

Abstract
Development of effective methods to screen binary interactions obtained by rigid-body protein–protein docking is key for structure prediction of complexes and for elucidating physicochemical principles of protein–protein binding. We have derived empirical knowledge-based potential functions for selecting rigid-body docking poses. These potentials include the energetic component that provides the residues with a particular secondary structure and surface accessibility. These scoring functions have been tested on a state-of-art benchmark dataset and on a decoy dataset of permanent interactions. Our results were compared with a residue-pair potential scoring function (RPScore) and an atomic-detailed scoring function (Zrank). We have combined knowledge-based potentials to score protein–protein poses of decoys of complexes classified either as transient or as permanent protein–protein interactions. Being defined from residue-pair statistical potentials and not requiring of an atomic level description, our method surpassed Zrank for scoring rigid-docking decoys where the unbound partners of an interaction have to endure conformational changes upon binding. However, when only moderate conformational changes are required (in rigid docking) or when the right conformational changes are ensured (in flexible docking), Zrank is the most successful scoring function. Finally, our study suggests that the physicochemical properties necessary for the binding are allocated on the proteins previous to its binding and with independence of the partner. This information is encoded at the residue level and could be easily incorporated in the initial grid scoring for Fast Fourier Transform rigid-body docking methods.
Keywords: protein–protein docking, rigid-body docking, docking scoring, statistical potentials, knowledge-based potentials, protein–protein interactions, docking-components of statistical potentials, transient interactions, permanent interactions, binding-site prediction
Introduction
Understanding the mechanisms of control, response, and regulation of the cell implies a deep knowledge on the relationships between the biochemical components of a biological network. With further interest on human health, protein interaction networks are a useful tool for characterizing diseases caused by malfunctions in genes or proteins1 and identifying novel cancer gene candidates with differential expression between the metastatic cells and their parental cells.2–5 Protein interaction maps can be used to infer the function of proteins,6 to calculate the number of binding sites of a protein,7 and to identify them on the protein surface.8,9 Knowledge of the precise structures of macromolecules could provide insights about quantitative parameters or help to elucidate functional networks. Recent efforts to gain knowledge on the structure of protein–protein complexes have been tackled at high-throughput level.10,11 Mosca et al.10 provided models for over 3000 protein–protein interactions in the yeast interactome and assessed the use of homology models for computational docking experiments too.
Computational docking methods aim to elucidate the structure of a binary interaction of biomolecules (e.g., two proteins) when experimental data regarding the structure of the complex are lacking but is present for the interacting partners. Docking methods were introduced in 1978.12 Since then, docking algorithms have substantially improved, with a breakthrough in algorithm speed given by the introduction of the fast Fourier transform (FFT)13,14 (e.g., FTDock,15 ZDock,16 and PIPER17) and by some other very successful geometry-based methods (e.g., FRODOCK,18 Hex,19 and MolFit13).
The general procedure for predicting the 3D structure of a protein–protein interaction using docking consists of an initial rigid-body exhaustive search. In this step, one performs a screening of a very large set of possible binary complex conformations obtained by rotating and translating one of the proteins around the other and by not allowing atom mobility. Next, a refinement step follows on some selected structures,20 which accounts for changes in the conformation of the two proteins. The final goal is to provide a near-native structure, that is, a structure close to the native one.
Typically, a rigid-body docking algorithm returns a long list of possible structures, which includes many false interactions. Hence, a crucial point after this initial step is the selection of a few structures that will be further analyzed. A common strategy is to re-rank the docking poses by means of a scoring function. The accurate scoring of rigid-body docking orientations represents one of the major difficulties in protein–protein docking prediction. Overall, good discrimination of near-native docking poses from the very early stages of rigid-body protein docking is an essential step before applying more costly interface refinement to the correct docking solutions. Some advances in this direction include the use of desolvation to predict the binding site area (e.g., pyDock21), the use of Monte Carlo simulations,22,23 the use of low-frequency normal modes, and side chain flexibility,24,25 or the use of energy evaluation during or after the docking generation phase, like Haddock,26 ClusPro/SmoothDock,27,28 RosettaDock,29 or ATTRACT.30
Scoring functions are usually built upon different properties of protein–protein interactions observed in known binary complexes. These properties include physical and chemical characteristics of the binding site, at the level of residue or atomic contacts. Among these scoring functions, statistical potential is a term that refers to a knowledge-based scoring function that depends on specific properties of known protein–protein interactions stored in some database. Their common structure is the sum over all interacting pairs of a score given to each pair of interacting residues or suitable atom types. This score is usually based on chemical, physical, or biological properties. They have been used, with different degree of success, as ranking scoring functions.16,17,31 Initially, statistical potentials were derived in order to distinguish a correct protein fold (i.e., near-native) of a model from a plethora of generated solutions. A vast amount of statistical potentials have been described and tested.32 Specific potentials were derived for the interaction between macromolecules in order to assess protein–protein interactions (e.g., M-TASSER,33 MULTIPROSPECTOR,34 and InterPrets35) or DNA-protein interactions.36−38
In a recent work, we provided a decomposition of knowledge-based potentials for protein folds into different energy terms that reflected different levels of detail of the residue-residue interactions.39 This decomposition allowed for a better characterization of the structural features that contribute to the greatest extend of highly discriminative potentials. We derive and split here empirical potential functions39 for protein–protein interactions. The purpose is, in the first place, to elucidate the properties of the standard residue-pair potential that account for its success in ranking docking poses, and, in the second place, to obtain a new ranking of poses that improves current near-native selection success rate. We are eager to uncover the most important features that characterize the native binding interface in comparison to the other a priori possible binding modes.
We have obtained four new statistical potentials, depicting the geometry of the interaction and the energetic component of placing some residues in a particular secondary structure and surface accessibility. Their success has been evaluated in two different databases (the benchmark dataset and a set of permanent interactions) and from different points of view. We have considered in the first place their ability to select near-native poses, that is, structures differing from the native one at most 2.5 Å [computed in terms of interface root mean square deviation (I-RMSD)]. Second, we have analyzed the number of top-ranked false interactions (by means of ROC curves), and, finally, we have studied their capacity to top-rank the best pose available in the set.
Results
Split statistical potentials
The interaction between two residues can be statistically described by means of a potential of mean force.40,41 Given two interacting proteins, several statistical potentials are defined by considering potentials of mean force reflecting different characteristics of the residue–residue interaction. These statistical potentials are obtained by summing the potential of mean force PMF for each pair of interacting residues a, b of the two proteins A and B:
| (1) |
Let kB denote the Boltzmann constant and let T be the standard temperature (300 K).
A residue condition is a triplet of the form
In this text, we consider the following potentials of mean force:
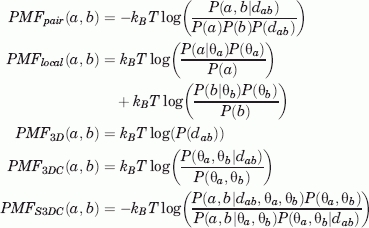 |
(see the Supplementary Material for details). Here, dab is the distance between the residues a and b (defined as the minimum of the distances between all pairs of atoms of the residues) and θa is the condition of residue a. The terms P(·) denote the probabilities of observing interacting pairs with some given characteristics. For instance, P(a,b|dab) is the conditional probability that residues a, b interact at distance smaller than or equal to dab, and P(dab) is the probability of finding any pair of residues interacting at distance smaller than or equal to dab. The other probabilities are defined similarly, and the details are given in the Supplementary Material. All probabilities P(·) are obtained from the relative frequencies in the selected database [3D interacting domains (3DID) in our case, see Methods].
The statistical potentials Epair, Elocal, E3D, E3DC, and ES3DC are defined using formula (1), with corresponding subindexes between E_ and PMF_. It was shown39 that Epair admits a decomposition of the form
where Ecmp is a residual energy term depending only on the conditions of the interacting residues that accounts for the reference state. This equation was initially derived for the scoring of protein folds, but it remains valid when applied to the residues in the interface between two interacting proteins.
