


Abstract
There are a variety of in vivo and in vitro methods to determine the genome-wide specificity of a particular trans-acting factor. However there is an inherent limitation to these candidate approaches. Most biological studies focus on the regulation of particular genes, which are bound by numerous unknown trans-acting factors. Therefore, most biological inquiries would be better addressed by a method that maps all trans-acting factors that bind particular regions rather than identifying all regions bound by a particular trans-acting factor. Here, we present a high-throughput binding assay that returns thousands of unbiased measurements of complex formation on nucleic acid. We applied this method to identify transcriptional complexes that form on DNA regions upstream of genes involved in pluripotency in embryonic stem cells (ES cells) before and after differentiation. The raw binding scores, motif analysis and expression data are used to computationally reconstruct remodeling events returning the identity of the transcription factor(s) most likely to comprise the complex. The most significant remodeling event during ES cell differentiation occurred upstream of the REST gene, a transcriptional repressor that blocks neurogenesis. We also demonstrate how this method can be used to discover RNA elements and discuss applications of screening polymorphisms for allelic differences in binding.
INTRODUCTION
A mechanistic understanding of gene expression requires the identification and mapping of protein–nucleic acid interactions. Transcription is mediated through complex interactions between transcription factors and chromatin that modulate the state or binding of basal transcription factors to core transcriptional elements. A variety of in vivo immunoprecipitation methods have been developed to determine occupancy of gene expression elements on a genomic scale (1,2). Chromatin immunoprecipitation (ChIP) has been used to study DNA–protein interactions (3). Co-immunoprecipitation of DNA with candidate trans-acting factors can be analyzed by array (ChIP-chip) or by high throughput sequencing (ChIP-seq).
Alternatively, there are high throughput in vitro approaches to identify target regions of a specific candidate factor. DNA binding arrays directly visualize binding events between fluorescently labeled recombinant proteins and doublestranded synthetic DNA ligands that are attached to a glass slide (4). The binding specificities of hundreds of factors have been reconstructed from these array measurements and are publicly available (5). A similar method, Selective Evolution of Ligand by EXponential enrichment (SELEX) is an iterative binding assay that selects high affinity ligands from a pool of random oligonucleotides (6). We have previously developed a method, MEGAshift, that is similar to SELEX but uses real sequence instead of starting with a randomized oligonucleotide pool (7). Specific DNA regions are resynthesized as a tiled oligonucleotide pool on a commercial DNA oligonucleotide array (8). The selected region is tiled across in 30 nt windows that shift by 10 bp, providing 10-nt resolution. These oligonucleotides are released from the slide and then used in a binding assay. A detection array complimentary to the synthesized array is used to measure the degree to which each oligonucleotide is enriched in the bound fraction. The array returns binding measurements for each oligonucleotide, which can be used to define biologically relevant motifs and to create binding maps.
While these approaches are useful in understanding target regions for a specific factor, they provide no opportunity to discover additional factors that can compete with or cooperate with the candidate factor to regulate the set of target genes. Mapping single factors does not provide information on how elements work together. This requires the development of new tools that map multiple factors on a limited region in order to determine spatial patterns among binding events. Recent efforts to model regulated transcriptional systems provide a useful conceptual framework for the study of cis elements (9,10). Previous studies of transcription have demonstrated how binding context influences function (11–13). For example, a protein can function as an activator or a repressor depending on the context in which it binds. Also, combinations of simple arrangements of overlapping and adjacent factor binding sites can result in elaborate cis-regulatory circuitry capable of integrating complex streams of environmental signals into a single transcriptional output. In order to define these complex dependencies experimentally a complete ‘parts list’ is required. The analysis of the sea urchin gene endo 16, one of the few systems studied with this level of rigor, revealed an elaborate cis-regulatory circuitry capable of responding to environmental inputs and propagating regulatory signals from distal elements (14).
A method that assays the binding affinity of all factors in cellular extract to an entire promoter or other nucleic acid regulatory regions would complement these existing technologies. DNA footprinting also returns a map of protein occupancy on a small region of nucleic acid. However, these assays are technically challenging and low throughput. High throughput FAIRE and DNAse sensitivity assays provides a map of unoccupied regions in vivo but at low resolution (hundreds of nucleotides). Here we present a high-throughput high resolution means of mapping occupancy on regions of interest that are defined by the user. Based on the MEGAshift protocol, the predefined sequences are incubated with cellular extract and nucleic acid-factor complexes are separated by a filter binding assay in order to determine where complexes form on DNA. Using this method we can compare complex formation between ligands, and cellular extract two different biochemical states.
