

Abstract
A new crystal structure of human ubiquitin is reported at 1.8 Å resolution. Compared with the other known crystal structure or the solution NMR structure of monomeric human ubiquitin, this new structure is similar in its overall fold but differs with respect to the conformation of the backbone in a surface-exposed region. The conformation reported here resembles conformations previously seen in complex with deubiquinating enzymes, wherein the Asp52/Gly53 main chain and Glu24 side chain move. This movement exposes the backbone carbonyl of Asp52 to the exterior of the molecule, making it possible to engage in hydrogen-bond contacts with neighboring molecules, rather than in an internal hydrogen bond with the backbone of Glu24. This particular crystal form of ubiquitin has been used in a large number of solid state NMR studies. The structure described here elucidates the origin of many of the chemical shift differences comparing solution and solid state studies.
Keywords: conformational switch, solid state NMR, conformational selection model, deubiquitinating enzyme, crystal contact
Introduction
Ubiquitin plays pivotal roles in the fate of the eukaryotic cells: proteins are tagged for degradation or localization when one or more copies of ubiquitin are added covalently to free amine groups. Its promiscuous interactions with myriad partners in the cell have led to some interest regarding ubiquitin's structural plasticity and the relation of plasticity to binding of partner molecules.
Felicitously, this key player in cellular biology has also been an ideal test system for NMR methods particularly for studies of conformational dynamics. Its compact globular form with mixed secondary structures and its inherent thermostability have made it a prime subject for pulse sequence and other methodological development.1 Microcrystalline ubiquitin has been the model system in important solid state NMR (SSNMR) method developments: ubiquitin was one of the first cases to be assigned by high resolution MAS methods, including the use of two-dimensional 13C-13C correlation experiments,2–4 1H-15N correlation experiments,5 and three-dimensional 15N-13C-13C correlation spectra.6 Several structure determination methods have been illustrated using microcrystalline ubiquitin.7–9 More recently, conformational dynamics studies characterizing backbone and side chain order parameters by use of 13C1Hx dipole interactions have been reported and compared with solution NMR.10 13C-13C double quantum spectroscopy has also been used to detect conformational dynamics.11
The majority of these studies used a specific crystal form of ubiquitin in a relatively high percentage (60%) of 2-methyl-2,4-pentanediol (MPD) as precipitant, because these conditions yield spectra with excellent resolution and no peak doubling due to conformational variants in the crystal. To date, however, no X-ray structure for this crystal form is available to offer detailed analysis of the SSNMR studies. Here, we report for the first time a high resolution X-ray structure for this form.
Results
The structure of a new crystal form of human ubiquitin, grown at pH 4.2 in the presence of 53% (v/v) MPD, has been determined at 1.8 Å resolution. The final R factor for the refined structural model is 18.3%, and the free R factor is 21.1%. The root-mean-square (RMS) deviation from ideal values for bond lengths is 0.011 Å and that for bond angles is 1.6°. The refined structural model contains residues 1-72 of ubiquitin and 91 solvent water molecules. This crystal form is in space group P3221, with unit cell parameters of a = b = 48.4 Å and c = 62.0 Å. The summary of crystallographic information was shown in Table I. In comparison, the crystal structure of monomeric human ubiquitin in the Protein Data Bank (PDB entry 1UBQ12 and 1UBI13) is in space group P212121, with unit cell parameters of a = 50.8 Å, b = 42.8 Å, and c = 29.0 Å, and the crystal structure of monomeric bovine ubiquitin (PDB entry 2ZCC, 100% sequence identity to human ubiquitin) is in space group P212121 and unit cell parameters a = 44.0 Å, b = 50.5 Å, and c = 93.9 Å. Therefore, the new crystal is unlikely to have any relationship to these earlier crystals. This new crystal structure is expected to correspond directly to the form of ubiquitin used in many SSNMR experiments that were carried out with microcrystals prepared in the presence of 51%–60% (v/v) MPD, pH 4.0–4.2. The range in MPD concentrations for the SSNMR studies results from the fact that although 60% MPD is generally used to prepare the crystals, additional cryoprotectants were used in some cases, resulting in a final concentration of approximately 51%.
Table I.