Note that the statistical potential ES3DC is a refinement of the residue-pair statistical potential Epair, in the sense that it takes into account not only the residues that interact but also the condition in which each of them sits. On the contrary, the statistical potential E3DC depends on the occurrence of interacting conditions, disregarding the specific interacting residues. The score Elocal is distance independent and it reflects the probability of placing a residue on a specific condition. Moreover, it splits into two terms, each of them depending only on the probability of placing a certain residue in some condition, for each chain separately. The energy term E3D concerns only the distance at which pairs of residues interact. Note that this score increases together with the number of interacting residue-pairs.
For our computations, we have considered that two residues interact if its minimum distance is below 5 Å. The reference state considered here is the one called mole-fraction,31 and its equivalent extensions for the condition-specific potentials of mean force.
The statistical potentials and native structure prediction on the 3DID dataset
We have undertaken a preliminary analysis of the statistical potentials by determining their capacity to correctly discriminate native structures from nonnative structures on the dataset of binary interactions 3DID. For each native structure in the dataset, 1000 nonnative structures were constructed by shuffling the residues of each protein sequence while fixing the structure.
The five potentials Epair, Elocal, E3D, E3DC, and ES3DC were analyzed using a five-fold approach. The dataset 3DID was split into five groups. The native structures in four of the groups were used to define the potentials (i.e., for the computation of the probabilities above), while the remaining group, together with the corresponding generated nonnative structures, was used for testing. This process was repeated for five times, so that each group was used as test group once. Additionally, in order to provide a better comparison of native and nonnative scores as well as of the different scores, each score was normalized with respect to the random distribution obtained by shuffling the residues in each sequence. That is, for each potential, a new score called Z-score was defined by subtraction of the mean score and division by the standard deviation among the randomized sequences.39
In Figure 1, the distribution of the Z-scores of native structures is shown. The distribution of Z-scores for nonnative structures is also given and is centered at zero (by construction). The deviation from the centered distribution of the Z-scores corresponding to native structures indicates that our statistical potentials discriminate to different extent native from nonnative structures and prove the validity to further use them for binary interactions structure prediction or validation. In this sense, Figure 1 shows that the most relevant scoring functions are Epair, Elocal, and ES3DC, while E3DC is probably the less useful potential for this task. Note that the Z-score of E3D vanishes because this score depends only on the coordinates of the interface residues that are fixed in the randomized sequences.
Figure 1.
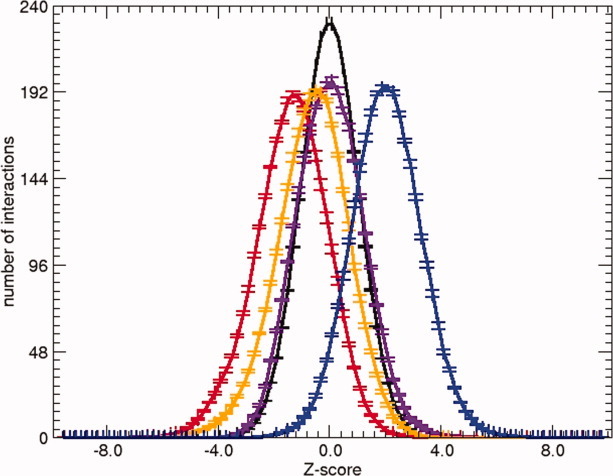
Average of the distribution of Z-scores using a five-fold approach plus the ranges of error. Z-scores are obtained with the statistical potentials Epair (red), ES3DC (orange), Elocal (blue), and E3DC (purple). Also the distribution of the Z-score of a random distribution, simulated by shuffling the sequences of the unbound proteins along the interface is shown in black.
Near-native decoy selection by the statistical potentials on the benchmark dataset
Each binary-complex conformation obtained by means of rigid-body docking from its two individual protein-chains will be referred as decoy. We denote by I-RMSD the interface Cα-RMSD42 from the native structure. A decoy is called near-native if I-RMSD < 2.5Å.31,43
We consider here the suitability of our scoring functions to single out near-native decoys from a pool of decoys. For that, we consider the benchmark dataset.44 It consists of a collection of binary complexes (124) with known structure (named targets) and a set of decoys for each of them (named target set). Note that there are 97 of the target sets in the dataset that contain at least one near-native decoy.
Each scoring function provides a ranking of the decoys in a target set. We call a target set a hit of a scoring function for a fixed number of allowed predictions m if the scoring function ranks at the top m at least one near-native decoy of the set. We build success curves of each scoring function by considering the percentage of hits in the dataset while varying the number of allowed predictions. We consider the success only up to 1000 predictions because for this number, the probability of finding at least one near-native decoy is around 0.9 for most target sets (see Methods). Therefore, it is essentially meaningful to analyze the behavior of scoring functions for small number of predictions. Moreover, the usual number of predictions provided by the uploaders in the CAPRI scoring function experiment (http://www.ebi.ac.uk/msd-srv/capri/) is 1000.
Figure 2(A) shows the success curves of the five statistical potentials. It can be seen that each of the potentials in which Epair was split (Elocal, E3D, E3DC, and ES3DC) has a lower near-native prediction capability than that of Epair. Surprisingly, the two statistical potentials that account for the most sophisticated conditions to take part in an interaction, ES3DC and E3DC, have less successful rate than Elocal and E3D potentials. With the exception of Epair, all other potentials show success curves below the random for more than 150 predictions [Fig. 2(A)].
Figure 2.
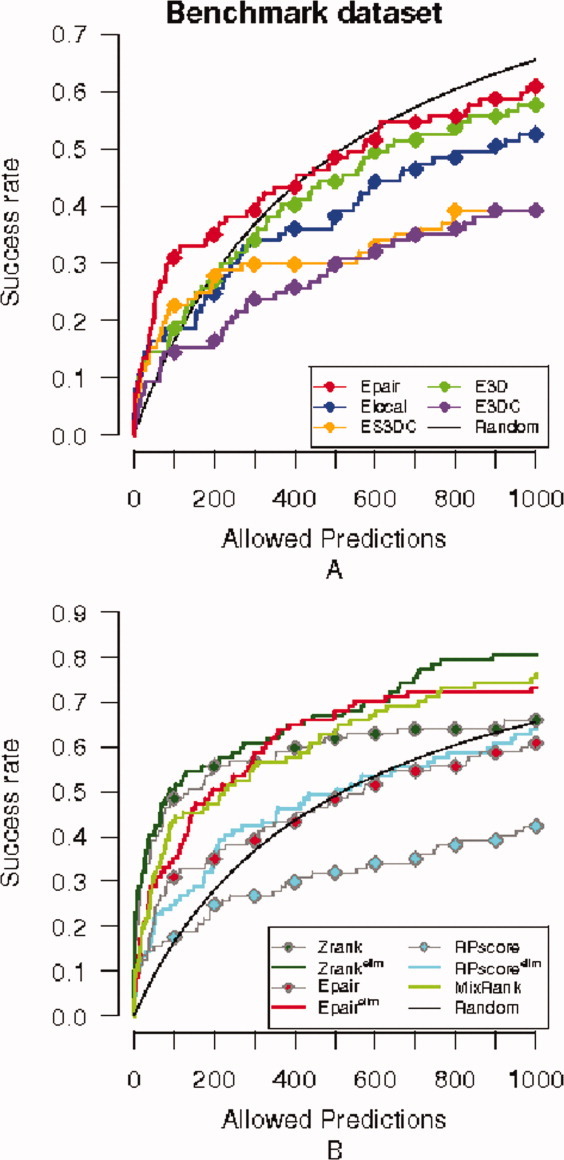
Success curves for the benchmark dataset. Success curves for the near-native criterion I-RMSD<2.5 Å are plotted. (A) Success curves for the five statistical potentials: Epair (red), ES3DC (orange), Elocal (blue), E3D (green), and E3DC (purple), plus the success curve expected by random (black). (B) Success curve of the MixRank strategy ranking and ranks with Epair, Zrank, RPScore scoring functions before and after application of the redundancy filter (superindex “elim” indicates the application of the filter).
