-splice site (1). SF1 recognizes the 7-nt BPS, the 65 kDa subunit of U2AF (U2AF65) recognizes the polypyrimidine tract (PPT), and the 35 kDa subunit of U2AF (U2AF35) recognizes the AG dinucleotide adjacent to the 3′-splice site (2,3). U2AF65 is believed to then recruit U2 snRNP to the pre-mRNA, where a conserved GUAGUA hexanucleotide in U2 snRNA hybridizes to the BPS, thereby releasing SF1 (4,5). The stable association of U2 snRNP with the 3′-splice site (A complex) is the first ATP-dependent step in the splicing pathway and specifies the 2′-OH of a bulged adenosine in the BPS as the nucleophile for the first trans-esterification reaction (6,7). However, trans-esterification only occurs after the formation of B complex through the joining of the U4/U5/U6 tri-snRNP to A complex and subsequent loss of U1 and U4 snRNPs along with conformational rearrangements to form the catalytically active B* complex (8).
The BPS in yeast introns is a nearly invariant UACUAAC sequence with the branch point adenosine (BP A) being the sixth nucleotide (highlighted). In contrast, the BPS is highly variable in metazoan introns, generally conforming to a YNCURAY consensus (BP A highlighted, Y is a pyrimidine, N is any nucleotide and R is a purine) (1). More recent work has demonstrated that the human BPS may be even more degenerate and represented by a 5-mer sequence, YUNAY, rather than a 7-mer (9). Consequently, it has been difficult to identify metazoan BPSs based on sequence alone. BPSs are frequently located 15–45 nt upstream of the 3′-splice site, but have also been found much further upstream, further complicating their identification (1,10,11). Introns with multiple BPSs also exist and may be spliced by unusual mechanisms, such as the recursive mechanism used to completely remove a single intron in Drosophila (11,12). BPS variations between isoforms of a single gene can affect splice site choice and result in variable exon inclusion, as observed in the third intron of the HLA-DQB1 gene (13,14). Such variations may provide an important source of transcript diversity. Furthermore, BPS mutations have been linked to several human genetic diseases (15–19), providing further incentive to understand how BPS variation contributes to splicing decisions.
SF1 binds the BPS using its KH domain (20,21) and forms a cooperative complex with U2AF (2), but its role in splicing has not been proven unambiguously. Conditional mutants of the Saccharomyces cerevisiae SF1 ortholog show reduced splicing of reporter genes containing weak splice sites but not strong splice sites, suggesting that SF1 may participate in the splicing of some, but not all, introns (22). Immunodepletion of SF1 from HeLa cell nuclear extracts does not generally block splicing but can affect the rate of spliceosome formation, dependent on the nature of the BPS. For example, a human IgM pre-mRNA engineered with the yeast consensus BPS recruits U2 snRNP three times faster than with its native BPS (23). RNAi-mediated SF1 depletion in HeLa cells showed little effect on splicing, as assayed by RT–PCR of a limited set of introns (24), but the activity of any SF1 remaining after such depletion is difficult to evaluate. Thus, SF1 can bind the BPS directly and the available evidence suggests that it may be required for the splicing of some introns, particularly those with weak splice sites, but no specific SF1-dependent introns have yet been identified.
Efforts to computationally identify BPSs in genomic sequence data have proven challenging given the short, degenerate nature of the BPS sequence motif, the limited number of experimentally defined BPSs, and the variable location of BPSs within introns, which limits the usefulness of sequence alignment tools. An analysis of BPS sequences in orthologous human and mouse introns revealed conservation of BPS location and identical sequences in only 32% of alternatively spliced and 3% of constitutively spliced exons (25). Previous methods relying on sequence-based models have met with some success (1,13,25,26), but a comparative model analysis has not been performed and BPS predictions have not been experimentally verified, making it difficult to gauge the relationship between statistical and biological success of the models. Here we report a complementary approach using SF1 RNA-binding affinity data to generate a BPS profile model, which is then iteratively refined using additional binding and branch point mapping data. We have combined this model with a PPT profile model based on sequences with high affinity for U2AF65, along with the distance between BPS and PPT binding sequences to approximate the cooperative interaction of SF1 and U2AF65, to examine a data set of nearly 120 000 human introns. The model can discriminate introns that bind SF1 from those that do not in high- and low-scoring cases, thereby identifying human intron candidates for SF1-dependent splicing.
MATERIALS AND METHODS
Intronic region data set
We utilized full-length mRNA sequences from a Homo sapiens RefSeq data set (NCBI, build 35.1) (27), using Spidey version 1.40 (28) to align mRNAs to the genome and determine intron positions. Intronic regions were defined as nucleotides −199 to +1 relative to the 3′-splice site, or extended upstream of the intron for shorter (<199 nt) introns. We eliminated redundancy in which 3′-splice sites were used in multiple mRNA isoforms, resulting in 117 499 sequences. Seven 3′-splice sites had a second upstream 3′-splice site within 200 nt, and 13 628 introns were short and had exons located within 200 nt. After constructing profile models, the data set was annotated with the sequence, location, and scores of the three highest scoring matches for the SF1 Affinity Model, and the highest scoring matches for the Literature BPS Model and U2AF65 Affinity Model in the expected PPT region (from −30 to −11). The data set also includes the distances between the BP A and U2AF65 Affinity Model site, the RefSeq mRNA and contig sequence identifiers and positions, the intron and the downstream exon lengths, and the type of splicing event (5′/3′, alternative/constitutive, first/last/middle exon, insertion/retention). These data are available upon request.
Construction of profile models
Sequence logos were generated from sequence alignments using Weblogo (29,30) and log-odds scoring matrices were generated using motifBS (31). Log-odds scores were represented in bits for each profile model. For all models except the SF1 Affinity Model described below, a uniform background frequency of 0.25 for all bases was assumed and a pseudocount of 1 was added for each nucleotide.
The Initial SF1 Binding Model was based on nine sequences previously known to bind SF1 (UACUAAC, UACUGAC, UAGUAAC, UAGUGAC, UACUAAU, CACUAAC, GACUAAC, UACUAAG, UGCUAAC, CACUGAC) (3,14), each represented once in a multiple sequence alignment (‘uniform weighting’). To incorporate SF1 binding data, each of 46 experimentally tested sequences (Figure 3) was represented multiple times in proportion to its affinity (‘affinity weighting’) (Supplementary Table S4), with UACUAAC, the tightest binding previously known SF1 site, present at 100 copies. Because adding multiple sequence copies did not add more information, we used pseudocount scaling to keep a constant ratio of 0.087 pseudocounts per sequence. During model refinement, we used an Interim SF1 Affinity Model to aid in selecting candidate BPSs and PPTs for branch point mapping experiments, using affinity data for 23 of the 46 sequences that eventually were used to construct the final SF1 Affinity Model.
Figure 3.