METHODS
Cell culture and cell extracts
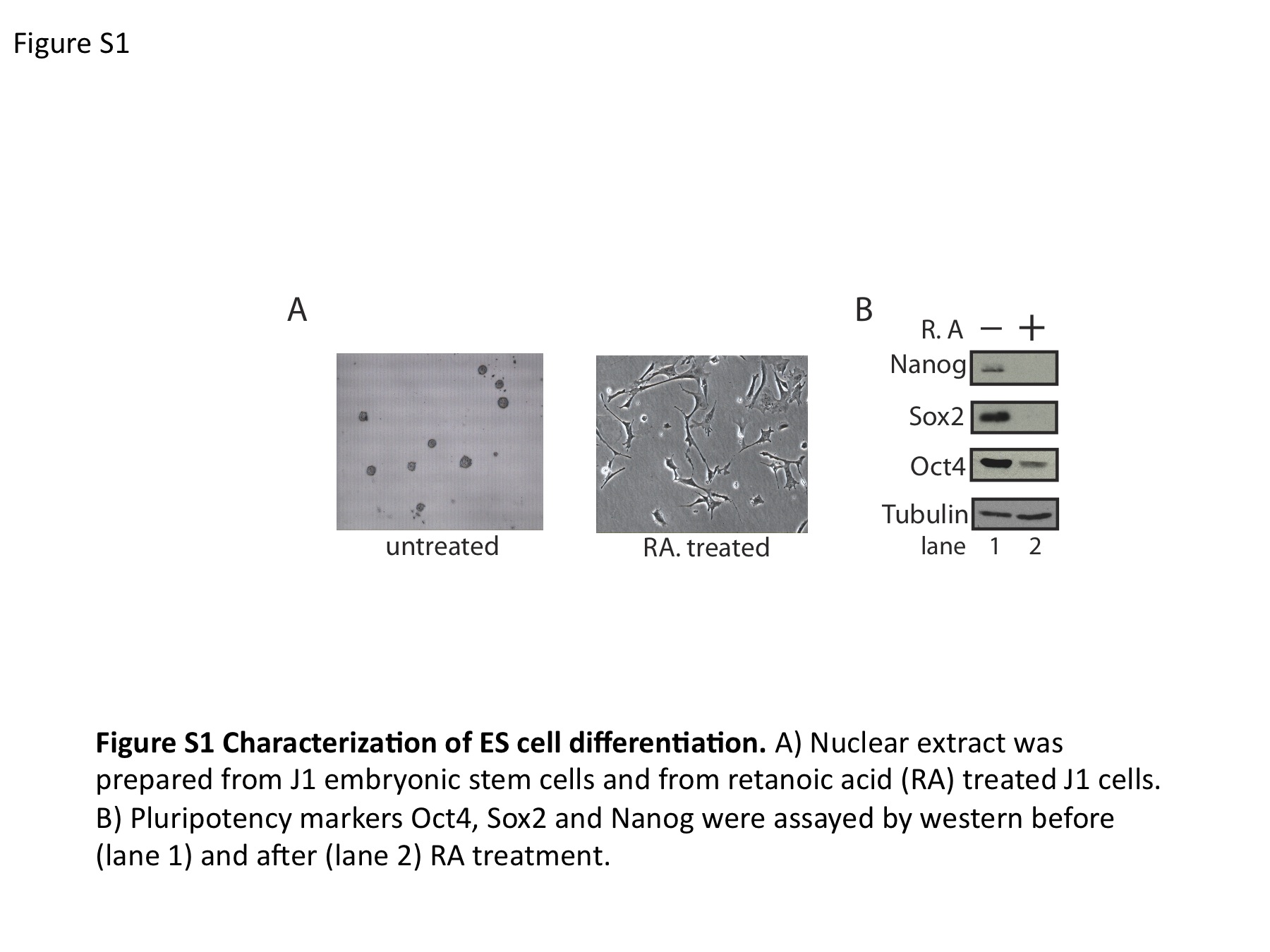
ES cells were cultured in DMEM + HEPES supplemented with 1 mM glutamine, 1 mM sodium pyruvate and 1 mM MEM non-essential amino acids (Invitrogen) plus 15% ES cell-qualified heat-inactivated fetal bovine serum (HyClone), 50 mM 2-mercaptoethanol (Sigma) and leukemia inhibitory factor (LIF/ESGRO, Chemicon). Differentiation of J1 ES cells occurred in the presence of 10–7 M [or 100 nM] retinoic acid (RA) over a 16-day period. Cellular extracts were obtained from J1 ES (male) undifferentiated and RA differentiated cells. Cells were pelleted, resuspended and incubated in extraction buffer (200 mM KCL, 100 mM Tris pH 8.0, 0.2 mM EDTA, 0.1% Igepal, 10% glycerol and 1 mM PMSF) for 50 min on ice. Cell debris was pelleted and extracts were frozen using liquid N2 and stored in the −80°C.
Library design and oligonucleotide synthesis
A complex pool of 60-mer oligonucleotides was synthesized to contain the union of the intersections of ChIP-ChIP fragments of Oct4, Nanog and Sox2 as found in Core Transcriptional Regulatory Circuitry in Human Embryonic Stem Cells (15). Using available ChIP-chip data, we downloaded 1554 genomic coordinates that define regions enriched for in vivo Nanog binding, 603 coordinates for Oct4 binding, and 1165 coordinates for Sox2 binding. The average size of the reported regions was ∼700 nt for all three experiments (757, 694 and 715 nt). This pool was synthesized as a custom oligonucleotide microarray. Nucleotides were liberated from the glass slide by boiling at 99°C for 1 h. Each oligonucleotide was designed as a tiled genomic 30-mer flanked by the common sequences CCAGTAGATCTGCCA and ATGGAGTCCAGGTTG that were used as the universal primer binding pair. DNA was recovered from synthesis arrays by adding 500 µl dH2O to the surface of the array and either thoroughly scouring and resuspending using a sterile 25-guage hypodermic needle or placing in a hybridization chamber and boiling for 1 h. The samples were then sonicated at 50% amplitude for three 5-s pulses in a Sonic Dismembrator Model 500 (Fisher). Pools were amplified by low cycle PCR (1 min at 94°C, 20 s at 55°C, 1:00 at 72°C first round; 10, 20, 10 s at each respective temperature for subsequent rounds; final elongation step of 5:00 at 72°C). Oligonucleotide sequences (library and P1–P5 and controls) can be downloaded at http://fairbrother.biomed.brown.edu/data/anonymous.
Dot Blots
Samples were prepared in 30 µl (0.6 × Buffer D, 50 ng/µl Poly dI•dC, 1 µg/µl BSA, 1 mM DTT, various amounts of sonicated herring sperm, 50 ng of probe and 5 µg of ES whole cell extract). Nitrocellulose was soaked in extraction buffer for 5 min. Dot Blots were blocked using 100 µl of 2 mg/ml sonicated herring sperm (Invitrogen) for 15 min. Samples incubated 30 in at room temperature and the volumes were raised to 100 µl with 0.6× extraction buffer. Samples were then loaded and blotted. Nitrocellulose was then washed for 15 min with cold 1× PBS at room temperature. Dot Blots were imaged using Phosphorimaging screens and the GE Typhoon™ 9410. Elution was carried out using a modified phenol:chloroform procedure in 7 M urea. Dots were cut out and placed in 1.5 ml eppendorf tubes. About 400 µl 7 M urea and 200 µl phenol were added to each tube and incubated at room temperature for 30 min. Following incubation, 200 µl chloroform was added. Samples were spun down and the aqueous layer was removed. A second phenol:chloroform extraction was then performed prior to using the samples for PCR and subsequent rounds of selection.
Microarrays
RNA probes were produced containing amino-allyl UTP using MEGAshortscript™ High-Yield Transcription kit (Ambion) after appending a T7 promoter to the oligonucleotides. Amino-Allyl UTP’s were then coupled to Cy3 and Cy5 dyes, through a labeling procedure. RNA was pelleted and resuspended in 1 M NA2CO3, monoreactive dyes were added and reaction was allowed to continue for 1 h at room temperature. About 4 M hydroxylamine was used to quench the reaction followed by phenol:chloroform and ethanol precipitation to remove remaining free nucleotides. An 8 × 15 k and a 2 × 104 k Custom Agilent oligonucleotide detection microarray were designed complimentary each oligonucleotide pool. Microarrays were hybridized for 3 h at 50°C using an optimized Agilent gene expression hybridization kit and protocol.
Microarrays were scanned at 5 µm using a GenePix 4000B scanner and analyzed using Feature Extraction Software from Agilent. Only sequences that scored one for the ‘gIsWellAboveBG’ value were used for analysis—this corresponds to sequences that showed green (starting) signal at least 2.6 standard deviations higher than the mean calculated background signal. Of 241 347 total sequences, 50 161 sequences fit this criterion or 20.7% of the array.
The enrichment values were visualized by mapping the data back to the UCSC Genome Browser. A wiggle-format custom track was created that performs the enrichment score averaging step across all the genomic regions (Figure 2). For each position, the log of the average enrichment score is taken for every oligonucleotide that overlaps that position, such that each 30-mer has data for 30 positions. A single false scaling data point is added to each continuous genomic region for the purpose of keeping the y-axis constant between different regions. Binding motifs were identified using the Gibbs Sampler (V 3.04.006).
Figure 2.