When the number of allowed predictions is lowered to 200, most P-values of the target sets in the benchmark dataset are small (Table SI). This indicates that near-native predictions are not likely to be solely due to random. The success curves of the scoring functions in Figure 2(A) are over the random curve for up to 200 predictions. In this case, 53 targets are hits for at least one scoring function and four of them are exclusive of EPair. We have observed that all but two of the hits of Elocal and E3D are also detected by EPair. On one hand, this indicates that Elocal and E3D do not incorporate new information to EPair, and, on the other, that most of the success of EPair is encoded in these two scoring functions. That is, the frequency of pairs of interacting residues observed in known binary interactions (measured by EPair) is essentially due to (a) the intrinsic geometry of the binding interface (E3D) and (b) the presence of some residues at certain locations, independently of the interacting partner (Elocal).
Although the scoring function ES3DC is less successful than EPair, it detects 14 hits (out of 27) where EPair failed, for 200 predictions. Also, the scoring function E3DC predicts six hits that were not found by EPair and four of them were also predicted by ES3DC. To emphasize the nonoverlap of the hits of EPair and ES3DC, we show in Figure S1 their success curves together with the percentage of common hits. We conclude that a relevant number of hits are different between ES3DC and EPair, independently of the number of allowed predictions.
For comparison, we have also plotted in Figure 2(B), the success curves of the scoring functions EPair, Zrank, and RPScore. RPscore is a pair potential scoring function, while Zrank encodes atomistic energy terms. We see that Zrank provides the best success curve in the benchmark dataset for the current near-native criterion. The differences between the scoring functions will be addressed later in the text.
Finally, to see the dependence of the results on the near-native decoy criterion, we show in the supplementary material (Figure S2), the success curves of our scoring functions obtained by considering a decoy to be near-native if I-RMSD < 5 Å.
ROC curves on the benchmark dataset
Success curves tell us about the number of hits of a scoring function in a dataset but provide no insight about the number of near-native decoys selected for each target set. This feature can be globally analyzed in the dataset by considering ROC curves. ROC curves are constructed here from the ratio of near-native decoys selected while varying the number of allowed predictions (see Methods). Our approach is equivalent to first compute the ROC curve for each target set and then average the curves over the target sets.
Figure 3 shows the ROC curves of the five statistical potentials (Epair, Elocal, E3D, E3DC, and ES3DC) together with those of the scoring functions Zrank and RPScore. The ROC curves of the five statistical potentials relate similarly to the relation among their success curves explained above.
Figure 3.
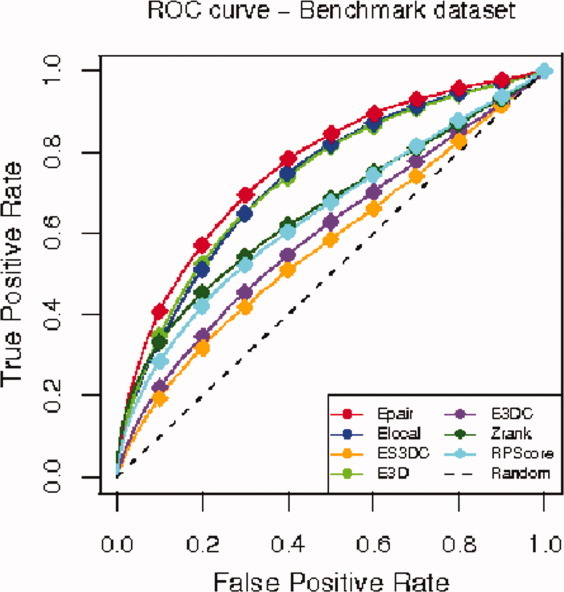
ROC curves for the benchmark dataset. We plot the ROC curves for each of the five statistical potentials Epair (red), ES3DC (orange), Elocal (blue), E3D (light green), E3DC (purple), the scoring functions Zrank (dark green) and RPScore (light blue), and the random classification ROC curve (dashed black).
Although the success curve of Zrank surpassed the curve of Epair (and hence of the other statistical potentials), it is remarkable that the statistical potentials Epair, Elocal, and E3D provide a bigger area under the ROC curve (AUC) than Zrank. This observation implies that these three statistical potentials tend to group near-native decoys together, providing a better separation of the two classes (near-native and non-near-native) than Zrank.
MixRank: A new strategy to rank the decoys of a target set
Based on the fact that ES3DC and EPair provide a fairly amount of nonoverlapping hits, we consider a new strategy to rank the decoys of a target set, called MixRank. It consists of first considering the lists of decoys ranked by the scoring functions ES3DC and EPair separately, and then alternatively selecting one decoy from each list. Additionally, in order to avoid repetitions, we apply a removal of redundant predictions.45 That is, we do not include decoys that are less than 5 Å of ligand-RMSD46 from an already selected decoy. This way of removal of redundancies was analyzed45 and was proved to provide better selection of near-native decoys. We wish to note that MixRank is not a scoring function but a method of ranking.
We have applied the same filter for redundant predictions to the scoring functions EPair, Zrank, and RPScore. Figure 2(B) shows the success curves of these scoring functions, with or without the redundancy filter, together with that of the MixRank.
It is noteworthy that the elimination of redundancy improves near-native prediction success for all scoring functions.45 Also, the MixRank strategy provides better hit-prediction in comparison with EPair when the number of selected predictions is small. This is due to the fact that this strategy adds hit-predictions of ES3DC without loosing hit-predictions of EPair. Note that even with the improvement of MixRank over EPair, Zrank (with elimination of redundancies) is the best ranking method for near-native selection on the benchmark dataset.
In order to understand the ratio of hits shared between decoys ranked with the MixRank strategy, Zrank and RPScore (after removal of redundancies), we show in Table I the name of the targets in the benchmark dataset for which each of the rankings include at least one near-native among the first 200 selected decoys. Zrank clearly provides the highest number of nonshared hits (17), but we observe that MixRank obtains a hit for nine of the targets where Zrank fails and RPScore for five (see Fig. 4 for the scores of target 1UDI, for which there is a near-native decoy ranked 1 with the MixRank).
Table I.
Hits by different ranking methods on the benchmark dataset
| Zrank and RPScore | Zrank | Zrank and MixRank |
| 1AY7, 1EWY, 1N8O, 1RLB, 2QFW, 2SIC, 2UUY | 1AY7, 1B6C, 1E96, 1HE1, 1I9R, 1IJK, 1IQD, 1JPS, 1KAC, 1KTZ, 1QFW, 1AZS, 1GLA, 2MTA, 2SNI, 2VIS, 2FD6 | 1AKJ, 1E6J, 1F34, 1F51, 1KXQ, 1ML0, 1MLC, 1NCA, 1GPW, 1K74, 1R0R, 1YVB, Z5Y, 1ZHI, 2HLE |
| Common for all | ||
| 1AHW, 1AVX, 1BJ1, 1BUH, 1BVN, 1DFJ, 1E6E, 1EAW, 1FSK, 1IQD, 1K4C, 1KXP, 1MAH, 1PPE, 1WEJ, 1T6B, 1XD3, 1Z0K, 2JEL, 2B42, 2CFH, 7CEI | ||
| RPScore | RPScore & MixRank | MixRank |
| 1FC2, 1J2J | 1CGI, 2BTF, 2I25 | 1EZU, 1I4D, 1TMQ, 1UDI, 2PCC, 2H7V |
We show the targets of the benchmark dataset that provide a near-native decoy among the first 200 predictions, for the rankings provided by the MixRank strategy, Zrank and RPScore, after applying the redundancy filter. Common hits are grouped. Total of structures: 71 (out of 97 possible).
Figure 4.