SF1 binding to BPS reporters using the SF1 affinity Tat-hybrid assay. (A) Tat-hybrid system showing the HIV-1 FFL reporter engineered with the BPS, PPT and AG dinucleotide. In the absence of Tat-fused SF1, transcription elongation is arrested. Recruitment of the Tat fusion through the SF1-BPS and U2AF-RNA interactions enhances transcription elongation and luciferase expression. Panels (B–F) SF1 affinity data from Tat-hybrid assays for BPSs predicted by the Initial SF1 Binding Model; (B) 10 of the high-scoring sequences; (C) 8 very low-scoring sequences; (D) 11 sequences with high frequency rankings relative to all profile matches in the expected BPS region above a score threshold (‘Materials and Methods’ section); (E) wild-type and mutant BPS variants found in genetic diseases: TH—tyrosine hydroxylase intron 11; LCAT—lecithin cholesterol acetyltransferase intron 4; ITGB4—integrin beta-4 intron 31; COL5A1—collagen 5A1 intron 32; (F) the 10 remaining UACUAAC point mutants not included in A, B or C. The dashed yellow lines in panels B–F represent an arbitrary threshold for SF1 binding, set at 50% of activation relative to UACUAAC. (G) Correlation between bit score calculated using the Initial SF1 Binding Model and measured SF1 affinities (expressed as percent activation relative to UACUAAC) for the combined set of high and low scoring sequences as well as for all 46 sequences assayed. ‘R’ represents the correlation coefficient between bit score and SF1 affinity. (H) Correlation between bit score calculated using the SF1 Affinity Model and measured SF1 affinities as in (G).
In addition to the two SF1-based models, we generated: a ‘YNCURAY Model’ to represent base frequencies of the mammalian BPS consensus (1), using uniform weighting of all sequences conforming to the YNCURAY consensus sequence; a ‘Pentamer BPS Model’ to represent a previous model based on the 3′-pentamer portion of BPSs from several species (26) by extracting nucleotide frequencies from the publication and converting them to log-odds scores; and a ‘Literature BPS Model’ using uniform weighting of 14 initially defined BPSs (32) and 13 additional experimentally validated sites (15–19,33–36). In addition, we created a ‘U2AF65 Affinity Model’ to represent the PPT using uniform weighting of sequences from a manually edited multiple alignment of in vitro selected high affinity U2AF65 binding sequences (37). The sequence logo from this U2AF65 alignment suggested a dimeric motif, where each monomer had similar information content (Supplementary Figure S1). Therefore only the 3′-half of this putative motif, corresponding to positions from 13 to 23 of the alignment, was used to construct the U2AF65 Affinity Model. We considered the first nucleotide from the 5′-end of the PPT to be the location at which the model matched the intron.
Evaluation of profile models
To evaluate each model, we first established bit score thresholds by generating all possible sequences of the same length as the profile model, scoring each (in bits), and setting a threshold corresponding to the top 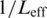 of all scores (∼0.5%), where
of all scores (∼0.5%), where 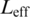 is the number of possible sites in a sequence (194 for a 200-nt sequence and 7-mer motif) and thus the number of independent trials needed for a Bonferroni correction. To identify best matches within the introns, we corrected for tied scores by weighting each as 1/n, where n is the total number of matches for a given score in a given intron. The number of ‘best matches’ at a position is the sum of weightings of each of the best matches above the threshold at a particular position.
is the number of possible sites in a sequence (194 for a 200-nt sequence and 7-mer motif) and thus the number of independent trials needed for a Bonferroni correction. To identify best matches within the introns, we corrected for tied scores by weighting each as 1/n, where n is the total number of matches for a given score in a given intron. The number of ‘best matches’ at a position is the sum of weightings of each of the best matches above the threshold at a particular position.
To estimate background values (Mbg), we averaged the number of profile matches for the first 100 nt of each intron region (from −189 to −90), all above the score thresholds. Mean backgrounds in the expected regions were defined as 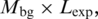 where
where 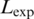 is the length of the expected region (30 nt from −45 to −16 for the BPS, and 20 nt from −30 to −11 for the PPT). The fraction of best matches in the expected BPS region at each position was therefore calculated by dividing the number of best matches in the peak region after subtracting background matches, by the total number of best matches anywhere in the intron after subtracting background matches.
is the length of the expected region (30 nt from −45 to −16 for the BPS, and 20 nt from −30 to −11 for the PPT). The fraction of best matches in the expected BPS region at each position was therefore calculated by dividing the number of best matches in the peak region after subtracting background matches, by the total number of best matches anywhere in the intron after subtracting background matches.
Signal-to-noise ratios (SNRs) were calculated for the BPS and PPT profile models in their respective expected regions. Noise was defined as the expected number of mean background matches in the expected regions  and signal as the total number of matches in the expected region minus those expected to be noise, with SNR calculated using
and signal as the total number of matches in the expected region minus those expected to be noise, with SNR calculated using
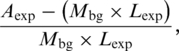 |
where 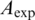 is the number of ‘best matches’ in the expected region. The SNR and information content are not necessarily expected to have a direct relationship because the values used for background subtraction and significance threshold differ between models and thus are not directly comparable.
is the number of ‘best matches’ in the expected region. The SNR and information content are not necessarily expected to have a direct relationship because the values used for background subtraction and significance threshold differ between models and thus are not directly comparable.
In vitro branch point mapping
Candidate BPS and PPT pairs for branch point mapping were selected from 1667 introns in 242 genes highly expressed in HeLa cells, identified using microarray data from the NCBI Gene Expression Omnibus (data set accession GDS885; http://www.ncbi.nlm.nih.gov/geo/gds/gds_browse.cgi?gds=885) (38). Four replicate microarray data sets were used (GEO sample accessions: GSM23372, GSM23373, GSM23377, GSM23377) from an experiment using the Affymetrix GeneChip Human Genome U133 Array (HG-U133A) platform (39). Candidates for the assay included only genes present in all data sets, having mean signal intensities >1 Z-score (relative to the mean signal intensity of all gene features in all replicates), and with standard deviations of signal intensities <1 Z-score (relative to the standard deviation of signal intensity of all gene features in all replicates). From the resulting set of genes, seven introns were chosen using the following criteria: high (>10 bits) or medium (4–8 bits) PPT scores; high (>7 bits) BPS scores using an interim SF1 affinity-weighted profile model; ≤35 nt spacing between the BP A and the start of the PPT; and intron location near the 3′-end of the mRNA to facilitate in vitro reverse transcription.
The BPS and PPT of a single intron in a Drosophila melanogaster Ftz pre-mRNA was replaced with BPSs and PPTs of the seven chosen introns by cloning PCR products into a Ftz-expressing vector (40). Variant plasmids were linearized with XhoI and runoff transcription was performed to generate uniformly 32P-labeled pre-mRNAs (41). The variant pre-mRNAs were gel purified and incubated in HeLa nuclear extracts for 45 min under previously described splicing conditions (42). Splicing products were resolved on a denaturing 12% polyacrylamide gel and RNAs corresponding to lariat/3′-exon intermediates were purified. A portion of the RNA was enzymatically debranched (43) and analyzed on a 6% denaturing polyacrylamide gel to confirm the identity of the lariat/3′-exon intermediate. To map branch sites by primer extension, 50 pmol of a synthetic oligonucleotide primer complementary to the 3′-exon of Ftz (5′-AGCGGGTGTACGTCTGACGGG-3′) was 5′-end-labeled, aliquots (∼1 × 106 counts) were annealed to either branched or debranched lariat/3′-exon RNAs, and RNAs were reverse transcribed using Superscript II (Stratagene) as previously described (36). After removing RNA by alkaline hydrolysis, primer extension products were analyzed on a denaturing 6% polyacrylamide gel, using Ftz plasmid DNA sequenced with the same primer as markers.