A map of cis-element in the upstream control region of the REST gene. (A) UCSC genome browser view of the REST gene displays the results of the binding assay on four specific TFBSs (Oct1, Oct4, Sox2 and Nanog) and the anonymous complexes. Oligonucleotide enrichments in the filter binding assays performed after incubation in undifferentiated ES cell extract (horizontal gray wiggle plot) and differentiated extract (horizontal green wiggle plot) are aligned for comparison. The location of gel shift probes (P1, P2, P3, P4 and P5) is indicated. (B) Gel shift assay validates array. Radiolabelled probes P1–5 and control probes C1 and C2 were incubated with extract and analyzed by EMSA. Apparent examples of non-specific binding (i.e. bands seen across multiple experimental and control lanes) are indicated with an arrow.
Electrophoretic Mobility Shift Assay
Oligonucleotides were prepared for electrophoretic mobility shift assay (EMSA) by end labeling PCR products with g-32P-ATP. Samples were prepared in 20 µl (0.6 × Buffer D, 50 ng/µl Poly dI•dC, 1 µg/µl BSA, 1 mM DTT and 20 ng of probe). Samples were incubated at room temperature for 30 min in extract or recombinant Oct4GST fusion proteins. Native 4% polyacrylamide gels (29:1 acrylamide:bisacrylamide, 1% glycerol, 0.5 × TBE) were pre-run for 1 h at 80 V, samples were loaded and run for 1.75 h at 80 V.
Reporter Assay
Sequences were cloned into a luciferase reporter vectors. All transfections were performed in the mouse J1 ES cell line by FuGENE HD Transfection Reagent (Roche) with and without differention. Enhancer activities were normalized by using empty expression vectors. After harvesting cells, luciferase activity was measured by using the standard Dual Luciferase Assay Reporter Assay System, according to manufactures instructions (Promega). The comparison of activity in differentiated and undifferentiated cells was presented as a ratio in Figure 3.
Figure 3.

Functional analysis of transcriptional elements in the upstream control region of the REST gene. (A) Subregions of the REST gene were selected for dual luciferase reporter assays. Elements were characterized by their binding behavior. ‘Diff’ bound complex in differentiated extract (blue), ‘undiff’ bound in undifferentiated (red) and pink represents elements that form complexes in both extracts. (B) Endogenous REST expression in whole cell extract from differentiated and undifferentiated ES assayed by western blotting and quantitative PCR. (C) Subregion containing variable numbers of predicted cis-elements was cloned into a luciferase reporter construct and transfect into ES or differentiated ES cells. The ratio of expression in undifferentiated versus differentiated cells normalized for transfection efficiency was recorded for each luciferase construct.
Western blotting and immunodetection
Following SDS–PAGE separation, proteins were transferred electrophoretically onto a nitrocellulose membrane for 1 h at 30 V following western blotting. Antibodies used to blot the membrane were anti-REST (Millipore), beta-Tubullin (Novus Biological) and goat anti-rabbit, HRP-linked (Cell Signaling Technology). REST results, were visualized by a chemiluminescent reaction using Pierce ECL Substrate Western blot detection (Pierce). For blot development, the membranes were exposed to radiography film for 30 s.
Remodelling diagrams
Position weight matrices (PWMs) from JASPAR (16) and UniPROBE (4) were used by Patser (17) to annotate binding motifs in the oligonucleotide pool. Transcription factor binding sites (TFBS) centroids were calculated by averaging the enrichments of the 100 best-matching oligonucleotides to a particular transcription factor (TF) PWM. We averaged the enrichment over these 100 oligonucleotides in both the differentiated and undifferentiated states to generate our data points, ending with one data point per TF. The same operation, performed on arbitrarily sampled sets of 100 nt, was used to calculate confidence intervals. To detect GO terms specifically enriched in significantly bound centroids (Supplementary Table S1), the complete set of TFBS centroids was divided into the enriched set that scored above the 95% confidence threshold (i.e. the top 2.5%) and second set consisting of the remaining oligonucleotides that scored in the bottom 97.5%. We then input these two groups into Gene Set Enrichment Analysis annotation to generate GO terms (18). We removed from this list the GO terms common to both groups, and placed into Supplementary Table S1 only those terms exclusive to the top centroids. This process was repeated for stringencies of centroid definition (i.e. top 10, 20, 50 100 and 200) and a total of 100 TFBS centroids were enriched in all conditions tested.
The reaction between a single transcription factor and an oligonucleotide can be represented as 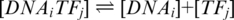 where [DNAi]is the concentration of oligonucleotide i, [TFj] is the concentration of transcription factor j; [DNAiTFj] is the concentration of the joint TF-oligonucleotide complex. The dissociation constant, KD for a particular TF and oligonucleotide is defined as:
where [DNAi]is the concentration of oligonucleotide i, [TFj] is the concentration of transcription factor j; [DNAiTFj] is the concentration of the joint TF-oligonucleotide complex. The dissociation constant, KD for a particular TF and oligonucleotide is defined as:
The dissociation constant is proportional to the energy of binding, ΔG: log2(KD)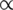 ΔG. For a given contiguous sequence of positions within an oligo, the binding energy is related to the information content of those positions as measured by a position weight matrix, Rijw=
ΔG. For a given contiguous sequence of positions within an oligo, the binding energy is related to the information content of those positions as measured by a position weight matrix, Rijw=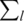 (2 + log2( f(b, l))), where Rijw represents the information content of position w in oligonucleotide i for a particular TFj; f(b, l)is the frequency of the base b found at position l. The information content is thus related to the binding energy as Rijw∝−ΔG (16).
(2 + log2( f(b, l))), where Rijw represents the information content of position w in oligonucleotide i for a particular TFj; f(b, l)is the frequency of the base b found at position l. The information content is thus related to the binding energy as Rijw∝−ΔG (16).
Since the proteins are assumed to bind DNA at any position in a random collision model, the discriminating term in the rate constant is the rate at which a protein leaves the complex, koff. Thus, we assume that kon is constant and Rijw∝−log2(koff).
In order to identify the bound transcription factors, candidate position weight matrices (PWMs) representing TFBS motifs were obtained from JASPAR (19) and UNIPROBE (5). Transcription factor concentrations were estimated from the NCBI GEO database (17). PWMs corresponding to non-vertebrate species and those for which GEO data were unavailable were pruned from the data set, resulting in a final count of 457 PWMs.
The top 5% of oligonucleotides by enrichment were scanned with each PWM using Patser (20) and a score Rij=maxw(Rijw) was calculated for each TF and oligo. Scores were scaled by concentration to yield a binding score sij=cj2Rij where cj is the concentration of TFjobtained from GEO data. For each oligonucleotide i in the top 5%, the top 5 scoring TFs were used to record a description of the putative binding event (Supplementary Table S2). Graphical diagrams display the top scoring factor with options to list the other top factors. The full set of remodeling diagrams can be viewed at http://fairbrother.biomed.brown.edu/data/anonymous. Source code and data used to generate the binding diagrams are available at the same web site.
RESULTS
In this article we present a high throughput filter binding assay for identifying DNA protein complexes on 316 pluripotency control regions. Pluripotency control regions were defined as genomic regions that interact in vivo with the transcription factors: Oct4, Sox2 and Nanog (15). However, unlike SELEX, this assay is performed on specifically designed oligonucleotide pools and binding measurements for each oligonucleotide is returned. This assay detects the degree to which a genomic region is occupied by factors without regard for the identity of the components in the complex (Figure 1A scheme). We refer to the genomic regions that become enriched in the filter as anonymous complexes, since the identity of binding partner is unknown. The output of this assay, a map of occupied versus unoccupied DNA, is similar to DNA footprinting when projected onto genomic coordinates (Figure 1B).