Summary of crystallographic information
| Maximum resolution (Å) | 1.8 |
| Space group | P3221 |
| Unit cell parameters (Å, °) | a = b = 48.4, c=62.0 |
| α = β = 90, γ = 120 | |
| Number of observations | 34,649 |
| Rmerge (%)a | 7.1 (31.1) |
| I/σI | 17.8 (5.1) |
| Redundancy | 4.3 (4.3) |
| Resolution range used for refinement (Å) | 30–1.80 |
| Number of reflections | 7857 |
| Completeness (%) | 99 (100) |
| R factor (%) | 18.3 (22.1) |
| Free R factor (%) | 21.1 (25.1) |
| RMS deviation in bond lengths (Å) | 0.011 |
| RMS deviation in bond angles (°) | 1.6 |
The numbers in parentheses are for the highest resolution shell (1.86–1.8 Å).
Alignment of the Cα atoms of this structure with respect to those of the previously reported structure (1UBQ) gives an RMS distance deviation of 0.43 Å (residues 1-72), as calculated using PyMOL.14 It is known that ubiquitin may adopt a stable, partially folded state (the A-form15) at low pH values and high concentrations of MPD. As elaborated previously6 and discussed below, differences in the chemical shifts comparing the crystalline form and the solution state signals did not show any particular pattern that would suggest the presence of the A form or any other unfolded or refolded form. Knowledge of the X-ray structure of ubiquitin under these crystallization conditions clearly supports our prior assumption that this crystalline form is unrelated to the partially unfolded A-form and is indeed in a normally folded ubiquitin form.
Despite their good overall agreement, there are interesting structural differences between this new structure of ubiquitin and the other structure of monomeric human ubiquitin available in the PDB, 1UBQ. One local structural difference involves the main chain of Asp52 and Gly53. In the new ubiquitin structure, the amide plane connecting the carbonyl group of Asp52 and amine group of Gly53 is flipped relative to its conformation in 1UBQ. The simulated annealing omit electron density map around Asp52/Gly53 is shown in Figure 1, clearly indicating the flip of this peptide bond. The carbonyl group of Asp52 is thereby exposed to solvent and neighboring molecules and the backbone NH of Gly53 forms an internal hydrogen bond with side chain of Glu24, which also flips due to this interaction. This is in contrast to the conformation in 1UBQ (as well as most ubquitin complex and multimeric structures) where the carbonyl group of Asp52 is engaged in an internal hydrogen bond with the backbone amide of Glu24 (Fig. 2).
Figure 1.
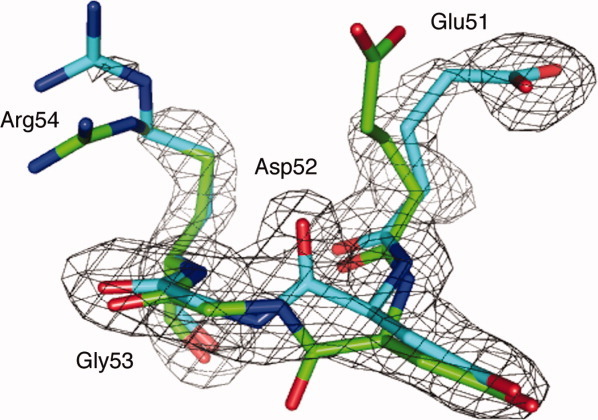
Simulated annealing omit Fo-Fc map for residues 51–54 and superposition of residues 51 to 54 of our new ubiquitin structure (cyan) and 1UBQ (green). The electron density map is contoured at 3σ. The peptide bond between Asp52 and Gly53 is flipped in the new structure with respect to 1UBQ. All structure figures in this paper were produced by PyMOL (http://www.pymol.org).
Figure 2.

The two ubiquitin structures [1UBQ (green) and our new structure (cyan)] have different hydrogen bonds capping the N-terminal α-helices (shown in red) and crystal contacts. (A) The Asp52 carbonyl of 1UBQ forms an internal hydrogen bond with backbone of Glu24. Gly53 forms an intermolecular hydrogen bond with Asp32 of a neighboring ubiquitin molecule. (B) The Gly53 amide of our new ubiquitin structure forms an internal hydrogen bond with the Glu24 side chain; the Asp52 carbonyl forms an intermolecular hydrogen bond with Lys63 of a neighboring ubiquitin molecule through a water molecule.