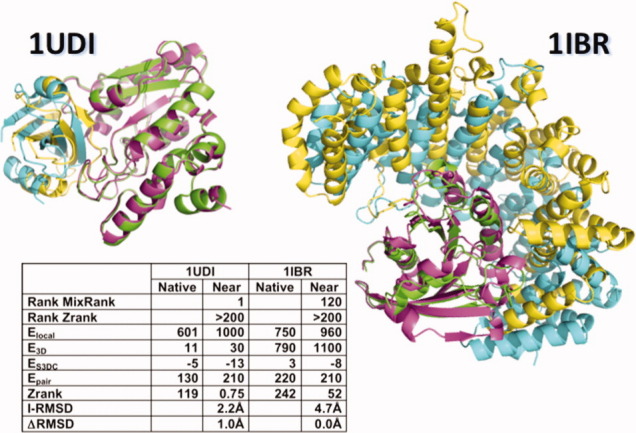
Comparison of the native conformation and one decoy for easy and difficult targets. The native conformation and a near-native decoy of 1UDI (easy case) and of a good decoy of 1IBR (difficult case) are plotted in ribbons. Decoy conformations obtained by rigid-body docking of the unbound proteins are shown in pink and yellow. Their respective chains in the native conformation of the binary complex are shown in green and cyan. Additional information of the scores (Zrank, Elocal, Epair, ES3DC, and E3D) calculated with the native and selected decoys are shown in the inner table. Also, for the decoys of both targets, the table includes the I-RMSD and the ranking by Zrank scores and the MixRank strategy.
Similar success in near-native decoy prediction was obtained by considering a MixRank strategy with the combination of ES3DC, Elocal, and E3D, or E3DC and EPair, or E3DC, Elocal, and E3D as above. Alternatively, we also tried to find a new scoring function by combining the five statistical potentials Epair, Elocal, E3D, E3DC, and ES3DC. This was attempted by using artificial intelligence methods (support vector machine and neural networks among others) without success. Failure was mainly due to the fact that all artificial intelligence methods tended to ignore success cases of ES3DC and E3DC in favor of the three most successful scoring functions EPair, Elocal, and E3D. As already observed, the last three scoring functions share most of the hits, and therefore, there is no gain in their combination.
Selection of the best decoy in a target set
We have been concerned with the selection of near-native decoys in a target set. However, only 97 of the target sets of the benchmark dataset contain decoys satisfying our criterion of near-native. We analyze here the capacity of the scoring functions and MixRank ranking to top-rank the best available decoy. To introduce some flexibility, we consider a decoy good if its I-RMSD differs less than 0.5 Å from the lowest I-RMSD among all the decoys in the target set. Note that this concept depends on the other decoys in the target set, and it is not a property of the decoy conformation in comparison to the native structure.
We have constructed new success curves [Fig. 5(A)] by considering the percentage of target sets in the benchmark dataset for which a good decoy is top-ranked, while varying the number of allowed predictions. Note that this definition of success allows us to test all 124 target sets. We observe that the relation between our five statistical potentials is analogous to that depicted in Figure 2(A). However, in this case, the most successful scores are better than random.
Figure 5.
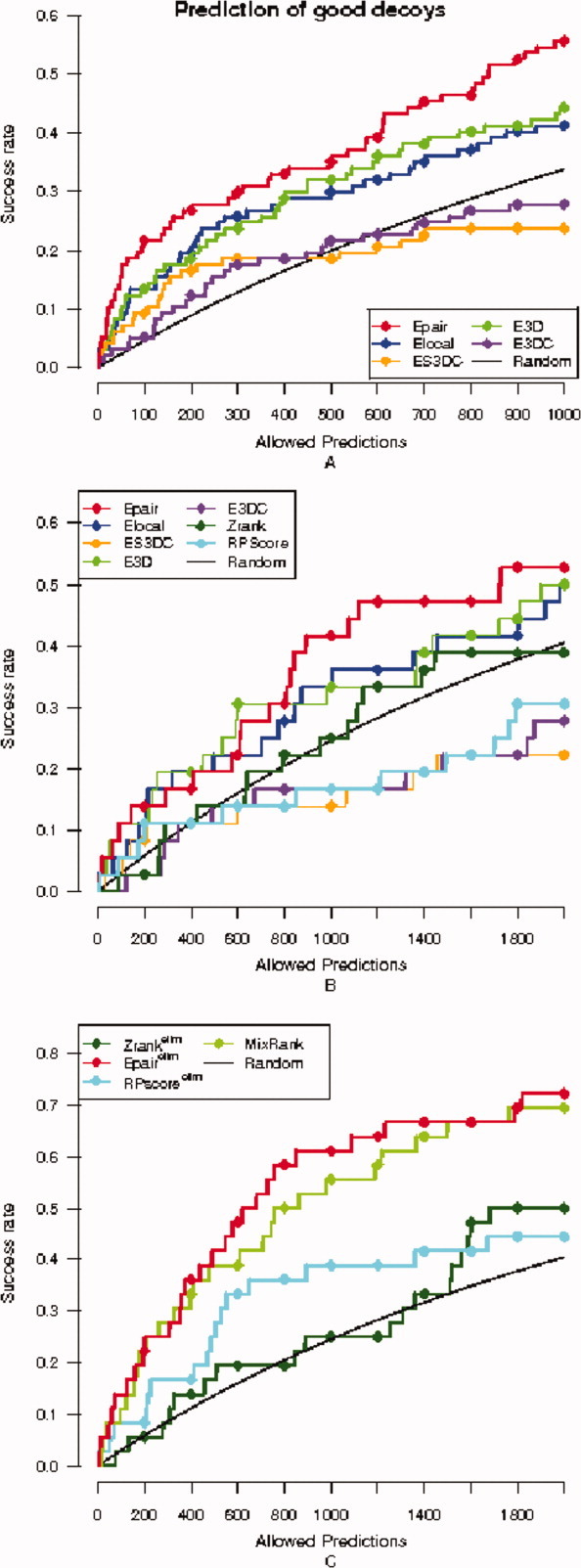
Success curves for good decoys in the benchmark dataset. (A) Success curves on the whole benchmark dataset are plotted for the five statistical potentials Epair (red), ES3DC (orange), Elocal (blue), E3D (light green), and E3DC (purple). (B) Success curves for good decoys for the five statistical potentials together with Zrank (dark green) and RPScore (cyan), only with the medium and difficult cases of the benchmark dataset (no redundancy filter applied). (C) Success curves are plotted after removal of redundant solutions for the MixRank strategy, Epair, Zrank and RPScore scoring functions, and also compared with the success curve expected by random (black), only with the medium and difficult cases of the benchmark dataset.
Determination of the best (or good) decoys in the target set allows us to take a closer look to the behavior of the scoring functions on the medium and difficult cases of the benchmark dataset (see Methods). For most targets in these two classes, there is no decoy in their target set satisfying our near-native criterion. The goal for the docking community is to obtain the best pose, but our purpose here concerns only the evaluation of scoring functions that are aimed to select the decoys that will be further processed, and we do not allow conformational changes at this step. Therefore, the success in these types of targets is to select the best decoy available.
Figures 5(B) and 5(C) provide the analysis of success when restricted to medium and difficult cases. Figure 5(B) depicts the success curves related to good decoys for medium and difficult cases, for the scoring functions Epair, Elocal, E3D, E3DC, and ES3DC and Zrank and RPScore. We observe that the statistical potentials Epair, E3D, and Elocal have a similar success in the selection of the best decoy [compare to Fig. 5(A)]. Additionally, these three statistical potentials surpass Zrank for these cases. Figure 5(C) provides the success curves for the MixRank strategy together with the ranks provided after the application of the redundancy filter to Epair, Zrank, and RPScore. The MixRank strategy and the rank provided by Epair are better than Zrank at predicting the best decoy in the dataset of medium and difficult cases.