Figure 1.

Comparison of DNA/Protein complex formation in extract from differentiated and undifferentiated ES cells. (A) An RNA or DNA oligonucleotide pool corresponding to specific sequences (e.g. genomic or pre-mRNA sequences, predicted binding sites, polymorphic regions) is designed, synthesized and incubated with cellular extract. This mixture is separated into a bound and unbound fraction by nitrocellulose gel filtration. The bound fraction is further enriched by repeating this cycle or analyzed by two color microarray. (B) Red/Green array signals that measure the degree of oligonucleotide binding are projected onto a genomic coordinate. Average enrichment values are calculated and uploaded to UCSC genome browser tracks (wiggle format). (C) The degree of each oligonucleotide’s enrichment in the bound fraction relative to the starting pool is plotted as Log10 red/green ratios for the binding assay performed in differentiated (x-axis) and undifferentiated (y-axis) extract. The darkened oligonucleotides correspond to the region upstream of the REST locus that undergo extreme shifts. The ‘x’ data points represent oligonucleotides that fall within the top 1% of enrichment in both experiments.
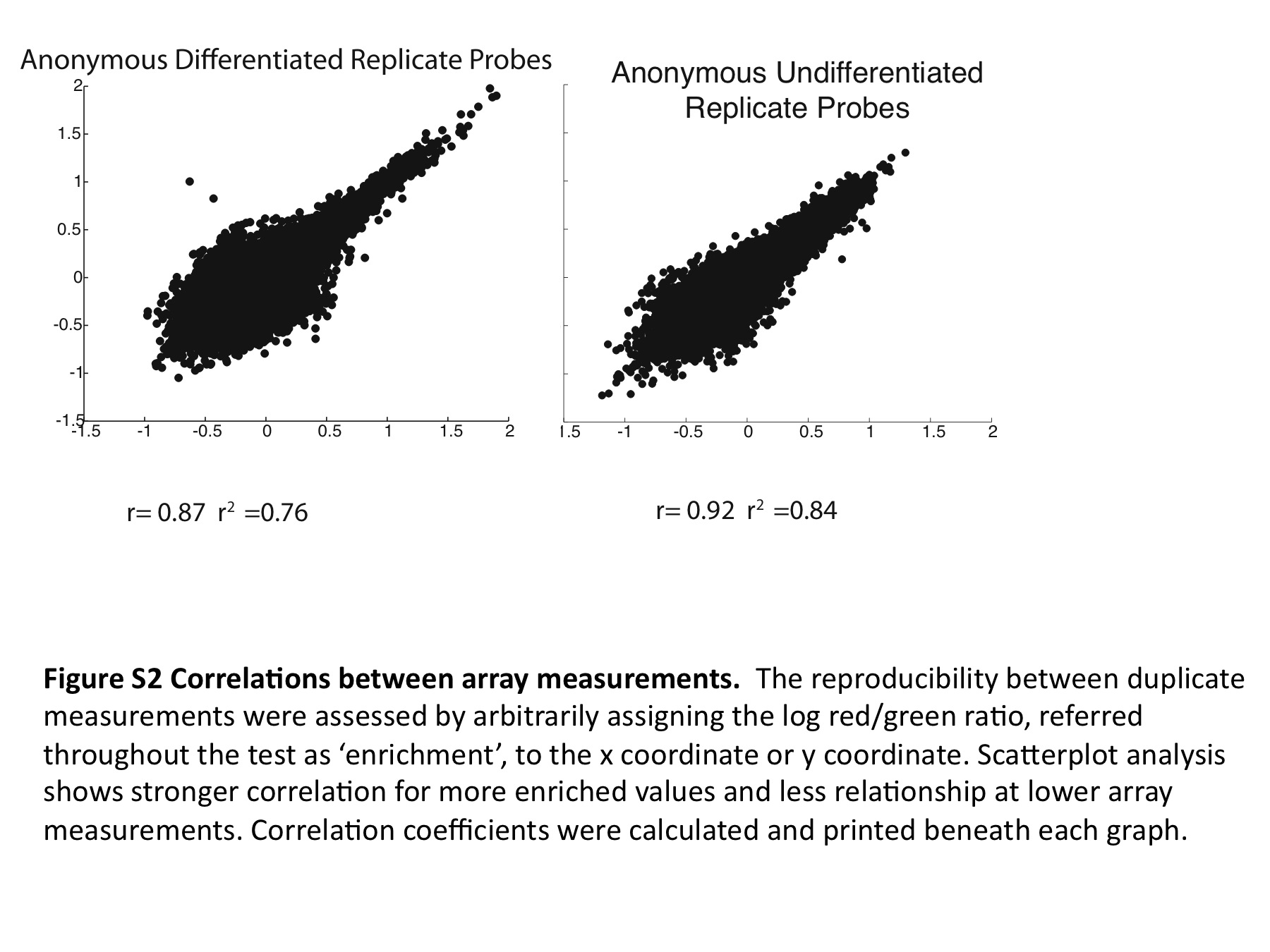
To identify these anonymous complexes, the in vivo Oct4/Sox2/Nanog ChIP enriched binding regions were resynthesized as an oligonucleotide pool. These regions were tiled across by advancing a 30-nt window in 10 nt increments through 316 pluripotency control regions. The result was a resynthesis of 400 Kb of genomic sequence as overlapping oligonucleotides at 3 × coverage. These oligonucleotides were incubated in extract of ES cells and differentiated ES cells at a gradient of increasing non-specific competitor (Supplementary Figure S1, characterization of differentiation). The stably bound complexes that formed in the presence of competitor were separated from the mixture by filtration through nitrocellulose paper. Each genomic 30-mer in the pool is flanked by universal primer binding sites, which allow for the amplification of the oligonucleotide fraction extracted from the nitrocellulose filter paper. The starting oligonucleotide pool and the bound oligonucleotides are differentially labeled and applied to the detection array, which was designed to hybridize to the oligonucleotides in the starting pool. The ratios detected by array are proportional to the degree to which each oligonucleotide was retained in the filter. The log of this red/green ratio is defined as enrichment. Enrichment scores between replicates was well correlated (Supplementary Figure S2; r2 = 0.76, 0.84). As these oligonucleotides represent tiled windows of genomic sequence, the enrichment from all probes overlapping each nucleotide position can be averaged at each nucleotide coordinate to make a map of anonymous complexes on genomic DNA (Figure 1B).
Performing this assay in extract from ES cells and differentiated ES cells enriched a distinct population of sequences on the filter. A clear positive relationship between enrichments was visualized across all 45 793 oligonucleotides in an enrichment scatterplot (positive slope Figure 1C). Thirty percent of the oligonucleotides significantly enriched (z-score > 2.33) in the differentiated bound fraction were also significantly enriched in the undifferentiated bound fraction (Figure 1C, data points marked ‘x’). Cross-species analysis strongly suggests these elements are functional. In each experiments, the top one percent of regions enriched in the filter were conserved at nearly three times the background level across 17 species of vertebrates (PhasCon scores = 2.7, 2.9 times background).