Besides the Asp52/Gly53/Glu24 “switch” region, several other residues also show difference from their positions in 1UBQ. The backbone of loop β1–β2 [residues 8 to 11, the nomenclature for all the α-helices, β-sheets, and loops are shown in Fig. 3(A)] and loop α1–β3 (residues 37 to 40) have above average RMS deviations (Cα, N, C′ and O atoms RMS distances) when optimally aligned with the 1UBQ structure. In strand β2, the side chains of residues 13 to 16 also have large RMS structural deviations (all side chains atoms except protons) with respect to 1UBQ. These regions were previously shown to have above average amplitude of motion (lower order parameters).10,16–18 Figure 3(B) highlights side chains with large RMS deviations between the two structures; many are solvent-exposed, ionizable residues (e.g., glutamate, aspartate, and arginines), and most of them are not located near crystal contacts. We speculate these structural differences result from the variation in pH (4.2 versus 5.6) and ionic strength for these two structures.
Figure 3.
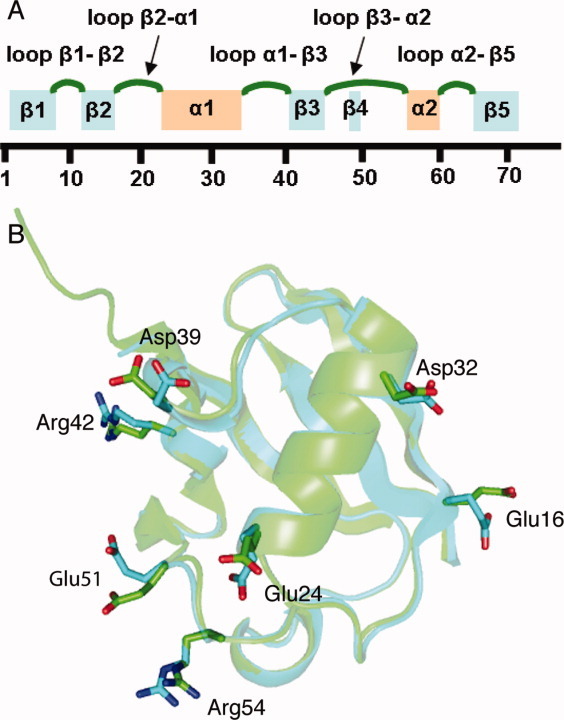
(A) The nomenclatures used here for α-helices, β-sheets, and loops. (B) Superposition of our new ubiquitin structure (cyan) and 1UBQ (green), highlighting glutamates, aspartates, and arginines with greater side chain RMS deviations (side chain RMS derivations greater than average RMS derivation).
Discussion
The new structure highlights a new conformational switch in unbound ubiquitin
We describe the structure of ubiquitin in a crystalline form previously used for many SSNMR studies. This form exhibits an alternative conformation or switched orientation of the Asp52/Gly53 peptide plane, along with a change in the orientation of the Glu24 side chain. This switched orientation in the Asp52/Gly53 peptide plane is also found in some of solid state NMR structure ensembles (discussed further later in this paper). This alternative conformer is likely to have functional significance, because the Asp52/Gly53/Glu24 switched conformer is also found in structures of ubiquitin, ubiquitin aldehyde, or diubiquitin in complex with deubiquitinating enzymes (e.g., PDB entries 2G45, 2HD5, 2IBI, 1NBF, 3I3T, 3IHP, 3NHE, 3MHS, and proximal ubiquitin of 2ZNV, which are all discussed further below). In contrast, the “unswitched” conformer is seen in essentially all other ubiquitin structures, including the previous structures for monomeric ubiquitin, di- and tetra-ubiquitin, and complexes with other kinds of enzymes. To our knowledge, this conformational switch and its relation to deubiquitination have not yet been discussed in the literature. Figure 4 compares our new ubiquitin structure with several other ubiquitin structures in the region of Asp52/Gly53.
Figure 4.