Predicted SF1–BPS interactions tested using the Tat-hybrid assay
To determine whether the SF1 Affinity Model could be used to identify SF1-binding introns, we chose 25 introns with high- and low-scoring BPSs for testing in the Tat-hybrid assay. The following criteria defined an initial set of candidate introns: a predicted BP A based on the 5′-most best match to the SF1 Affinity Model which was ≤50 nt upstream of the 3′-splice site and ≤10 nt upstream of the start of the PPT as inferred from the U2AF65 Affinity Model, and a PPT defined as the top scoring 5′-most match to the U2AF65 Affinity Model with a starting position in the region from −30 to −11, with high or low PPT scores. To describe the scores of the selected intronic regions relative to distributions of profile model scores, we calculated profile model Z-scores based on the mean and standard deviation of the distribution of bit scores for all possible 7-mers for the SF1 model and all 11-mers for the PPT model. For the SF1 model, we also computed percentiles based on the distribution of scores of all above-threshold matches, or all best matches if the score was below threshold, in the expected BPS region in the set of 117 499 introns. Ten introns with high BPS scores (≥7.1 bits, average Z-score = 3.6, and 12th percentile) were chosen from the high-scoring PPT set (≥11.7 bits, average Z-score = 4.4), 10 with the lowest BPS scores (≤2.4 bits, average Z-score = 1.9, and bottom first percentile) were chosen from the high-scoring PPT set (≥10.0 bits, average Z-score = 3.9), and five with low-BPS scores (≤1.9 bits, average Z-score = 1.8, and bottom first percentile of all matches) were chosen from the low scoring PPT set (≤1.0 bits, average Z-score = 1.8). For the high BPS/high PPT set, we chose predicted BPSs that also matched sequences used to construct the Literature BPS Model to increase the likelihood that the site would be a functional BPS in addition to binding SF1.
For each of the 25 introns chosen, HIV-1 LTR reporters containing 100 nt immediately upstream of the 3′-splice sites were constructed in an IRES firefly luciferase (FFL) plasmid (3). The inserted sequence of the BPS reporter, beginning at the 5′-end of the transcript and encompassing the BPS, AdML PPT and AG dinucleotide (shown in boldface, with ‘X’ representing any nucleotide within the BPS) is 5′-GGTCTCTCTGGCTTAAGTTCGXXXXXXXCCTGTCCCTTTTTTTTCCACAGCAAGCTT-3′, with the AflII and HindIII sites underlined (3). Plasmids expressing Tat (residues 1–72) fused to SF1 (residues 2–307) or U2AF65 (residues 2–475) with a linker of three glycines were described previously (3). For Tat-hybrid assays, 25 ng of a BPS FFL reporter plasmid typically was cotransfected with 25 ng of an HIV-1 TAR Renilla luciferase (RL) reporter and 5 ng of a Tat-fusion expressor plasmid into HeLa cells using Polyfect (Qiagen) or Lipofectamine 2000 (Invitrogen). FFL and RL activities from triplicate transfections were measured after 48 h using Dual-Glo luciferase assays (Promega). FFL values were normalized to RL values, which independently monitor Tat-fusion expression levels by activation of the HIV-1 TAR reporter by the Tat portion of the fusion, and to FFL values from parallel transfections without the expressor to determine fold activation levels over basal transcription. Data are presented as percent of UACUAAC activity or fold activation over reporter alone.
RESULTS
An initial BPS profile model based on SF1 binding sequences
The sequence requirements for the BPS overlap with the SF1 binding site to an unknown but significant degree, as suggested by the high affinity of yeast and mammalian SF1 for the UACUAAC yeast BPS (20). We reasoned that this overlap might be exploited to help identify BPSs, particularly those that utilize SF1 for splicing, and constructed a BPS profile model using sequences bound by SF1 with varying affinities. An initial model (initial SF1 binding model) was constructed using nine equally-weighted experimentally-determined SF1 binding sequences, five of which also are experimentally validated BPSs (see ‘Materials and Methods’ section for sequences) (3,14). Nucleotide frequencies were incorporated into a log-odds scoring matrix, which was used to predict BPSs in a set of 117 499 unique human intronic regions, each consisting of 199 nt upstream of the 3′-splice site and the first exonic nucleotide. To enrich for high-scoring matches, we applied a bit score threshold such that no more than one random match would be expected per intronic region. The top-scoring matches, corresponding to putative BPSs scoring above threshold, are distributed across the intronic region (Figure 1). A peak is observed from positions −45 to −16, corresponding to a region of frequently observed BPSs (1,10,11) and referred to as the expected BPS region, with a similar distribution to that previously observed (44). A plateau of matches is observed for all analyzed positions between −50 and −199. A peak is also observed at the −4 position that may reflect overlapping sequence preferences between the BP adenosine and the PPT (Figure 1 legend). Two other models (SF1 Affinity Model and Literature BPS Model) described below, as well as the mammalian YNCURAY consensus, show similar distributions (Figure 1). The distribution of matches to a U2AF65 Affinity Model, generated using sequences that are tightly bound by U2AF65 (37), shows the expected location of PPTs directly upstream of 3′-splice sites.
Figure 1.

Comparison of SF1 binding models to other profile models. Results of profile model searches in 117 499 human intronic regions (nucleotides from −199 to +1 relative to the 3′-splice site). For BPS models, the number of best matches above threshold (‘Materials and Methods’ section) was plotted based on the location of the predicted BP A. For the U2AF65 Affinity Model the number of best matches above threshold was plotted using the 5′-start position of the predicted PPT. The dotted box represents the expected BPS region (from −45 to −16). The area under the curve in the expected region, in excess of the number of background matches estimated from the number of matches at positions from −198 to −90, was used to evaluate and compare the models (‘Materials and Methods’ section). An unexpected sharp peak at position −4 was observed in all BPS models. This is the single position where the BP A does not conflict with the preferred pyrimidine preferences of the PPT. Indeed, a BP A has been mapped to position −4 of intron 3 of the human XPC gene, which also has a second BP A at position −24. Mutation of either BPS results in a variably penetrant form of familial xeroderma pigmentosum (18). The BPS with a BP A at position −24 is the top-scoring match for the SF1 Affinity Model while the BPS with BP A at position −4 also scores above threshold. The inset shows the distribution of profile matches with the y-axis scaled to display the complete profile match distribution for the U2AF65 Affinity Model.
Of the 117 499 human introns, 65 813 introns (56%) were found to have at least one best profile match with a score above threshold, using the Initial SF1 Binding Model (‘Materials and Methods’ section). We considered only the best match of the model to the intron, accounting for tied scores proportionately at each matched position, leading to a fractional measure of best matches at each site of a predicted BP A. We next calculated an average background level of best matches at each position in the range from −189 to −90 from the 3′-splice site. After subtracting this background value from the actual number of best matches, 88.4% of remaining best matches above threshold identified by the Initial SF1 Binding Model were located in the expected BPS region, from −45 to −16. To evaluate profile model performance, we calculated the SNR ratio. The signal value was defined as the number of best matches above threshold in the expected BPS or PPT region minus the background estimate, while the noise was defined as the estimate of background matches (‘Materials and Methods’ section). The resulting SNR value of 0.81 suggests low enrichment for BPSs but may be an underestimate, in part because the estimate of background matches includes functional BPSs that occur upstream of the expected BPS region.