In comparing the difference in oligonucleotide representation in the bound fraction between differentiated and undifferentiated ES cells, the most extreme changes were observed within the REST control region. REST, a transcription factor implicated in repressing neuronal differentiation, plays an important role in the stem cell pluripotency network (21,22). Here, we found that 5 of the top 10 oligonucleotide enrichments in the bound fraction of differentiated extract mapped back to the REST promoter (dark circles Figure 1C, n = 45 793). Rendering these array enrichments into a genomic coordinate system allows for the comparison of these anonymous complexes with other annotation such as binding locations for specific transcription factors that are known to bind these regions (Figure 2A compare anonymous complex locations to binding locations of Sox2, Oct1, Oct4 and Nanog). Similar maps of enrichment in the bound fraction from all 316 pluripotency control regions covering >300 Kb of genomic sequence were written as UCSC custom genome browser tracts and can be downloaded at (http://fairbrother.biomed.brown.edu/data/anonymous.).
Gel shifts in ES cell extracts confirmed array predictions of complex formation on a small panel the oligonucleotides that map to the remodeled portion of the REST upstream control region (gel shifts, Figure 2B; probe locations, Figure 2A). The oligonucleotides P1, P2, P3 are significantly shifted in EMSA whereas only background levels of binding are seen in P4 and P5 and control oligonucleotides arbitrarily selected from other genomic regions (Figure 2B). The largest changes in oligonucleotide enrichment were DNA complex formation upon differentiation. As REST expression decreases during differentiation, we hypothesize the formation of a repressive complex develops to allow for neurogenesis. To organize sequence elements according to their binding properties, we annotated all locations that support complex formation in the repressed differentiated state as ‘diff’ (Figure 3A, blue circles) and all locations that support complex formation in the transcriptionally active undifferentiated state as ‘undiff’ and those complexes that appear to form in both states as ‘both’ (Figure 3A). All three types of elements may contribute to the regulation of the endogenous (i.e. a higher relative expression of REST in ES cells prior to differentiation).
Panels of luciferase reporter assays that include variable lengths of the REST upstream control region were used to assay the differential expression observed between these states. The REST gene product can be detected in ES cells in the undifferentiated state but not after differentiation. Real-time PCR indicates that endogenous REST is expressed in undifferentiated ES cells at more than three times the level of expression observed after differentiation (Figure 3B). The luciferase construct containing the four elements identified by high throughput binding analysis is also expressed at about three time the level of observed after differentiation (Figure 3C). In general, the luciferase reporters that contained portions of the REST promoter had modest activity, but reflected the behavior of the endogenous gene when transfected into differentiated and undifferentiated J1 ES cells. Like the endogenous gene activity, fragments cloned into the luciferase vector conferred suppressed transcription in differentiated cells. While the dual luciferase assay is not ideal for detecting repression, the ratio of normalized activities in undifferentiated versus differentiated ES cells appears to scale with the number of elements contained within the promoter fragment. In other words, the list of subregions ranked by their relative ability to drive expression in ES cells (i.e. B < A < AB ≤ full length REST promoter; Figure 3B and C) corresponds to regions ranked by the number of elements annotated by this assay (B, 2 elements < A, 3 elements < AB, 4 elements ≤ REST promoter, all elements; Figure 3A). While a detailed functional analysis of the REST promoter is beyond the scope of this report, this result demonstrates the utility of this method for mapping transcriptional complexes and tracking remodeling events in a differentiating system like ES cells. For each of the 316 promoters included in the library, the identity of the transcription factors complexes will eventually need to be determined. We have described an approach that identifies bound cis-elements leaving a variety of options for identifying the cognate trans-acting factors. Since a biochemical purification of these complexes would be practical for only a few cases, we employed a strategy that uses probabilistic binding models within a thermodynamic framework to predict the identity of proteins bound to the DNA oligonucleotides. For the purpose of this analysis we consider an oligonucleotide ‘bound’ if it falls within the top 5% enrichment in the nitrocellulose filter.
In order to identify the bound transcription factors, candidate position weight matrices (PWMs) representing TFBS motifs were obtained from JASPAR (19) and UNIPROBE (4). Oligonucleotide sequences were scored with Patser (20) to estimate binding affinity between candidate transcription factors and each oligo. Comparing the average binding enrichment of the top 100 matches of each of the PWMs to a randomly selected set of 100 oligonucleotides demonstrate that oligonucleotides that contain a strong match to a TF motifs are significantly more likely to be enriched in the bound fraction (Figure 4A, note excess of TF motifs outside the 95% C.I. in the upper and right subregions). Each TF PWM was scored against each oligonucleotide position, w, giving an information score which is related to the disassociation rate constant:
For each oligo, the score value of the maximum scoring position was taken as the information score Rij, for that (16). This equation determines to what degree sequences that partially match a TFBS would have a higher dissociation rate constant. This rate constant in combination with a transcription factor’s concentration in differentiated or undifferentiated ES cells allowed us to estimate the degree to which any transcription factor (j) is likely to form a complex at any position (w) on any oligonucleotide (i). Estimated transcription factor concentrations were inferred from the transcript levels reported in the NCBI GEO database (17). Binding affinities were estimated as sij=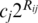 where cj is the concentration of the transcription factor TFj.
where cj is the concentration of the transcription factor TFj.
Figure 4.

The computational reconstruction of remodeling events that occur during ES cell differentiation. Transcription factor binding models were downloaded from UNIPROBE and JASPAR and used to score the oligonucleotide pool. (A) For each transcription factor, TFBS centroids were constructed by averaging the binding enrichment scores of oligonucleotides that contained the top 100 matches to the transcription factor binding model. Centroid enrichment in the bound fraction of differentiated and undifferentiated extract were plotted over 95% confidence intervals (dashed gray line). (B) Remodeling events were inferred from measurements of protein/DNA complex formation performed with 33663 genomic oligonucleotide sequences and nuclear extract from undifferentiated and differentiated ES cells. The example of binding enrichments projected onto genomic coordinates is shown for the TSC22 d1 gene. The y-axis represents array enrichment the x-axis represents genomic position. Regions enriched above the 5% threshold were called ‘bound’ and annotated with binding events predicted to occur in the differentiated and undifferentiated state (see ‘Methods’ section). This prediction was graphically displayed below. Oligonucleotides enriched in the top 5% of the immunoprecipitate of Oct4 (yellow), Nanog (blue) or Sox2 (red).
By examining the top possibilities for a given highly enriched region, we inferred remodeling events between differentiated and undifferentiated ES cells. The databases used in this study include 514 binding models and many transcription factors are missing or of lower quality (e.g. Nanog and Sox2). The severity of these defects are evident in considering the three transcription factors most associated with pluripotency: Nanog lacks a binding model, Sox2′s specificity is poorly defined and for Oct4 and Oct1, only one of the multiple binding models are included. However despite these limitations, this method improves existing analysis options by providing unbiased testable hypothesis for potential regulators that has been supported by physical measurements. These predictions can be combined with conservation filters or clustering algorithms or other methods designed to reduce false positives (23). We present these predictions with the mapping results for Oct4, Sox2 and Nanog on these regions (Figure 4). The complete list of remodeling diagrams can be viewed at (http://fairbrother.biomed.brown.edu/data/anonymous). The output can be configured to return either the top predicted factor or a list of potential factors ranked by their predicted likelihood.