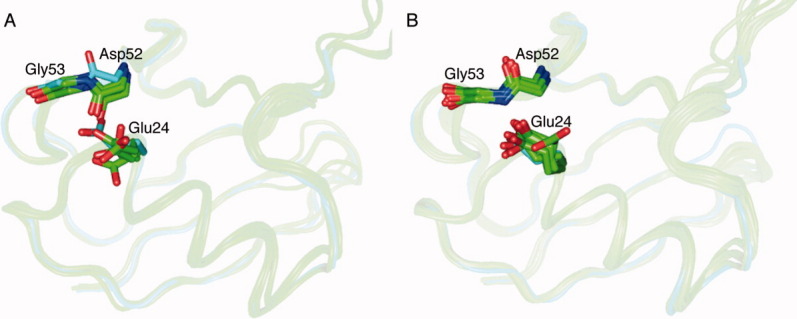
(A) Superposition of the new ubiquitin structure (cyan), the 1UBQ structure and some diubiquitin/tetraubiquitin structures (green). The Asp52/Gly53 peptide plane of the new ubiquitin structure flips to the opposite direction compared with other structures. The Glu24 side chain position also has greater structure deviation compared with the other structures. (B) The new ubiquitin structure (cyan) has a similar conformation as the ubiquitin-deubiquitinating enzyme complex structures 2G45, 2HD5, 2IBI, 1NBF, 3IHP, 3I3T, 3MHS, 3NHE, and 2ZNV (all colored green) in the Asp52/Gly53 peptide plane and Glu24 side chain positions. An interactive view is available in the electronic version of the article.
Ubiquitin has been an important experimental system for studies of intrinsic conformational dynamics of proteins, because it is known to undergo crucial conformational exchange processes during functional binding events with its many and varied binding partners. Ubiquitin-associated motifs (UBA), ubiquitin-interacting motifs (UIM), coupling of ubiquitin to endoplasmic reticulum degradation (CUE), and Ubiquitin-conjugating enzyme E2 variants (UEV) are the common domains found in ubiquitin-binding proteins. All of these motifs interact with ubiquitin residues around Leu8, Ile44, and Val70, but differ in some details regarding the binding modes and the conformations of ubiquitin they engage in binding. In examples, as E3 ubiquitin ligases [e.g., the UBA domain of Cbl-b,19 and the UBA domain of E3 isolated by differential display (EDD) protein20] or endosomal sorting complexes (e.g. Vps23 of ESCRT-I21), the primary contacts involve loop β1-β2 (Leu8), loop β3-α2 (Ile44), and residues around Val70.
Deubiquitinating enzymes, including ubiquitin C-terminal hydrolases (UCHs), ubiquitin-specific processing proteases (UBP and USP families), ovarian tumor (OUT) domain proteins, Machado-Joseph disease protease, and JAB1/MPN/Mov34 metalloenzymes (JAMM) catalyze the hydrolysis of the isopeptide bond in ubiquitin-protein conjugates. Deubiquitinating enzymes interact extensively with C-terminus (residues 71–76) of ubiquitin. Deubiquitinating enzymes also interact with residues around loop β1–β2 as do other ubiquitin binding partners. The motif is somewhat different from some other complexes in that the residues near Ile44 are not uniformly engaged, and the interactions at the C-terminus are more extensive. For example, in the UCH-L3/ubiquitin vinylmethylester complex, the C-terminus of ubiquitin makes interactions with the active site cleft of UCH-L3 and Leu8 and Thr9 also interact with UCH-L3.22 Superposition of ubiquitin from all these various bound complexes (above paragraph) shows that while loop β1–β2, loop β3–α2, and the C-terminal regions have a large RMS structural deviations throughout all ubiquitin complexes, the deviations for the deubiquitinating family of complexes are much less, and the structures are much more similar particularly the complexes that have switched conformer (Asp52/Gly53/Glu24) in ubiquitin (or ubiquitin aldehyde, diubiquitin) structures (Fig. 5).
Figure 5.