Branch point mapping validates BPS predictions
Given the low SNR of the Initial SF1 Binding Model, we performed in vitro branch point mapping (36,43) to help evaluate profile model predictions. We selected seven introns, all from genes highly expressed in HeLa cells, with high-scoring BPSs, intermediate- to high-scoring PPTs, and spacings between the BP A and the start of the PPT consistent with a cooperative SF1-U2AF65 interaction (‘Materials and Methods’ section; Supplementary Table S1). We engineered these BPS and PPT regions into a D. melanogaster fushi tarazu (Ftz) pre-mRNA, known to splice robustly in HeLa cell nuclear extracts (40), performed in vitro splicing reactions, and mapped the branch points from purified lariat and 3′-exon intermediates using primer extension (Figure 2). Six of the seven introns tested showed the BP A at the predicted position. The one exception (AP2M1 intron 11) mapped to an adenosine 1 nt upstream of the predicted BP A (UUCUAAC versus UUCUAAC). This result is consistent with the observations that U2 snRNA can hybridize to adenosines at either positions 5 or 6 of the BPS, bulging out the other (45), and that reverse transcriptase can stop within 1 nt of the branch point (46,47). Thus, it appears that all of the selected BPSs are indeed functional.
Figure 2.

Branch point mapping of initial model predictions. Seven introns were chosen for branch point mapping in the genes indicated (HUGO gene identifiers), with the predicted BPSs shown and the predicted BP A’s highlighted. Ftz WT corresponds to the D. melanogaster fushi tarazu pre-mRNA containing its native BPS and PPT while all others represent the Ftz pre-mRNA engineered with predicted BPSs and PPTs from: ZMPSTE24—zinc metalloproteinase STE24 intron 8; CDC20—cell division cycle 20 intron 8; HMGN3—high-mobility group nucleosomal binding domain 3 intron 5; PPS1—phosphoribosyl pyrophosphate synthase intron 6; snRNP F—small nuclear ribonucleoprotein F intron 2; MSN—moesin intron 11; AP2M1—adaptor-related protein complex 2, mu 1 intron 11. For each intron, DNA sequencing lanes were used to locate the branch point, observed as reverse transcriptase stops in primer extensions reactions. The ‘dbr’ lanes are primer extensions performed on purified lariat/3′-exon intermediates treated (+) or not treated (−) with debranching enzyme. Red dots correspond to predicted branch points and arrows correspond to mapped branch points. Of note, the mapped branch point for HMGN3 intron 5 is interpreted as the same as the predicted branch point. However, the darkest band on the gel corresponds to the location of the cytosine at position 7. This is in keeping with the observation that reverse transcriptase can stop within a nucleotide of the actual branch point, as described in the text.
Initial SF1 binding model predicts SF1 binding sites
We next wished to assess whether the Initial SF1 Binding Model could correctly identify SF1 binding sites. We selected a range of BPS scores, including 10 of the highest scoring sequences and 8 of the lowest scoring sequences (including 3 sequences tied for the lowest score). We also examined all of the sequences that were identified as best matching the model in the expected BPS region of each intron, and from these selected the 11 sequences found most frequently. Finally, we examined all of the sequences identified by the model in the expected BPS region of each intron and selected two frequently observed sequences (Supplementary Table S2). We engineered the sequences into the Tat-hybrid system, in which transcription is activated at the HIV promoter using a fusion between an RNA-binding protein and the HIV transcription activator, Tat (Figure 3A). In this case, a fragment of SF1 (residues 2–307) was fused to the C-terminus of Tat (residues 1–72). This fusion protein can be recruited to a HIV-1 promoter engineered with a BPS, adenovirus major late (AdML) PPT, and 3′-splice site AG dinucleotide, which replace the cognate HIV Tat trans-activating RNA (TAR), and thereby activate transcription of a FFL reporter (Figure 3A) (3). This cellular reporter system supports assembly of multi-protein complexes with endogenous proteins, including U2AF65, and has been used to demonstrate a requirement for cooperative interaction between SF1 and U2AF65 in binding to the BPS and PPT (3).
Of the 10 highest scoring sequences, 7 were at least 50% as active as the UACUAAC reporter, the best SF1 binder reported to date (Figure 3B) (3,20), whereas only 2 of the 8 lowest scoring sequences (AUAACCA and GUCUUGA) gave a minimum of 50% activity, and most are very poor binders (Figure 3C). Interestingly, AUAACCA does not conform to the YNCURAY mammalian consensus and lacks adenosine at the branch position, but is bound by SF1 comparably to UACUAAC. It is possible that AUAACCA may be an unusual BPS or may bind SF1 but does not function as a BPS. Alternatively, if the site were shifted by 1 nt to the adjacent UAACCAC sequence, the score would improve substantially (from −7.97 to 3.81). Similarly, shifting GUCUUGA by 1 nt to the adjacent UCUUGAC would increase the score from −3.11 to 2.74, shifting ACAGUCA to CAGUCAC would increase the score from −8.37 to 2.06, and shifting ACUACCA to CUACCAC would increase the score from −8.70 to −3.84. In contrast, several sequences that had low bit-scores and bound SF1 poorly did not have alternate registers with higher scores, such as CGAACAG (−4.34), ACGGUGG (−11.24), UGAGUAG (−3.30), ACGGCGG (−10.61) and UAGUGAC (−5.15). However it is difficult to systematically account for possible register shifts because we cannot directly ascertain whether the measured activity results from a shifted binding site. Of the 11 frequently observed sequences tested, SF1 binds seven with at least 50% the activity of the UACUAAC reporter (Figure 3D). As a control, no SF1 binding was observed with reporters containing a mutant PPT (data not shown), which eliminates U2AF65 binding and the cooperative interaction with SF1 (3). Thus, BPS scores calculated using the Initial SF1 Binding Model discriminate between binding and non-binding sequences.
We found four genes reported to have BPS mutations associated with genetic disease: tyrosine hydroxylase (TH) intron 11, lecithin cholesterol acetyltransferase (LCAT) intron 4, integrin beta 4 (ITGB4) intron 31, and collagen 5A1 (COL5A1) intron 32 (15–17,19). Each of the mutant BPSs had a lower score by the Initial SF1 Binding Model than its corresponding wild-type BPS. In the reporter assay, all wild-type sequences showed moderate to weak SF1 binding and three of the four showed a non-significant reduction in binding upon mutation, with the LCAT BPS mutant having the most significant decrease when the conserved U at position 4 was changed to C (Figure 3E). In contrast, the same U changed to G in the COL5A1 BPS resulted in an unexpected increase in SF1 binding. The relatively weak activities observed do not obviously support a role for SF1 in binding to these BPSs, but we note that the sites in these reporters were placed in the context of the strong adenovirus PPT and not in their natural intronic contexts.
To expand our data set of SF1 binding sequences and to allow us to further evaluate and refine the computational model, we determined the activities of UACUAAC point mutants at each of the seven positions. Of the 21 possible single nucleotide substitutions, 11 were already present in the set of high frequency and high-scoring sequences. Of the 10 remaining unique sequences, six were substituted in the conserved U at position 4 or the conserved A at position 6, suggesting that these would be bound poorly (21) and indeed, we observed that only 1 of these 10 sequences (AACUAAC) bound SF1 above the 50% UACUAAC value (Figure 3F). While these single point mutations can significantly reduce SF1 binding, their bit scores generally are close to those of the high-scoring sequences (Supplementary Table S2). Thus, while the Initial SF1 Binding Model discriminates reasonably well between tight- and weak-binding sequences, it performs relatively poorly for intermediate affinities. The Pearson correlation coefficient between bit scores and SF1 affinities for the highest and lowest scoring sequences (‘Materials and Methods’ section) is 0.47, but it is only 0.29 for the entire set of 46 sequences assayed (Figure 3G). The lack of a strong correlation is consistent with under-sampling of the 16 384 possible 7-mers, but the model performed particularly poorly when considering sequences with intermediate SF1 affinities and required further refinement.