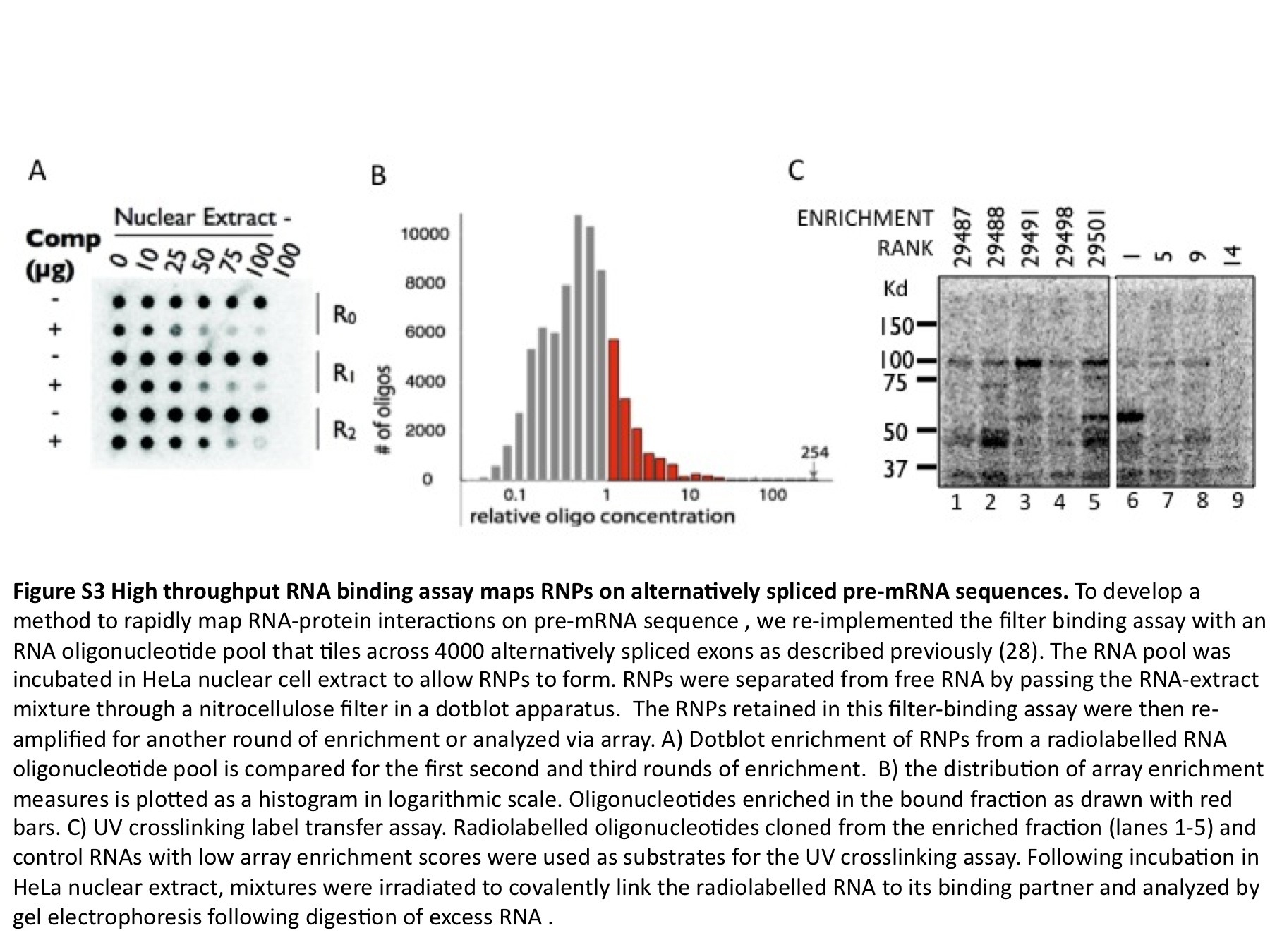
In addition to providing this resource, this study presents a highly flexible method that can be applied to the unbiased identification of cis-elements. While this study focuses on the transcriptional circuitry that maintains pluripotency elements, this method can also be used to identify RNA elements that control pluripotency. To demonstrate how this assay can be readily re-implemented on RNA, we have mapped RNA:protein complexes (RNPs) anonymously on pre-mRNA sequences around 4000 alternatively spliced exons, some of which may be involved in regulated developmental switches similar to the transcriptional regulation seen in the pluripotency control network (Supplementary Figure S3A). After three rounds of selection we isolated 100-fold enrichments (Supplementary Figure S3B) and demonstrated that ligands preferentially retained in the filter can be characterized by more RNA protein contacts in the UV crosslinking assay (Supplementary Figure S3C). The RNA elements and their enrichments can be downloaded as a UCSC genome browser track (http://fairbrother.biomed.brown.edu/data/anonymous/).
DISCUSSION
This study presents the first high-throughput method for biochemically defining binding sites without the regard to the identity of a specific trans-acting factor. We anticipate this method will be used for a variety of purposes. This versatile method can be used to pre-screen regions for cis-element architecture in order to identify targets for subsequent mutagenesis. As binding can be readily compared across experiments, this high-throughput assay could be used in drug/small molecule screening or to identify binding events that are modulated by environmental stimuli or changes in cellular state (24). Alternately, the library can be used to test how changes in cis-elements affect binding. For example, a single experiment could be used to screen thousands of simple nucleotide polymorphism (SNPs) to identify functional polymorphisms (25). Additionally, this assay could compliment existing location studies by defining the context in which transcription factors bind. We demonstrate that array measurements of filter binding can be recapitulated by EMSA and that elements defined by binding recapitulate the function of the endogenous gene when cloned upstream of the luciferase gene.