(A) Superposition of the Cα trace of our new ubiquitin structure (cyan) with other ubiquitin structures, as observed in complex with UBA, CUE, UIM, and UEV domains (gray green). Loop β1–β2, loop β3–α2, and the C-terminus have greater RMS deviations than the average for the protein. (B) Superposition of our new ubiquitin structure (cyan) with ubiquitin-deubiquitinating enzymes (gray green and 1NBF, 2G45, 2HD5, 2IBI, 3IHP, 3ITI, 3MHS, 3NHE, and 2ZNV are dark green). Loop β1–β2 and the C-terminus have greater RMS deviations than the average.
As described above, the Asp52-Gly53 peptide plane flips along with the Glu24 side chain in many of the deubiquitinating complex structures but not in the majority structures of other ubiqutin complexes. An internal hydrogen bond Glu24(NH)-Asp52(CO) is disrupted, and a new hydrogen bond appears between Gly53's amine group and the side chain of Glu24. The “switched” structures pertain to the family of ubiquitin-specific processing proteases (USP, represented by PDB entries 2G45,23 2HD5,24 2IBI, 1NBF,25 3I3T, 3IHP, 3NHE, and 3MHS26), and JAB1/MPN/Mov34 metalloenzymes (JAMM, represented by PDB entry 2ZNV27).
It is interesting that the switch occurs distal to the major intermolecular contact area. It is, therefore, not clear how the conformational switch is influenced by the deubiquinating enzyme. The Asp52 carbonyl of 2IBI forms a weak water mediated hydrogen bond with a deubiquitinating enzyme (side chain of Arg349 of USP2). Also, the Gly53 of 3IHP and 3I3T have some contacts (< 5 Å) with USPs (for 3IHP, Gly53 has contacts with Arg730 of USP5; Gly53 of 3I3T has contacts with Tyr297 of USP21). For other structures, the contacts are less clear. It is noteworthy that another study of ubiquitin also apparently undergoes a binding-induced structural perturbation on a surface that is distal to its intermolecular contact area. Sgourakis et al. found UIM binding-induced conformational exchange processes in Ser20 and Asn60, which are distal to the interaction surface with the UIM binding partner and are also different conformational switches than the one discussed here. These authors discussed possible functional significance of the switch in the recognition process.28 What causes the Asp52/Gly53 switch to occur in this new monomeric form? One possibility is that the difference between this structure and 1UBQ in this region is related to crystal contacts. The carbonyl of Asp52 is engaged in a water-mediated hydrogen bond with the side chain of Lys63 from a neighboring ubiquitin molecule; in contrast, 1UBQ, the amide group of Gly53 is engaged in a water-mediated hydrogen bond with side chain of Asp32 in a neighboring ubiquitin molecule. Nevertheless, the clear pattern of conformational switch upon binding to a specific class of enzymes would suggest that this switch is directly related to protein's function. To our knowledge, this is the first discussion of the possibility that this particular switch is related to a particular biological function.
Inherent plasticity has been assumed to be important for the binding promiscuity of ubiquitin. Ubiquitin participates in many different molecular interactions, generally engaging the same set of surface exposed residues: loop β1–β2, the C-terminus, and region around Ile44. Superimposition of the crystal structures of the complexes of ubiquitin with all of these various motifs demonstrates that loop β1–β2, loop β3–α2, and the C-terminal regions have greater RMS deviation as compared with other regions (Fig. 5). Our observation that both forms of this newly described switch can be adopted by monomeric ubiquitin, in absence of the deubiquinating enzyme partner, directly demonstrates that the exchange or switch must be intrinsic to the monomeric form. It has been previously shown that conformational changes exhibited on binding can sometimes occur in the native apoprotein, in advance of contact with the binding partner. Conformational selection models invoke a facile conformational change in the apoprotein and posit that the conformational switch precedes binding in a sequential fashion.29,30 Indeed, many other conformational switches required for binding can be observed in absence of the binding partners, either as minor populations or induced by subtle environmental factors, including the case of ubiquitin. Lange et al.16 recently proposed that an inherent, large amplitude, collective “pincer-like” motion of loop β1–β2 and loop α1–β3 is responsible for interface adaption in the context of a conformational selection model for ubiquitin's binding to a wide variety of partner molecules. These authors noted that regions of ubiquitin exhibiting greater variability in structure (comparing among various complexes) sometimes also correspond to regions of higher B factor in apo-ubiquitin X-ray structures and greater mobility as indicated by solution NMR measurements16 or computational methods.31 From this perspective, inherent plasticity of the protein can be useful for its range of binding functions. The native plasticity around Asp52/Gly53 probably plays an important role in ubiquitin-deubiquitinating enzyme recognition and binding. In the ensembles of the free monomer, the plasticity around Asp52/Gly53 was also seen.