An SF1 affinity-weighted BPS profile model and model comparisons
The branch point mapping and SF1 binding data encouraged us to test whether the model could be refined by explicitly incorporating affinity data. We chose to use data from the Tat-hybrid assays rather than in vitro binding data, as Tat-hybrid assays more likely represent in vivo assembly and cooperative U2AF complexes. We generated an SF1 Affinity Model in which the 46 sequences tested for binding were proportionally weighted according to their measured affinities (‘Materials and Methods’ section). This model produced a similar location distribution of profile matches as the other models (Figure 1). Of the set of 117 499 human intron sequences, 65 018 (55%) had profile matches with scores above threshold. After subtracting the average background from each position as before, 87.9% (9868) of remaining best matches were located in the expected BPS region (‘Materials and Methods’ section) (Figure 4).
Figure 4.

Evaluation of BPS and PPT profile models. Sequence logos representing each BPS and PPT profile model [Weblogo (29)] are shown with statistics for each model: ‘% of Introns with Above Threshold Matches’ is the percent of introns with any profile match above threshold (‘Materials and Methods’ section) in the entire intronic region from −199 to +1; ‘% of Above-Background Best Matches in Expected Region’ was determined by first calculating an average background level of best matches at each position in the range from −189 to −90 from the 3′-splice site, subtracting this background value from number of best matches at each position, and comparing the number of these residual best matches in the expected BPS region (BP A in −45 to −16) or PPT region (5′-PPT start of match in the region from −30 to −11) to the number of expected background matches in the expected region (‘Materials and Methods’ section); ‘Signal-to-Noise Ratio’ is the improvement over random based on the residual number of best matches in the expected region relative to the background estimate; ‘Correlation with SF1 Affinity’ is the Pearson correlation coefficient between profile model bit scores and SF1 affinities for the 46 sequences assayed.
We compared our SF1 affinity-based models to three others: the mammalian YNCURAY consensus, a previously published pentamer model (26), and a ‘Literature BPS’ model based on equal weighting of 27 experimentally verified BPSs from the literature (15–19,32–36). The BPS profile models identified variable numbers of introns with above-threshold matches ranging from 36 to 63%. However, all BPS models identified a peak in the expected BPS region. After subtracting the average background for each model in the range from −189 to −90, from 86 to 90% of remaining best matches localized to this region (Figure 4). We also included a well-established U2AF65 affinity model describing the PPT(37) (Figure 4). The U2AF65 Affinity Model had above-threshold matches in 88% of introns, although a direct comparison to the BPS profile models is precluded due to the more constrained location of the PPT relative to the 3′-splice site and the lower sequence complexity of this motif.
Our SF1 Affinity Model had an improved SNR (defined in ‘Materials and Methods’ section) of 1.10 compared to the Initial SF1 Binding Model (0.81) but this SNR is lower than those of the YNCURAY (1.63), Pentamer BPS (1.56) and Literature BPS (1.19) models. Given that not all introns are expected to bind SF1, our SF1-based model is not expected to perform as well as others in predicting all types of BPSs. However, the low SNR also may reflect the inability of the SF1 Affinity Model to fully represent SF1 binding affinity preferences due to the relatively limited number of sequences with affinity data used for model construction.
Pearson correlation coefficients between model scores and SF1 affinity data provide another way to compare models. As expected, the correlation coefficient for the SF1 Affinity Model (0.39) is highest (Figure 4), although all BPS models have correlation coefficients of at least 0.29. The U2AF65 Affinity Model (−0.14) serves as an uncorrelated control. A comparison of scatter plots between the initial and final models (Figure 3G and H) shows the relatively weak correlations and slight improvement in the final SF1 Affinity Model. Of note, substituting the register-shifted BPSs described above for the originally selected BPSs results in higher correlation coefficients for both the Initial SF1 Model (0.38) and SF1 Affinity Model (0.47) (data not shown).
As a further test of model quality, we evaluated how well the models predict a set of independently validated BPSs in 50 introns from 20 human housekeeping genes initially evaluated by Gao et al. (9). Each BPS was scored using our models and the fraction correctly predicted was calculated. No model showed high agreement (Supplementary Table S5), but our SF1 Affinity Model performed best (0.36) in the expected BPS region and in the Gao et al. BPS range, suggesting that model refinement using SF1 binding data enhances identification of BPSs, whether or not they depend on SF1 for splicing. While it is interesting that all BPS models exhibit some correlation with SF1 binding affinities, even those derived purely from sequence preferences, the generally weak performance of the SF1 Affinity Model suggests that other criteria are needed to enhance BPS predictions, supporting the view that the BPS itself is only one of several factors that specify the location of the branch site (25). Across the BPS models, C at position 3, U at position 4 and A at position 6 have the highest information content (Figure 4). The sequence preferences of the Initial SF1 Binding Model are close to the yeast UACUAAC sequence, but with more degeneracy. This in part may be due to the limited and biased sequence data used to construct the model. Nevertheless, it is interesting that the preferences of the SF1 Affinity Model gravitate towards the mammalian YNCURAY consensus, as all but the first two positions of the SF1 Affinity Model consensus sequence appear more degenerate than the YNCURAY consensus, suggesting that a subset of SF1 binding sites may function as BPSs, especially when localized to the expected BPS region.
Identification of introns with potential SF1 binding sites
SF1 may not be a constitutive splicing factor but may act at specific introns or participate in regulated or alternative splicing (24). No SF1-dependent introns have yet been identified nor have objective criteria been developed to identify candidate introns. Our SF1 Affinity Model may provide a tool for this purpose, particularly in conjunction with the U2AF65 Affinity Model and spacing parameters for the BPS and PPT that might approximate the cooperative interaction of SF1 and U2AF65 when bound to RNA. Combining BPS and PPT sequence-based models with location criteria has been found to enhance BPS identification (25). To test whether our affinity-weighted models could help identify candidate SF1-dependent introns, we searched our 148 643 intron data set to identify 10 introns with high-BPS and high-PPT scores and with BPS matches that have previously been experimentally validated as functional BPSs (high BPS–high PPT, most likely to bind SF1), 10 with low-BPS and high-PPT scores (low BPS–high PPT, less likely to bind SF1), and 5 with low BPS and low PPT scores (low BPS–low PPT, least likely to bind SF1) (see ‘Materials and Methods’ section for detailed selection parameters and Supplementary Table S3 for list of introns) (Figure 5A). The distribution of scores for all SF1 Affinity Model best matches scoring above threshold in the expected BPS region is shown as the histogram inset in Figure 5A. For high- and low-BPS matches, respectively, we selected intronic regions with SF1 Affinity Model scores in the top 12th percentile and bottom 1st percentile of scores for all best-matches scoring above threshold in the expected BPS region.
Figure 5.