We attribute the differences to the fact that Zrank takes into account detailed atomic features (fine-grain) and hence it is likely to fail in predicting decoys that are not close enough to the native structure. On the contrary to Zrank, our statistical potentials are less dependent on small variations of the conformation of the binding interface (coarse-grain). This observation was also pointed out in a previous work.45
Figure 4 shows the scores and ranks for the best near-native decoy of target 1IBR predicted as solution at rank 120 according to MixRank and at rank 3722 according to Zrank (applying the redundancy filter). The value of I-RMSD of this decoy is 4.71 Å. In this example, it is shown that the Zrank score of the native conformation of the binary complex of target 1IBR is significantly different to the Zrank score of this near-native decoy, while the differences using Epair scores are not that significant. Using the scoring functions Elocal or E3D alone, we also mistake the ranking. Therefore, Zrank can only be used if the structure of the binary complex after rigid-docking is optimized and correctly modified, while this example shows how Epair, Elocal, and E3D can be combined to obtain a good ranking of rigid-docking poses.
Near-native decoy selection by the statistical potentials on the permanent interactions dataset
Next, we analyze the success of our statistical potentials Epair, Elocal, E3D, E3DC, and ES3DC in the detection of near-native decoys on the permanent dataset. We observe that the success curves in this case (Fig. 6) are similar to the success curves obtained for the benchmark set (Fig. 2). It has to be noted that most scoring functions are over the random curve in the permanent interactions dataset and that Epair overcomes the rest of the potentials [Fig. 6(A)].
Figure 6.
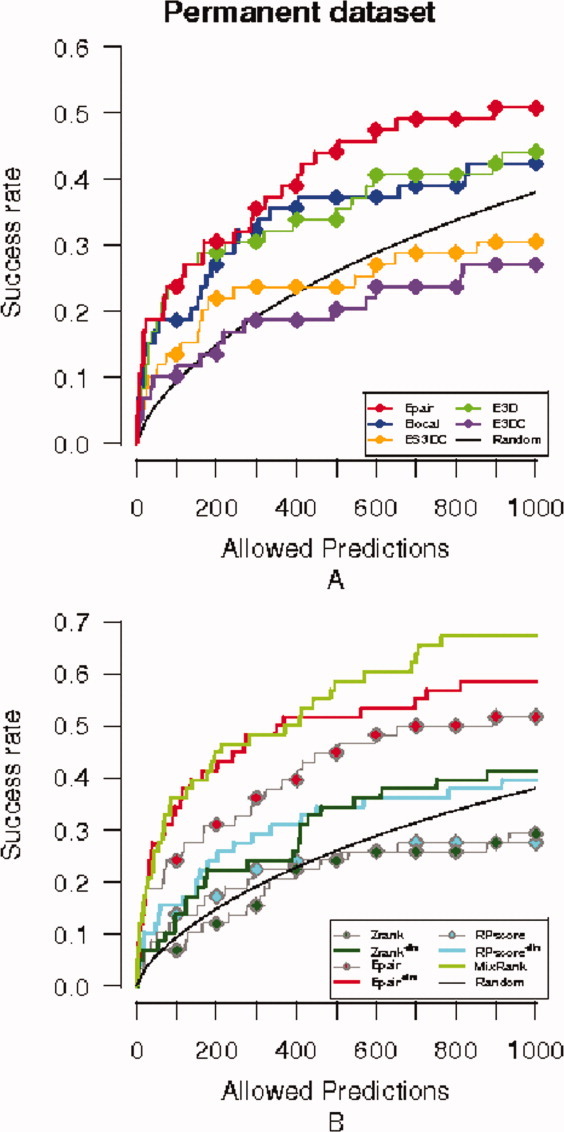
Success curves for the permanent dataset. Legend colors of potentials as in Figure 2. (A) Success curves for the five statistical potentials: Epair, ES3DC, Elocal, E3D, and E3DC, plus the success curve expected by random. (B) Success curve of the MixRank strategy ranking and ranks with Epair, Zrank, RPScore scoring functions before and after application of the redundancy filter.
As we did with the benchmark dataset, we have analyzed the overlap among the hits of the different statistical potentials when allowing 200 predictions. We observe that all hits of Elocal and all but one hit of E3D are hits of Epair, while seven hits of ES3DC are not found by the rest of scoring potentials. This suggests that the MixRank strategy can improve the rank given by Epair for permanent interactions. For the analysis of the statistical significance of the hits, we show in Table SII the probability of finding at least one near-native decoy among the 200 top-ranked decoys of each target set. P values in the table show similar difficulties to guess a near-native decoy by chance to those found for the benchmark dataset (Table SI).
Based on the previous observation, we have further compared the near-native prediction success of MixRank strategy to that of the rank given by Epair, Zrank, and RPScore after applying the redundancy filter. The corresponding success curves are given in Figure 6(B). We note that, contrary to the scenario shown in Figure 2(B) for the benchmark dataset, Zrank has lower success rate than the MixRank and Epair rankings (both with the redundancy filter). We are apparently reflecting some property of the permanent dataset not present in the benchmark dataset. Zrank is a scoring function that is a linear combination of energy terms, whose weights are obtained by training on the cases of the benchmark dataset of the first version,47 which is not included in the later versions of the dataset that were used for testing. We believe that this makes Zrank more specific to transient interactions, because the ratio of targets being transient interactions in the benchmark dataset is higher than in the permanent dataset, and this is why it has a reduced success rate when scoring permanent interactions.
Table II provides the name of the targets of the permanent interaction dataset for which the ranks given by the MixRank strategy, Zrank, and RPScore produced at least one near-native decoy in the first 200 predictions.
Table II.
Hits by different ranking methods on the permanent dataset
| RPScore & MixRank | MixRank | Zrank & MixRank |
| Dehydratase_LU#Dehydratase_SU | ATP-synt#ATP-synt_DE_N | ATP-synt_DE#ATP-synt_Eps |
| Fumarate_red_C#Fumarate_red_D | Cloacin#Cloacin_immun | MCR_beta#MCR_gamma |
| NHase_alpha#NHase_beta | COX4#COX7B | |
| Ribosomal_L11_N#Ribosomal_L12 | MCR_alpha#MCR_gamma | |
| RNA_pol_A_bac#RNA_pol_Rpb1_3 | MHC_II_alpha#MHC_II_beta | |
| RNA_pol_Rpb1_4#RNA_pol_Rpb2_3 | PA28_alpha#PA28_beta | |
| TFIIF_alpha#TFIIF_beta | Ribosomal_S10#Ribosomal_S3_N | |
| RuBisCO_large#RuBisCO_small | ||
| TP_methylase#TP_methylase | ||
| tRNA-synt_2e#tRNA-synt_2e | ||
| Urease_alpha#Urease_beta | ||
| RPScore | Common for all | Zrank |
| RNA_pol_L#RNA_pol_Rpb1_3 | Como_LCP#Como_SCP | COX1#COX3 |
| COX6C#COX7B | DNA_pol3_beta#DNA_pol3_beta_3 | |
| LigA#LigB | Glyco_hydro_11#Glyco_hydro_18 | |
| Me-amine-dh_H#Me-amine-dh_L | Ribosomal_S18#Ribosomal_S6 | |
| Ribosomal_S2#Ribosomal_S5 | SRP14#SRP9 | |
| Trp_syntA#Trp_syntA |
We show the targets of the permanent interactions dataset that provide a near-native decoy among the first 200 predictions with rankings provided by the MixRank strategy and the scoring functions Zrank and RPScore, after applying the redundancy filter. All hits that are shared between Zrank and RPScore are also hits for the MixRank.
We have also analyzed the prediction of good decoys using the split statistical potentials (Fig. S3). We obtained similar success curves for Epair, Elocal, and E3D. We further checked the consistency of our approach if decoys of permanent interactions were obtained using the unbound structures. We found only one case in 3DID (RibosomalS2#RibosomalS8) with the unbound structures of both partners (see Table SIII for the ranks of the first good decoy in this case).