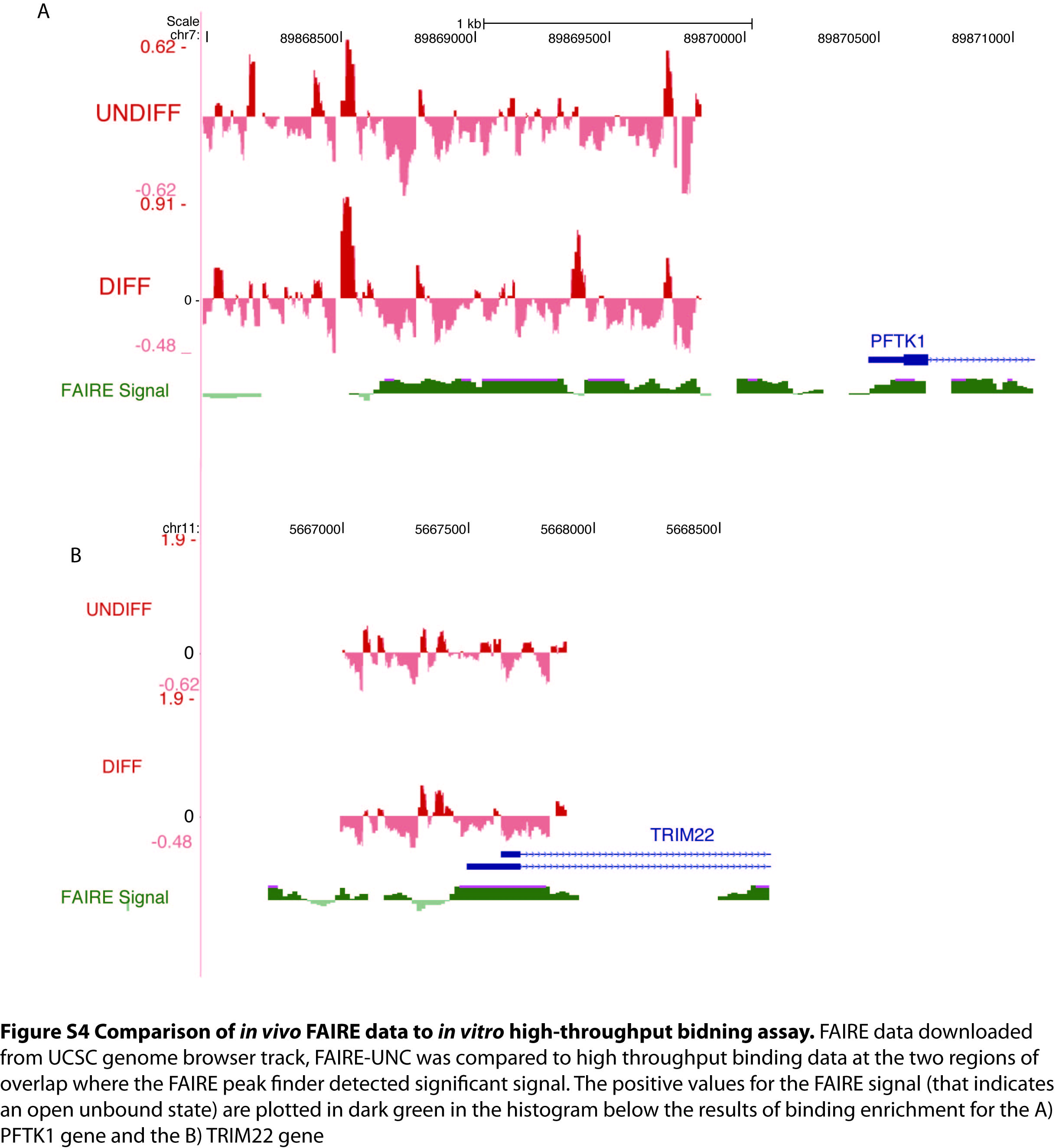
The occupancy data reported here could be used as another means of indentifying functional elements—perhaps in conjunction with other methods such as cross species conservation (23). This study utilizes pre-existing binding models to computationally screen the identified complexes for TFBSs. The thermodynamic implementation of this TFBS screen attempts to account for the quality of the site and the availability of the transcription factor. The maps generated predict which trans-acting factors recognize which cis elements. These mappings are preliminary and almost certainly contain false positives for reasons mentioned above. Long range interactions, such as DNA looping, or natural modifications of DNA such methylation could influence occupancy in a way that would not be detected in vitro. While higher order structures such as chromatin could render an in vitro measure of binding an incomplete reflection of the events that occur in vivo, specificity in gene expression must ultimately be driven by sequence determinants and the ability to separate sequence into a bound and unbound fraction narrows the search for functional elements. The observation that conservation levels were almost three times higher in the bound fraction than background argues strongly that this method is identifying functional elements. The fact that oligonucleotides that contain matches to TF motifs are enriched in the bound fraction further argues that binding enrichment is a useful tool for detecting transcriptional complex. The 100 transcription factors with sites found in the bound fraction are associated with GO terms that are consistent with the biology of ES cells being differentiated into a neuronal fate (Supplementary Table S1). Furthermore, by examining the few regions of overlap between this high throughput binding study and existing FAIRE studies we observe the expected negative relationship between FAIRE-enriched sequences and oligonucleotides predicted to bind protein (Supplementary Figure S4) (26,27). The success of this experiment could be due to the fact that the oligonucleotide library was designed to tile through regions that have been preselected for transcription factor accessibility (e.g. the library used in this study binds Nanog, Sox2 and Oct4 in vivo). Regions defined as relatively free of chromatin by nuclease-sensitivity or FAIRE would be a natural starting point for further study by this method.
Another future direction of these studies will be to identify RNA sequences that bind in an allelic fashion. Here, we have demonstrated the feasibility of mapping complexes onto RNA (Supplementary Figure S3). Several recent studies have found numerous examples of allele-specific RNA processing using technologies that are well suited to detect the phenomena but poorly suited to find the causative allele (28). Numerous genome wide association studies have identified regions that contribute to genetic risk for common diseases. Estimates of the fraction of disease mutations that affect splicing range from 15 (29) to 62% (30). Transcript analysis of genotyped cell lines has discovered numerous cases of allelic splicing, demonstrating that polymorphisms also disrupt splicing (28). Variations that alter splicing likely account for a large fraction of genetic risk for complex disease and could be a target of molecular intervention. In future studies, this high-throughput binding assay could be used to both identify causative alleles that disrupt binding of trans factors and then screen small molecules that restore allelic balance to their recognition by trans-acting factors.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institutes of Health Award Number (R21HG004524); National Human Genome Research Institute; General Medicine (R01GM095612); National Science Foundation (MCB-1020552). NSF UBM-Group for summer support (to M.J.L. and A.P.S.); the SURF-EPSCoR program (DUE-0734234 to M.J.L.); a CCMB Scholarship Award (to W.G.F.) and the Brown University Research Seed Fund Program (to W.G.F). Funding for open access charge: National Institutes of Health Award Number (R21HG004524); National Human Genome Research Institute; General Medicine (R01GM095612); National Science Foundation (MCB-1020552).
Conflict of interest statement. None declared.
ACKNOWLEDGEMENTS
The authors also thank NSF UBM-Group for summer support to M.J.L. and A.P.S.