SF1 binding to intronic region reporters using the Tat-hybrid assay. (A) Twenty-five 100-nt intronic regions were chosen from the intron database based on SF1 Affinity Model criteria and were tested for binding using LTR FFL reporters in Tat-hybrid assays. HUGO gene names are shown. High BPS–high PPT, low BPS–low PPT and low BPS–high PPT describe the intron selection criteria detailed in ‘Materials and Methods’ section. Fold activation represents the activity of the Tat72-SF1 fusion relative to the reporter alone. The UACUAAC PPT reporter was used as a positive control and calibration standard for SF1 binding in all experiments. The UACUAAC mPPT reporter was used to estimate background levels of activation. The histogram in the inset shows the SF1 Affinity Model score distribution for high scoring matches in the expected region. (B) Correlation between bit score calculated using the SF1 Affinity Model and measured SF1 affinities as in Figure 3H.
To evaluate SF1 binding in a relatively native sequence context, we measured activities of Tat-fused SF1 on HIV LTR reporters containing 100 nt upstream of each 3′-splice site. Of the 10 intronic regions with high-BPS and high-PPT scores (‘Materials and Methods’ section), 7 were bound by SF1 (compare to the mutant PPT negative control; Figure 5A) whereas only one each of the low BPS–low PPT and low BPS–high PPT sets showed activity above that seen with the negative control. The correlation between BPS scores and fold activation in this data set (Figure 5B) produced a Pearson correlation coefficient of 0.73, and there were no obvious sequence similarities among the highly active sequences. Binding of Tat-fused U2AF65 showed similar activities across all reporters (data not shown), consistent with the need for cooperative SF1/U2AF binding to activate in these assays (3). Thus, SF1 and U2AF65 affinity parameters appear to provide useful tools to identify candidate SF1-binding introns.
DISCUSSION
We constructed an SF1 affinity-weighted BPS profile model using an iterative computational and experimental approach and have used it as a tool to experimentally identify candidate SF1-binding introns. The model identifies possible SF1 binding sites located in the expected BPS region with a frequency similar to that of other BPS models that are based solely on sequence information. Although none of the models perform particularly well, the SF1 Affinity Model is able to predict BPSs with higher accuracy than the other models, based on a comparison using an independent set of validated BPSs, suggesting that the requirements for SF1 binding may be dictated by factors other than nucleotide sequence. This is further highlighted by the low SNRs exhibited by all models, which reflect in part the high degeneracy of the sequence motifs and is consistent with the suggestion that the BPS is one of just several features needed to specify the branch site (25). However, when the SF1 Affinity Model is combined with a U2AF65 Affinity Model and spacing of the sites is taken into account to approximate the cooperative protein–protein interaction on the RNA (‘Materials and Methods’ section), it is possible to successfully identify SF1-binding introns. This approach is related to one in which the BPS and PPT were scored based on sequence preferences, without affinity data, and spaced only within 6 nt of each other (25). That study evaluated a smaller set of introns and achieved some statistical success, but predicted BPSs were not examined experimentally and the involvement of SF1 was not explicitly considered a factor.
The SF1 Affinity Model identified sequences scoring above a threshold in 55% of introns. Within the expected BPS region, we found 87.9% of best matches above threshold, after subtracting average background. These estimates are consistent with previous suggestions that not all human introns require SF1 for splicing (22,24). Most high-scoring sequences outside the expected region are likely to be false positives (26), but some branch points have been found far upstream (35,48,49) and our model correctly predicts the BPS as the highest scoring site in three of four introns with known distant BPSs (human PTB intron 10, rat α-actinin intron 18, and rat α-tropomyosin intron 1; data not shown).
Our model is limited by several factors: the profile model treats each position of the BPS independently and thus cannot capture pairwise or higher order correlations between nucleotides, such as base stacking preferences for SF1 recognition (21). The SF1 affinity data used reflect a cooperative interaction with U2AF65 and not the SF1–BPS interaction alone, although it more faithfully represents the in vivo context. The model was constructed with a relatively small sequence set biased toward the tight-binding UACUAAC sequence. Some sequences that bind SF1 have been incorporated into the model but may not function as BPSs, such as AUAACCA and GUCUUGA. Such sequences may represent binding sites for other proposed SF1 functions, such as transcriptional repression or nuclear mRNA retention (22,50), and it is interesting that neither AUAACCA nor GUCUUGA show a preferential distribution in the expected BPS region (data not shown). Despite the lack of a BP A, these sequences could still function as BPSs, especially if located near a PPT. For example, a C located 28 nt upstream of the 3′-splice site is used as the branch point in intron A of human growth hormone, and a U located 23 nts upstream of the 3′-splice site is the branch point in intron 4 of calcitonin/CGRP-I (33,34). Alternatively, low-scoring sequences apparently bound by SF1 in our reporter experiments (Figure 3) might actually reflect binding to higher scoring sequences shifted in register by one or more nucleotides, as highlighted by increased correlation coefficients calculated using high-scoring shifted sequences. Nevertheless, it is difficult to assess which cases may reflect a register shift, but the number is expected to be small and therefore not substantially change the SF1 Affinity Model.
Of the ten 100-nt intronic regions containing sequence profile matches with high BPS and PPT scores tested using the Tat-hybrid intronic context assay, three bound SF1 poorly (Figure 5). Despite their predicted high BPS scores, two of these intronic regions, RNF41 and DOK5, have predicted BPSs (Supplementary Table S3) that also were bound poorly by SF1 in the context of the strong adenovirus PPT (Figure 5). The third, TPK1, has a BPS that was comparable to UACUAAC when assayed with the adenovirus PPT, suggesting that binding in its native context may be affected by the flanking RNA sequence or structure or by the binding of other proteins. Furthermore, other regions of the intron or surrounding exons, not included in the reporters, may influence SF1 complex formation. Despite these limitations, our model accurately identified introns bound by SF1 in the majority of cases.
The sequences bound by SF1 are largely representative of both the mammalian consensus BPS, YNCURAY (20), as well as the recently published human consensus BPS, YUNAY (9), suggesting that many of these sites function as BPSs. Other sequences that conform to the consensus but bind SF1 poorly may function as BPSs through an SF1-independent mechanism or may require additional factors to stabilize the SF1–BPS interaction not recruited in the Tat-hybrid assay. Indeed, it has been proposed that the U2 snRNP-BPS interaction may be mediated predominantly by the SF3a and SF3b integral U2 snRNP proteins, using sequences surrounding the BPS (51,52). The p14 subunit of SF3b has been shown to crosslink to the BP A of pre-mRNA, and the SF3b155 subunit of SF3b interacts directly with U2AF65 (53). RNAi knockdown of SF3a also causes a splicing defect in several genes (24). It will be interesting to distinguish which introns splice in SF1-dependent and -independent modes and to compare their underlying splicing mechanisms and possible roles in regulating splicing.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institutes of Health grants GM47478 (ADF), GM61987 (DCR) and P20 GM068136, K22 HG00056, GM071655 (SEB); a Sloan Research Fellowship, Searle Scholars Program 01-L-116 (SEB); a University of California Quantitative Biology Institute Initiative grant; a University of California Graduate Research and Education in Adaptive Biotechnology (GREAT) pre-doctoral fellowship 2004-21 (to A.W.P.). Funding for open access charge: Department of Biochemistry and Biophysics, University of California, San Francisco.
Conflict of interest statement. None declared.
ACKNOWLEDGEMENTS
We thank Richard Green for the mRNA–genome and mRNA–mRNA alignments, and splice site data, and Richard Green, Gavin Crooks, Maki Inada, Donna Hendrix, Liana Lareau, Jocelyn Grunwell and Valerie Calabro for helpful discussions.