Discussion
Statistical potentials have often been used to study protein folding and protein–protein interactions. Since they were first defined in the 1970s, they have been the focus of several studies. Their accuracy has improved substantially thanks to the increase of information available in the databases and the newly developed machine learning algorithms. When studying protein–protein interactions, it has been pointed out by several works14,16,17 that the integration of statistical potentials into the FFT framework increases the number of detected near-native structures during a rigid-body docking search.
In this study, we have tested a series of novel residue-pair statistical potentials for scoring protein–protein interactions, following the methodology of our previous work in protein folds.39 The main focus of study has been the ability to discriminate near-native decoys from a collection of decoys obtained by rigid-body docking experiments. Four new statistical potentials have been considered: ES3DC and E3DC, concerning the frequency of interacting residues, including the specific conditions (secondary structure, exposure and polar character) in which they sit; Elocal, measuring the probability of placing the residues involved in the binding interface within a specific condition; and E3D, based exclusively on the geometry of the binding interface.
We have noticed that the information carried by the standard residue-pair statistical potential Epair is mainly a mixture of two facts: first, the tendency of some residues that are at some specific conditions to be at the binding interface (encoded by Elocal), and, second, some geometric constraints (encoded by E3D). The fact that the scoring function Elocal contains the largest part of the information required to predict an interaction implies that the physico-chemical properties necessary for the binding are allocated on the proteins previous to its binding and with independence of the partner. This is the first main conclusion of this work, however, given previous results in the literature, it is not entirely unexpected. The usage of desolvation and optimal docking area to predict binding sites has been described in the literature21,48 and has already been used to predict protein–protein interactions. In our case, we have proved the relevance of certain regions, formed by residues located near the surface of the protein and within a specific secondary structure, to later interact with a protein and to become buried in the protein–protein interface. We have to note that Elocal is computed as a sum of two terms, each of them obtained as the sum of statistical energies of the residues of the binding interface in one of the chains. This suggests that it might be worth introducing Elocal into the FFT procedure, as initial grid scores, in order to improve the docking surface search.
When studying the rest of statistical potentials, we found that ES3DC correctly detected some targets that escaped residue-pair statistical potential selection. ES3DC is a refinement of EPair that takes into account the frequency by which two residues interact and the local features in which the residues sit. Based on this observation, we elucidated a ranking strategy of rigid-docking poses, MixRank, that combined the selection power of the statistical potentials EPair and ES3DC in a way that its success curve was better than that of the standard residue-pair potential for a small number of predictions. This improvement was noticed with the benchmark dataset and the permanent interactions dataset, and also when searching near-native decoys or the best decoys available in the dataset. Our strategy to combine both scores seems plausible to follow if: (i) the hits given by independent scores are different; and (ii) we do not know a priori the most successful scoring function. We wish to note that we do not obtain a new scoring function but a strategy to rank the decoys.
Similarly, in ClusPro,28 the authors follow the strategy to select 2000 decoys by picking the top-ranked decoys according to two different scores (desolvation free energy and electrostatics energy). They proceed with the clustering of the selected decoys and the centers of the biggest clusters are further refined. Our approach here is slightly different, because the selection of decoys is not based on the size of the cluster but keeps the initial order of the individual scores.
ROC curves for EPair, Elocal,E3D, and Zrank on the benchmark dataset suggested that predictions derived from the first three scoring functions included less non near-native decoys than Zrank. This was valid for a large ratio of false-positives (non near-native decoys being considered near-native), and it depends on the total number of allowed predictions. This was particularly relevant for medium and difficult cases, there were success curves for EPair, Elocal, and E3D surpassed Zrank and RPScore functions, and stressed when removing redundant solutions and using the MixRank strategy. This drives us to the second main conclusion of this work, that is, we have generated a ranking based on statistical potentials able to compete with the best available methods (i.e., Zrank) in successful rates, unless there is evidence that only small conformational changes occur upon binding. Besides, because this method is defined from residue-pair statistical potentials and does not require an atomic level description, it can also surpass Zrank when scoring rigid-docking decoys in cases where the unbound partners of an interaction have to endure conformational changes upon binding.
We have tested our methods in two different datasets: the standard benchmark set and a more specific set of permanent interactions. First, the benchmark decoy dataset44 was used to analyze the success of our scoring functions. However, this dataset contains many transient interactions. Therefore, in order to test whether different features would appear in other types of interactions, we constructed a new dataset consisting of only permanent interactions. We wish to note that for the permanent interactions dataset, the MixRank strategy and Epair scoring function surpassed the success rate of Zrank and RPScore when filtering out redundant poses. Therefore, the third main conclusion from this work is that we have obtained a good methodology to rank protein–protein interactions of permanent complexes. This is particularly relevant to tackle the next challenge in protein docking, which is to ensemble higher-order structures (i.e., multiprotein complexes) from their individual components.49,50 These molecular machines are often constituted by a central core of interactions, which are permanent and confer the main function to the complex, decorated by some others with regulatory roles.51 Thus, a good set of potentials that predict the conformation of stable complex cores will be paramount.52 Besides, although it can be argued that the statistical potentials derived from domain–domain interactions are more suitable to the task of detecting permanent interactions, the fact that our methods predicted medium and difficult cases of rigid-docking with better or similar rate of success than other methods validates its generalized use for rigid-docking.
Methods
Databases
We have considered three databases, one to calculate the knowledge based pair potentials (3DID) and two to test the scoring functions (a benchmark decoy set and the permanent interactions set).
3D Interacting Domains (3DID)
We have considered a nonredundant set of interacting domains extracted from the database 3DID.53 The database 3DID consists of a nonredundant collection of domain–domain interactions in proteins for which high-resolution three-dimensional structures are known. Interactions in this database are labeled by the PFAM code of each of the interacting domains. This database has been used for the computation of the frequencies required in the statistical potentials definition.
Benchmark decoy dataset
We have considered the benchmark decoy dataset of Weng and coworkers.44 This dataset is based on a set of nonredundant real interactions for which both the complex 3D structure and the individual chain structures are available. We consider the 54,000 decoys generated using the rigid-body docking algorithm ZDock3.016 from the individual chain structures. The set of binary-complex conformations of a rigid-body prediction are classified according to the expected difficulties to obtain a near-native solution of the target. They deal with three types named: easy, medium, and difficult cases. This classification scheme is based on the degree of conformation changes as measured by I-RMSD and the fraction of non-native residue contacts.44 In Figure 4, we compare the native conformations of targets 1UDI (easy case) and 1IBR (difficult case) to selected decoys (with I-RMSD 2.23 and 4.71 Å, respectively).
In total, the dataset consists of 124 cases, 88 of which are straight forward for rigid-body docking, 19 are medium and 17 are difficult cases for which further conformational changes are required upon binding. Only 97 of them (88 rigid-body and nine medium) fit into our near-native decoy criterion.
Permanent interactions dataset
We have collected a subset of permanent interactions and its accompanying docking decoys, by selecting from 3DID one representative structure of all those interactions whose interacting partner components can only function in their complex form, and thus are unlikely to exist in isolation. Each binary complex has been decomposed in two unbound domains and used to generate a set of decoys of binary interactions. The procedure to obtain the unbound structure of the interacting domains is as follows: First, for each binary complex, we searched in the PDB54 for the structures containing the same domains without its interacting partner. Second, if the unbound domain was not found in the PDB, we searched for homologous proteins with solved structures in the PDB containing the unbound domain and we used them as templates to model the desired unbound domain with MODELLER.55 And third, we constructed the rest of unbound domains of the permanent dataset by extracting the backbone of each unbound domain and remodeling the side-chains with MODELLER. In this way, the dataset contains 143 targets of binary complexes and its unbound structures.
Finally, a total of 54,000 decoys for each target were created using ZDock 3.0 with a 6° sampling. After the sampling, 59 of the targets produced at least one decoy satisfying the I-RMSD < 2.5Å.