REFERENCES
- 1.Ule J, Jensen K, Mele A, Darnell RB. CLIP: a method for identifying protein-RNA interaction sites in living cells. Methods. 2005;37:376–386. doi: 10.1016/j.ymeth.2005.07.018. [DOI] [PubMed] [Google Scholar]
- 2.Buck MJ, Lieb JD. ChIP-chip: considerations for the design, analysis, and application of genome-wide chromatin immunoprecipitation experiments. Genomics. 2004;83:349. doi: 10.1016/j.ygeno.2003.11.004. [DOI] [PubMed] [Google Scholar]
- 3.Solomon MJ, Larsen PL, Varshavsky A. Mapping protein-DNA interactions in vivo with formaldehyde: evidence that histone H4 is retained on a highly transcribed gene. Cell. 1988;53:937–947. doi: 10.1016/s0092-8674(88)90469-2. [DOI] [PubMed] [Google Scholar]
- 4.Badis G, Berger MF, Philippakis AA, Talukder S, Gehrke AR, Jaeger SA, Chan ET, Metzler G, Vedenko A, Chen X, et al. Diversity and complexity in DNA recognition by transcription factors. Science. 2009;324:1720–1723. doi: 10.1126/science.1162327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Newburger DE, Bulyk ML. UniPROBE: an online database of protein binding microarray data on protein-DNA interactions. Nucleic Acids Res. 2009;37:D77–D82. doi: 10.1093/nar/gkn660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tuerk C, Gold L. Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science. 1990;249:505–510. doi: 10.1126/science.2200121. [DOI] [PubMed] [Google Scholar]
- 7.Tantin D, Gemberling M, Callister C, Fairbrother W. High-throughput biochemical analysis of in vivo location data reveals novel distinct classes of POU5F1(Oct4)/DNA complexes. Genome Res. 2008;18:631–639. doi: 10.1101/gr.072942.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Watkins KH, Stewart A, Fairbrother W. A rapid high-throughput method for mapping ribonucleoproteins (RNPs) on human pre-mRNA. J. Vis. Exp. 2009;2:34. doi: 10.3791/1622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Istrail S, Davidson EH. Logic functions of the genomic cis-regulatory code. Proc. Natl Acad. Sci. USA. 2005;102:4954–4959. doi: 10.1073/pnas.0409624102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kuhlman T, Zhang Z, Saier MH, Jr, Hwa T. Combinatorial transcriptional control of the lactose operon of Escherichia coli. Proc. Natl Acad. Sci. USA. 2007;104:6043–6048. doi: 10.1073/pnas.0606717104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Botquin V, Hess H, Fuhrmann G, Anastassiadis C, Gross MK, Vriend G, Scholer HR. New POU dimer configuration mediates antagonistic control of an osteopontin preimplantation enhancer by Oct-4 and Sox-2. Genes Dev. 1998;12:2073–2090. doi: 10.1101/gad.12.13.2073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ma J. Crossing the line between activation and repression. Trends Genet. 2005;21:54–59. doi: 10.1016/j.tig.2004.11.004. [DOI] [PubMed] [Google Scholar]
- 13.Zhao R, Gish K, Murphy M, Yin Y, Notterman D, Hoffman WH, Tom E, Mack DH, Levine AJ. Analysis of p53-regulated gene expression patterns using oligonucleotide arrays. Genes Dev. 2000;14:981–993. [PMC free article] [PubMed] [Google Scholar]
- 14.Yuh CH, Bolouri H, Davidson EH. Genomic cis-regulatory logic: experimental and computational analysis of a sea urchin gene. Science. 1998;279:1896–1902. doi: 10.1126/science.279.5358.1896. [DOI] [PubMed] [Google Scholar]
- 15.Boyer LA, Lee TI, Cole MF, Johnstone SE, Levine SS, Zucker JP, Guenther MG, Kumar RM, Murray HL, Jenner RG, et al. Core transcriptional regulatory circuitry in human embryonic stem cells. Cell. 2005;122:947–956. doi: 10.1016/j.cell.2005.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shultzaberger RK, Roberts LR, Lyakhov IG, Sidorov IA, Stephen AG, Fisher RJ, Schneider TD. Correlation between binding rate constants and individual information of E. coli Fis binding sites. Nucleic Acids Res. 2007;35:5275–5283. doi: 10.1093/nar/gkm471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Barrett T, Troup DB, Wilhite SE, Ledoux P, Rudnev D, Evangelista C, Kim IF, Soboleva A, Tomashevsky M, Marshall KA, et al. NCBI GEO: archive for high-throughput functional genomic data. Nucleic Acids Res. 2009;37:D885–D890. doi: 10.1093/nar/gkn764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl Acad. Sci. USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sandelin A, Alkema W, Engstrom P, Wasserman WW, Lenhard B. JASPAR: an open-access database for eukaryotic transcription factor binding profiles. Nucleic Acids Res. 2004;32:D91–D94. doi: 10.1093/nar/gkh012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hertz GZ, Stormo GD. Identifying DNA and protein patterns with statistically significant alignments of multiple sequences. Bioinformatics. 1999;15:563–577. doi: 10.1093/bioinformatics/15.7.563. [DOI] [PubMed] [Google Scholar]
- 21.Singh SK, Kagalwala MN, Parker-Thornburg J, Adams H, Majumder S. REST maintains self-renewal and pluripotency of embryonic stem cells. Nature. 2008;453:223–227. doi: 10.1038/nature06863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jorgensen HF, Chen ZF, Merkenschlager M, Fisher AG. Is REST required for ESC pluripotency? Nature. 2009;457:E4–5. doi: 10.1038/nature07783. discussion E7. [DOI] [PubMed] [Google Scholar]
- 23.Wasserman WW, Sandelin A. Applied bioinformatics for the identification of regulatory elements. Nature Rev. 2004;5:276–287. doi: 10.1038/nrg1315. [DOI] [PubMed] [Google Scholar]
- 24.Chang B, Levin J, Thompson WA, Fairbrother WG. High-throughput binding analysis determines the binding specificity of ASF/SF2 on alternatively spliced human pre-mRNAs. Com. Chem. High T. Scr. 2009;13:242–252. doi: 10.2174/138620710790980522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bond GL, Hu W, Bond EE, Robins H, Lutzker SG, Arva NC, Bargonetti J, Bartel F, Taubert H, Wuerl P, et al. A single nucleotide polymorphism in the MDM2 promoter attenuates the p53 tumor suppressor pathway and accelerates tumor formation in humans. Cell. 2004;119:591–602. doi: 10.1016/j.cell.2004.11.022. [DOI] [PubMed] [Google Scholar]
- 26.Giresi PG, Lieb JD. Isolation of active regulatory elements from eukaryotic chromatin using FAIRE (formaldehyde assisted isolation of regulatory elements) Methods. 2009;48:233–239. doi: 10.1016/j.ymeth.2009.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Giresi PG, Kim J, McDaniell RM, Iyer VR, Lieb JD. FAIRE (formaldehyde-assisted isolation of regulatory elements) isolates active regulatory elements from human chromatin. Genome Res. 2007;17:877–885. doi: 10.1101/gr.5533506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kwan T, Benovoy D, Dias C, Gurd S, Provencher C, Beaulieu P, Hudson TJ, Sladek R, Majewski J. Genome-wide analysis of transcript isoform variation in humans. Nat. Genet. 2008;40:225–231. doi: 10.1038/ng.2007.57. [DOI] [PubMed] [Google Scholar]
- 29.Stenson PD, Ball EV, Mort M, Phillips AD, Shiel JA, Thomas NS, Abeysinghe S, Krawczak M, Cooper DN. Human Gene Mutation Database (HGMD): 2003 update. Hum. Mutat. 2003;21:577–581. doi: 10.1002/humu.10212. [DOI] [PubMed] [Google Scholar]
- 30.Lopez-Bigas N, Audit B, Ouzounis C, Parra G, Guigo R. Are splicing mutations the most frequent cause of hereditary disease? FEBS Lett. 2005;579:1900–1903. doi: 10.1016/j.febslet.2005.02.047. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}