REFERENCES
- 1.Burge CB, Tuschl T, Sharp PA. Splicing of Precursors to mRNAs by the Spliceosomes. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1999. [Google Scholar]
- 2.Berglund JA, Abovich N, Rosbash M. A cooperative interaction between U2AF65 and mBBP/SF1 facilitates branchpoint region recognition. Genes Dev. 1998;12:858–867. doi: 10.1101/gad.12.6.858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Peled-Zehavi H, Berglund JA, Rosbash M, Frankel AD. Recognition of RNA branch point sequences by the KH domain of splicing factor 1 (mammalian branch point binding protein) in a splicing factor complex. Mol. Cell. Biol. 2001;21:5232–5241. doi: 10.1128/MCB.21.15.5232-5241.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wu J, Manley JL. Mammalian pre-mRNA branch site selection by U2 snRNP involves base pairing. Genes Dev. 1989;3:1553–1561. doi: 10.1101/gad.3.10.1553. [DOI] [PubMed] [Google Scholar]
- 5.Wu JA, Manley JL. Base pairing between U2 and U6 snRNAs is necessary for splicing of a mammalian pre-mRNA. Nature. 1991;352:818–821. doi: 10.1038/352818a0. [DOI] [PubMed] [Google Scholar]
- 6.Bindereif A, Green MR. An ordered pathway of snRNP binding during mammalian pre-mRNA splicing complex assembly. Embo J. 1987;6:2415–2424. doi: 10.1002/j.1460-2075.1987.tb02520.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Parker R, Siliciano PG, Guthrie C. Recognition of the TACTAAC box during mRNA splicing in yeast involves base pairing to the U2-like snRNA. Cell. 1987;49:229–239. doi: 10.1016/0092-8674(87)90564-2. [DOI] [PubMed] [Google Scholar]
- 8.Rino J, Carmo-Fonseca M. The spliceosome: a self-organized macromolecular machine in the nucleus? Trends Cell Biol. 2009;19:375–384. doi: 10.1016/j.tcb.2009.05.004. [DOI] [PubMed] [Google Scholar]
- 9.Gao K, Masuda A, Matsuura T, Ohno K. Human branch point consensus sequence is yUnAy. Nucleic Acids Res. 2008;36:2257–2267. doi: 10.1093/nar/gkn073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lim LP, Burge CB. A computational analysis of sequence features involved in recognition of short introns. Proc. Natl Acad. Sci. USA. 2001;98:11193–11198. doi: 10.1073/pnas.201407298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Norton PA. Polypyrimidine tract sequences direct selection of alternative branch sites and influence protein binding. Nucleic Acids Res. 1994;22:3854–3860. doi: 10.1093/nar/22.19.3854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Burnette JM, Miyamoto-Sato E, Schaub MA, Conklin J, Lopez AJ. Subdivision of large introns in Drosophila by recursive splicing at nonexonic elements. Genetics. 2005;170:661–674. doi: 10.1534/genetics.104.039701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kralovicova J, Houngninou-Molango S, Kramer A, Vorechovsky I. Branch site haplotypes that control alternative splicing. Hum. Mol. Genet. 2004;13:3189–3202. doi: 10.1093/hmg/ddh334. [DOI] [PubMed] [Google Scholar]
- 14.Zhuang YA, Goldstein AM, Weiner AM. UACUAAC is the preferred branch site for mammalian mRNA splicing. Proc. Natl Acad. Sci. USA. 1989;86:2752–2756. doi: 10.1073/pnas.86.8.2752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Burrows NP, Nicholls AC, Richards AJ, Luccarini C, Harrison JB, Yates JR, Pope FM. A point mutation in an intronic branch site results in aberrant splicing of COL5A1 and in Ehlers-Danlos syndrome type II in two British families. Am. J. Hum. Genet. 1998;63:390–398. doi: 10.1086/301948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chavanas S, Gache Y, Vailly J, Kanitakis J, Pulkkinen L, Uitto J, Ortonne J, Meneguzzi G. Splicing modulation of integrin beta4 pre-mRNA carrying a branch point mutation underlies epidermolysis bullosa with pyloric atresia undergoing spontaneous amelioration with ageing. Hum. Mol. Genet. 1999;8:2097–2105. doi: 10.1093/hmg/8.11.2097. [DOI] [PubMed] [Google Scholar]
- 17.Janssen RJ, Wevers RA, Haussler M, Luyten JA, Steenbergen-Spanjers GC, Hoffmann GF, Nagatsu T, Van den Heuvel LP. A branch site mutation leading to aberrant splicing of the human tyrosine hydroxylase gene in a child with a severe extrapyramidal movement disorder. Ann. Hum. Genet. 2000;64:375–382. doi: 10.1046/j.1469-1809.2000.6450375.x. [DOI] [PubMed] [Google Scholar]
- 18.Khan SG, Metin A, Gozukara E, Inui H, Shahlavi T, Muniz-Medina V, Baker CC, Ueda T, Aiken JR, Schneider TD, et al. Two essential splice lariat branchpoint sequences in one intron in a xeroderma pigmentosum DNA repair gene: mutations result in reduced XPC mRNA levels that correlate with cancer risk. Hum. Mol. Genet. 2004;13:343–352. doi: 10.1093/hmg/ddh026. [DOI] [PubMed] [Google Scholar]
- 19.Li M, Pritchard PH. Characterization of the effects of mutations in the putative branchpoint sequence of intron 4 on the splicing within the human lecithin:cholesterol acyltransferase gene. J. Biol. Chem. 2000;275:18079–18084. doi: 10.1074/jbc.M910197199. [DOI] [PubMed] [Google Scholar]
- 20.Berglund JA, Chua K, Abovich N, Reed R, Rosbash M. The splicing factor BBP interacts specifically with the pre-mRNA branchpoint sequence UACUAAC. Cell. 1997;89:781–787. doi: 10.1016/s0092-8674(00)80261-5. [DOI] [PubMed] [Google Scholar]
- 21.Liu Z, Luyten I, Bottomley MJ, Messias AC, Houngninou-Molango S, Sprangers R, Zanier K, Kramer A, Sattler M. Structural basis for recognition of the intron branch site RNA by splicing factor 1. Science. 2001;294:1098–1102. doi: 10.1126/science.1064719. [DOI] [PubMed] [Google Scholar]
- 22.Rutz B, Seraphin B. A dual role for BBP/ScSF1 in nuclear pre-mRNA retention and splicing. Embo J. 2000;19:1873–1886. doi: 10.1093/emboj/19.8.1873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Guth S, Valcarcel J. Kinetic role for mammalian SF1/BBP in spliceosome assembly and function after polypyrimidine tract recognition by U2AF. J. Biol. Chem. 2000;275:38059–38066. doi: 10.1074/jbc.M001483200. [DOI] [PubMed] [Google Scholar]
- 24.Tanackovic G, Kramer A. Human splicing factor SF3a, but not SF1, is essential for pre-mRNA splicing in vivo. Mol. Biol. Cell. 2005;16:1366–1377. doi: 10.1091/mbc.E04-11-1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kol G, Lev-Maor G, Ast G. Human-mouse comparative analysis reveals that branch-site plasticity contributes to splicing regulation. Hum. Mol. Genet. 2005;14:1559–1568. doi: 10.1093/hmg/ddi164. [DOI] [PubMed] [Google Scholar]