Scoring functions
We have compared the performance of our five scoring functions (Epair, Elocal, E3D, E3DC, and ES3DC) with Zrank43 and RPScore.31 Zrank is the scoring function included in ZDock, obtained as a linear weighted sum of van der Waals and electrostatic energies and desolvation. RPScore is a knowledge-based pair potential scoring function included in FTDock.
Statistical assessment of hits
For the assessment of the statistical significance of predictions, we have used the P-value computation and the random expected curve.45
I-RMSD and Ligand-RMSD
I-RMSD (interface root mean square deviation)31,43 of a decoy refers to the pairwise RMSD of corresponding Cα-atoms of the residues in the interface of the native conformation.
Ligand-RMSD46 between two decoys (obtained from rigid-body docking) is computed as the RMSD between corresponding Cα-atoms of all the residues in the ligand. In rigid-body docking, the protein that is rotated and translated around the other protein is called the ligand.
Measures for correct predictions
Depending on the approach taken, we consider correct predictions those being either near-native decoys or good decoys. A decoy is called near-native if I-RMSD<2.5 Å and good if its I-RMSD differs less than 0.5 Å from the lowest I-RMSD among all the decoys in the target set.
ROC curves
The ROC curve is the plot of the false positive rate (FPR) versus the true positive rate (TPR) calculated while varying the selection threshold of a scoring function:
Here, Pos and Neg are the total number of positive and negative objects, respectively, TP is the number of correctly predicted positive objects, and FP is the number of objects incorrectly predicted to be positive.
Acknowledgments
We acknowledge the ZDock team for helping us on the use of their database and the helpful comments of Dr. Fernández-Recio. E.F. acknowledges the help of the rest of members of Structural Bioinformatics Group (GRIB-UPF), in particular J. Bonet and O. Fornés for their scripts. E.F. wants to thank the Bioinformatics Research Centre in Aarhus (Denmark) where part of this manuscript was elaborated during a visit in spring 2010.
References
- 1.Goh KI, Cusick ME, Valle D, Childs B, Vidal M, Barabasi AL. The human disease network. Proc Natl Acad Sci USA. 2007;104:8685–8690. doi: 10.1073/pnas.0701361104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kar G, Gursoy A, Keskin O. Human cancer protein-protein interaction network: a structural perspective. PLoS Comput Biol. 2009;5:e1000601. doi: 10.1371/journal.pcbi.1000601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Aragues R, Sander C, Oliva B. Predicting cancer involvement of genes from heterogeneous data. BMC Bioinform. 2008;9:172–190. doi: 10.1186/1471-2105-9-172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Martin B, Aragues R, Sanz R, Oliva B, Boluda S, Martinez A, Sierra A. Biological pathways contributing to organ-specific phenotype of brain metastatic cells. J Proteome Res. 2008;7:908–920. doi: 10.1021/pr070426d. [DOI] [PubMed] [Google Scholar]
- 5.Martin B, Sanz R, Aragues R, Oliva B, Sierra A. Functional clustering of metastasis proteins describes plastic adaptation resources of breast-cancer cells to new microenvironments. J Proteome Res. 2008;7:3242–3253. doi: 10.1021/pr800137w. [DOI] [PubMed] [Google Scholar]
- 6.Fornes O, Aragues R, Espadaler J, Marti-Renom MA, Sali A, Oliva B. ModLink+: improving fold recognition by using protein-protein interactions. Bioinformatics. 2009;25:1506–1512. doi: 10.1093/bioinformatics/btp238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Aragues R, Sali A, Bonet J, Marti-Renom MA, Oliva B. Characterization of protein hubs by inferring interacting motifs from protein interactions. PLoS Comput Biol. 2007;3:1761–1771. doi: 10.1371/journal.pcbi.0030178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kundrotas PJ, Vakser IA. Accuracy of protein-protein binding sites in high-throughput template-based modeling. PLoS Comput Biol. 2010;6:e1000727. doi: 10.1371/journal.pcbi.1000727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tuncbag N, Gursoy A, Guney E, Nussinov R, Keskin O. Architectures and functional coverage of protein-protein interfaces. J Mol Biol. 2008;381:785–802. doi: 10.1016/j.jmb.2008.04.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mosca R, Pons C, Fernández-Recio J, Aloy P. Pushing structural information into the yeast interactome by high-throughput protein docking experiments. PLoS Comput Biol. 2009;5:e1000490. doi: 10.1371/journal.pcbi.1000490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kundrotas PJ, Zhu Z, Vakser IA. GWIDD: genome-wide protein docking database. Nucleic Acids Res. 2010;38:D513–517. doi: 10.1093/nar/gkp944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wodak SJ, Janin J. Computer analysis of protein-protein interaction. J Mol Biol. 1978;124:323–342. doi: 10.1016/0022-2836(78)90302-9. [DOI] [PubMed] [Google Scholar]
- 13.Katchalski-Katzir E, Shariv I, Eisenstein M, Friesem AA, Aflalo C, Vakser IA. Molecular surface recognition: Determination of geometric fit between proteins and their ligands by correlation techniques. Proc Natl Acad Sci USA. 1992;89:2195–2199. doi: 10.1073/pnas.89.6.2195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pons C, Grosdidier S, Solernou A, Pérez-Cano L, Fernández-Recio J. Present and future challenges and limitations in protein-protein docking. Proteins. 2010;78:95–108. doi: 10.1002/prot.22564. [DOI] [PubMed] [Google Scholar]
- 15.Gabb HA, Jackson RM, Sternberg MJ. Modelling protein docking using shape complementarity, electrostatics and biochemical information. J Mol Biol. 1997;272:106–120. doi: 10.1006/jmbi.1997.1203. [DOI] [PubMed] [Google Scholar]
- 16.Mintseris J, Pierce B, Wiehe K, Anderson R, Chen R, Weng Z. Integrating statistical pair potentials into protein complex prediction. Proteins. 2007;69:511–520. doi: 10.1002/prot.21502. [DOI] [PubMed] [Google Scholar]
- 17.Kozakov D, Brenke R, Comeau SR, Vajda S. Piper: an fft-based protein docking program with pairwise potentials. Proteins. 2006;65:392–406. doi: 10.1002/prot.21117. [DOI] [PubMed] [Google Scholar]
- 18.Garzon JI, Lopez-Blanco JR, Pons C, Kovacs J, Abagyan R, Fernandez-Recio J, Chacon P. FRODOCK: a new approach for fast rotational protein-protein docking. Bioinformatics. 2009;25:2544–2551. doi: 10.1093/bioinformatics/btp447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ritchie DW, Kemp GJ. Protein docking using spherical polar Fourier correlations. Proteins. 2000;39:178–194. [PubMed] [Google Scholar]
- 20.Vajda S, Kozakov D. Convergence and combination of methods in protein-protein docking. Curr Opin Struct Biol. 2009;19:164–170. doi: 10.1016/j.sbi.2009.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cheng TMA, Blundell TL, Fernández-Recio J. pyDock: electrostatics and desolvation for effective scoring of rigid-body protein-protein docking. Proteins. 2007;68:503–515. doi: 10.1002/prot.21419. [DOI] [PubMed] [Google Scholar]
- 22.Gray JJ, Moughon S, Wang C, Schueler-Furman O, Kuhlman B, Rohl CA, Baker D. Protein-protein docking with simultaneous optimization of rigid-body displacement and side-chain conformations. J Mol Biol. 2003;331:281–299. doi: 10.1016/s0022-2836(03)00670-3. [DOI] [PubMed] [Google Scholar]