- 26.Harris NL, Senapathy P. Distribution and consensus of branch point signals in eukaryotic genes: a computerized statistical analysis. Nucleic Acids Res. 1990;18:3015–3019. doi: 10.1093/nar/18.10.3015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pruitt KD, Tatusova T, Maglott DR. NCBI reference sequences (RefSeq): a curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res. 2007;35:D61–D65. doi: 10.1093/nar/gkl842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wheelan SJ, Church DM, Ostell JM. Spidey: a tool for mRNA-to-genomic alignments. Genome Res. 2001;11:1952–1957. doi: 10.1101/gr.195301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Crooks GE, Hon G, Chandonia JM, Brenner SE. WebLogo: a sequence logo generator. Genome Res. 2004;14:1188–1190. doi: 10.1101/gr.849004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schneider TD, Stephens RM. Sequence logos: a new way to display consensus sequences. Nucleic Acids Res. 1990;18:6097–6100. doi: 10.1093/nar/18.20.6097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Blanchette M, Green RE, Brenner SE, Rio DC. Global analysis of positive and negative pre-mRNA splicing regulators in Drosophila. Genes Dev. 2005;19:1306–1314. doi: 10.1101/gad.1314205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nelson KK, Green MR. Mammalian U2 snRNP has a sequence-specific RNA-binding activity. Genes Dev. 1989;3:1562–1571. doi: 10.1101/gad.3.10.1562. [DOI] [PubMed] [Google Scholar]
- 33.Adema GJ, Bovenberg RA, Jansz HS, Baas PD. Unusual branch point selection involved in splicing of the alternatively processed Calcitonin/CGRP-I pre-mRNA. Nucleic Acids Res. 1988;16:9513–9526. doi: 10.1093/nar/16.20.9513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hartmuth K, Barta A. Unusual branch point selection in processing of human growth hormone pre-mRNA. Mol. Cell. Biol. 1988;8:2011–2020. doi: 10.1128/mcb.8.5.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Helfman DM, Ricci WM. Branch point selection in alternative splicing of tropomyosin pre-mRNAs. Nucleic Acids Res. 1989;17:5633–5650. doi: 10.1093/nar/17.14.5633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Krainer AR, Maniatis T, Ruskin B, Green MR. Normal and mutant human beta-globin pre-mRNAs are faithfully and efficiently spliced in vitro. Cell. 1984;36:993–1005. doi: 10.1016/0092-8674(84)90049-7. [DOI] [PubMed] [Google Scholar]
- 37.Singh R, Valcarcel J, Green MR. Distinct binding specificities and functions of higher eukaryotic polypyrimidine tract-binding proteins. Science. 1995;268:1173–1176. doi: 10.1126/science.7761834. [DOI] [PubMed] [Google Scholar]
- 38.Barrett T, Troup DB, Wilhite SE, Ledoux P, Rudnev D, Evangelista C, Kim IF, Soboleva A, Tomashevsky M, Edgar R. NCBI GEO: mining tens of millions of expression profiles–database and tools update. Nucleic Acids Res. 2007;35:D760–D765. doi: 10.1093/nar/gkl887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Carson JP, Zhang N, Frampton GM, Gerry NP, Lenburg ME, Christman MF. Pharmacogenomic identification of targets for adjuvant therapy with the topoisomerase poison camptothecin. Cancer Res. 2004;64:2096–2104. doi: 10.1158/0008-5472.can-03-2029. [DOI] [PubMed] [Google Scholar]
- 40.Rio DC. Accurate and efficient pre-mRNA splicing in Drosophila cell-free extracts. Proc. Natl Acad. Sci. USA. 1988;85:2904–2908. doi: 10.1073/pnas.85.9.2904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Melton DA, Krieg PA, Rebagliati MR, Maniatis T, Zinn K, Green MR. Efficient in vitro synthesis of biologically active RNA and RNA hybridization probes from plasmids containing a bacteriophage SP6 promoter. Nucleic Acids Res. 1984;12:7035–7056. doi: 10.1093/nar/12.18.7035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ruskin B, Krainer AR, Maniatis T, Green MR. Excision of an intact intron as a novel lariat structure during pre-mRNA splicing in vitro. Cell. 1984;38:317–331. doi: 10.1016/0092-8674(84)90553-1. [DOI] [PubMed] [Google Scholar]
- 43.Ruskin B, Green MR. RNA lariat debranching enzyme as tool for analyzing RNA structure. Methods Enzymol. 1990;181:180–188. doi: 10.1016/0076-6879(90)81120-j. [DOI] [PubMed] [Google Scholar]
- 44.Zhang XH, Heller KA, Hefter I, Leslie CS, Chasin LA. Sequence information for the splicing of human pre-mRNA identified by support vector machine classification. Genome Res. 2003;13:2637–2650. doi: 10.1101/gr.1679003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Query CC, Moore MJ, Sharp PA. Branch nucleophile selection in pre-mRNA splicing: evidence for the bulged duplex model. Genes Dev. 1994;8:587–597. doi: 10.1101/gad.8.5.587. [DOI] [PubMed] [Google Scholar]
- 46.Rodriguez JR, Pikielny CW, Rosbash M. In vivo characterization of yeast mRNA processing intermediates. Cell. 1984;39:603–610. doi: 10.1016/0092-8674(84)90467-7. [DOI] [PubMed] [Google Scholar]
- 47.Zeitlin S, Efstratiadis A. In vivo splicing products of the rabbit beta-globin pre-mRNA. Cell. 1984;39:589–602. doi: 10.1016/0092-8674(84)90466-5. [DOI] [PubMed] [Google Scholar]
- 48.Southby J, Gooding C, Smith CW. Polypyrimidine tract binding protein functions as a repressor to regulate alternative splicing of alpha-actinin mutally exclusive exons. Mol. Cell. Biol. 1999;19:2699–2711. doi: 10.1128/mcb.19.4.2699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wollerton MC, Gooding C, Wagner EJ, Garcia-Blanco MA, Smith CW. Autoregulation of polypyrimidine tract binding protein by alternative splicing leading to nonsense-mediated decay. Mol. Cell. 2004;13:91–100. doi: 10.1016/s1097-2765(03)00502-1. [DOI] [PubMed] [Google Scholar]
- 50.Zhang D, Paley AJ, Childs G. The transcriptional repressor ZFM1 interacts with and modulates the ability of EWS to activate transcription. J. Biol. Chem. 1998;273:18086–18091. doi: 10.1074/jbc.273.29.18086. [DOI] [PubMed] [Google Scholar]
- 51.Will CL, Luhrmann R. Spliceosomal UsnRNP biogenesis, structure and function. Curr. Opin. Cell Biol. 2001;13:290–301. doi: 10.1016/s0955-0674(00)00211-8. [DOI] [PubMed] [Google Scholar]
- 52.Will CL, Schneider C, MacMillan AM, Katopodis NF, Neubauer G, Wilm M, Luhrmann R, Query CC. A novel U2 and U11/U12 snRNP protein that associates with the pre-mRNA branch site. EMBO J. 2001;20:4536–4546. doi: 10.1093/emboj/20.16.4536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Spadaccini R, Reidt U, Dybkov O, Will C, Frank R, Stier G, Corsini L, Wahl MC, Luhrmann R, Sattler M. Biochemical and NMR analyses of an SF3b155-p14-U2AF-RNA interaction network involved in branch point definition during pre-mRNA splicing. RNA. 2006;12:410–425. doi: 10.1261/rna.2271406. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.