- 23.Fernández-Recio J, Totrov M, Abagyan R. ICM-DISCO docking by global energy optimization with fully flexible side-chains. Proteins. 2003;52:113–117. doi: 10.1002/prot.10383. [DOI] [PubMed] [Google Scholar]
- 24.Dobbins SE, Lesk VI, Sternberg MJ. Insights into protein flexibility: The relationship between normal modes and conformational change upon protein-protein docking. Proc Natl Acad Sci USA. 2008;105:10390–10395. doi: 10.1073/pnas.0802496105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Król M, Chaleil RA, Tournier AL, Bates PA. Implicit flexibility in protein docking: across-docking and local refinement. Proteins. 2007;69:750–757. doi: 10.1002/prot.21698. [DOI] [PubMed] [Google Scholar]
- 26.Dominguez C, Boelens R, Bonvin AM. HADDOCK: a protein-protein docking approach based on biochemical or biophysical information. J Am Chem Soc. 2003;125:1731–1737. doi: 10.1021/ja026939x. [DOI] [PubMed] [Google Scholar]
- 27.Camacho CJ, Gatchell DW. Successful discrimination of protein interactions. Proteins. 2003;52:92–97. doi: 10.1002/prot.10394. [DOI] [PubMed] [Google Scholar]
- 28.Comeau SR, Gatchell DW, Vajda S, Camacho CJ. ClusPro: an automated docking and discrimination method for the prediction of protein complexes. Bioinformatics. 2004;20:45–50. doi: 10.1093/bioinformatics/btg371. [DOI] [PubMed] [Google Scholar]
- 29.Wang C, Schueler-Furman O, Andre I, London N, Fleishman SJ, Bradley P, Qian B, Baker D. RosettaDock in CAPRI rounds 6-12. Proteins. 2007;69:758–763. doi: 10.1002/prot.21684. [DOI] [PubMed] [Google Scholar]
- 30.Zacharias M. ATTRACT: protein-protein docking in CAPRI using a reduced protein model. Proteins. 2005;60:252–256. doi: 10.1002/prot.20566. [DOI] [PubMed] [Google Scholar]
- 31.Moont G, Gabb HA, Sternberg MJ. Use of pair potentials across protein interfaces in screening predicted docked complexes. Proteins. 1999;35:364–373. [PubMed] [Google Scholar]
- 32.Shen MY, Sali A. Statistical potential for assessment and prediction of protein structures. Protein Sci. 2006;15:2507–2524. doi: 10.1110/ps.062416606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen H, Skolnick J. M-TASSER: an algorithm for protein quaternary structure prediction. Biophys J. 2008;94:918–928. doi: 10.1529/biophysj.107.114280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lu L, Lu H, Skolnick J. MULTIPROSPECTOR: an algorithm for the prediction of protein-protein interactions by multimeric threading. Proteins. 2002;49:350–364. doi: 10.1002/prot.10222. [DOI] [PubMed] [Google Scholar]
- 35.Aloy P, Russell RB. InterPreTS: protein interaction prediction through tertiary structure. Bioinformatics. 2003;19:161–162. doi: 10.1093/bioinformatics/19.1.161. [DOI] [PubMed] [Google Scholar]
- 36.Gao M, Skolnick J. DBD-Hunter: a knowledge-based method for the prediction of DNA-protein interactions. Nucleic Acids Res. 2008;36:3978–3992. doi: 10.1093/nar/gkn332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ahmad S, Keskin O, Sarai A, Nussinov R. Protein-DNA interactions: structural, thermodynamic and clustering patterns of conserved residues in DNA-binding proteins. Nucleic Acids Res. 2008;36:5922–5932. doi: 10.1093/nar/gkn573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Temiz NA, Camacho CJ. Experimentally based contact energies decode interactions responsible for protein-DNA affinity and the role of molecular waters at the binding interface. Nucleic Acids Res. 2009;37:4076–4088. doi: 10.1093/nar/gkp289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Aloy P, Oliva B. Splitting statistical potentials into meaningful scoring functions: Testing the prediction of near-native structures from decoy conformations. BMC Struct. Biol. 2009;9:71–93. doi: 10.1186/1472-6807-9-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Melo F, Feytmans E. Novel knowledge-based mean force potential at atomic level. J Mol Biol. 1997;267:207–222. doi: 10.1006/jmbi.1996.0868. [DOI] [PubMed] [Google Scholar]
- 41.Miyazawa S, Jernigan RL. How effective for fold recognition is a potential of mean force that includes relative orientations between contacting residues in proteins? J Chem Phys. 2005;122:024901. doi: 10.1063/1.1824012. [DOI] [PubMed] [Google Scholar]
- 42.Vajda S. Classification of protein complexes based on docking difficulty. Proteins. 2005;60:176–180. doi: 10.1002/prot.20554. [DOI] [PubMed] [Google Scholar]
- 43.Pierce B, Weng Z. Zrank: Reranking protein docking predictions with an optimized energy function. Proteins. 2007;67:1078–1086. doi: 10.1002/prot.21373. [DOI] [PubMed] [Google Scholar]
- 44.Hwang H, Pierce B, Mintseris J, Janin J, Weng Z. Protein-protein docking benchmark version 3.0. Proteins. 2008;73:705–709. doi: 10.1002/prot.22106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Feliu E, Oliva B. How different from random are docking predictions when ranked by scoring functions? Proteins. 2010;78:3376–3385. doi: 10.1002/prot.22844. [DOI] [PubMed] [Google Scholar]
- 46.Lorenzen S, Zhang Y. Identification of near-native structures by clustering protein docking conformations. Proteins. 2007;68:187–194. doi: 10.1002/prot.21442. [DOI] [PubMed] [Google Scholar]
- 47.Chen R, Mintseris J, Janin J, Weng Z. A protein-protein docking benchmark. Proteins. 2003;52:88–91. doi: 10.1002/prot.10390. [DOI] [PubMed] [Google Scholar]
- 48.Fernández-Recio J, Totrov M, Skorodumov C, Abagayan R. Optimal docking area: a new method for predicting protein-protein interaction sites. Proteins. 2005;58:134–143. doi: 10.1002/prot.20285. [DOI] [PubMed] [Google Scholar]
- 49.Aloy P, Böttcher B, Ceulemans H, Leutwein C, Mellwig C, Fischer S, Gavin AC, Bork P, Superti-Furga G, Serrano L, Russell RB. Structure-based assembly of protein complexes in yeast. Science. 2004;303:2026–2029. doi: 10.1126/science.1092645. [DOI] [PubMed] [Google Scholar]
- 50.Alber F, Dokudovskaya S, Veenhoff LM, Zhang W, Kipper J, Devos D, Suprapto A, Karni-Schmidt O, Williams R, Chait BT, Rout MP, Sali A. Determining the architectures of macromolecular assemblies. Nature. 2007;450:683–694. doi: 10.1038/nature06404. [DOI] [PubMed] [Google Scholar]
- 51.Gavin AC, Aloy P, Grandi P, Krause R, Boesche M, Marzioch M, Rau C, Jensen LJ, Bastuck S, Dümpelfeld B, Edelmann A, Heurtier MA, Hoffman V, Hoefert C, Klein K, Hudak M, Michon AM, Schelder M, Schirle M, Remor M, Rudi T, Hooper S, Bauer A, Bouwmeester T, Casari G, Drewes G, Neubauer G, Rick JM, Kuster B, Bork P, Russell RB, Superti-Furga G. Proteome survey reveals modularity of the yeast cell machinery. Nature. 2006;440:631–636. doi: 10.1038/nature04532. [DOI] [PubMed] [Google Scholar]
- 52.Bravo J, Aloy P. Target selection for complex structural genomics. Curr Opin Struct Biol. 2006;16:385–392. doi: 10.1016/j.sbi.2006.05.003. [DOI] [PubMed] [Google Scholar]
- 53.Stein A, Panjkovich A, Aloy P. 3did Update: domain-domain and peptide-mediated interactions of known 3D structure. Nucleic Acids Res. 2009;37:D300–D3004. doi: 10.1093/nar/gkn690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The Protein Data Bank. Nucleic Acids Research. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Eswar N, Webb B, Marti-Renom MA, Madhusudhan MS, Eramian D, Shen MY, Pieper U, Sali A. Comparative protein structure modeling using MODELLER. Curr Protoc Protein Sci. 2007 doi: 10.1002/0471140864.ps0209s50. Chapter 2, Unit 2.9. [DOI] [PubMed] [Google Scholar]