Because both conformers are seen when engaged in complexes with biologically important partners, we posit that plasticity in this region is important for function. In support of the assertion of native plasticity of the Asp52/Gly53 peptide plane orientation, the region near the Asp52/Gly53/Glu24 conformational switch has been discussed in terms of exchange phenomena previously. Indeed, the residues around Asp52/Gly53 and Glu24 have elevated dynamics according to previous NMR studies. Massi et al.32 carried out 15N R1ρ relaxation experiments and identified a rate constant of 25,000 s−1 chemical exchange process affecting residues Ile23, Asn25, Thr55, and Val70 and described a very broad and weak peak in Glu24, presumably engaging in a related process. Exchange motions were hypothesized to involve low probability events in which the N-terminus of the α-helix capping hydrogen bonds are disrupted, including Ile23(NH)-Arg54(CO), Glu24(NH)-Asp52(CO), and Asn25(NH)-Thr22(OH). Relaxation studies by Lienin et al.33 showed that residues Asn25 and Asp52 have significantly lower 13C′ T2 values compared with all the other residues and that the Glu24 and Gly53 peaks have notably low sensitivity caused by line broadening. In addition, Majumdar et al.34 studied correlation motions between 2-spin pairs in ubiquitin and found correlation motions involving C′(Glu24)−Cα(Glu24)−N(Asn25), C′(Glu51)−N(Asp52), Cα(Glu51)−C′(Asp51), and Cα(Glu51)−Cα(Asp52) on the microsecond–millisecond timescale. Furthermore, SSNMR studies of conformational dynamics exhibited elevated dynamics in Gly5335 (although Asp52 and Glu24 were not reported because of spectral congestion). The work done by Lange et al.16 also showed flexibility in this region, although on a faster timescale (4 ns 50 μs); the ubiquitin structure ensemble extracted from residual dipolar coupling experiments showed elevated Cα RMS fluctuations around Glu24 and Asp52 and lower NH order parameters around Asp52. All of these studies indicate that Glu24 and Asp52/Gly53 probably have flexibility that is elevated above the majority of the protein. In our new ubiquitin structure, the Ile23-Arg54, Asn25-Thr22 are still engaged in the key capping hydrogen bonds, while the Glu24-Asp52 hydrogen bond is broken, which is clearly different from one of the models proposed for microsecond mobility in this region.32 The switch we discuss here, while not identical to any of the previously hypothesized models, is likely nevertheless to be closely related to the processes underlying these dynamical measurements. It is interesting to note that there is also evidence of the altered Asp52/Gly53 conformation in prior SSNMR structural studies: The ubiquitin microcrystalline structural ensemble determined by Manolikas et al. (PDB entry 2JZZ) shows some examples in which the Asp52/Gly53 peptide plane flips along with Glu24 side chain movement. Schneider et al. structurally characterized another ubiquitin microcrystalline (precipitated in polyethylene glycol instead of MPD) using SSNMR. This structure has greater derivations around Asp52/Gly53, and the Arg54-Ile23 hydrogen bonds are disrupted.11 The Asp52/Gly53 peptide plane of the structural mean in this study also appeared to be flipped as compared to the 1UBQ structure (but not exactly equivalent to this new ubiquitin structure reported herein).
In the context of native plasticity, it is interesting to discuss the thermal disorder in this new crystal form. The variation of Cα, Cβ, and CO B factors with respect to residue numbers have a qualitatively similar pattern in terms of which regions of the protein are most disordered: the B factors are higher for the loop regions, particularly loop β1–β2. This new ubiquitin structure, though, exhibits generally higher B factors as compared to those reported for 1UBQ. The crystal contacts (which of course differ for the two forms) do not appear to correspond to particularly high or low B factors for either monomeric form, with the possible exception of the N-terminal portion of the α-helix α1 (Glu24 and Asn25). The N-terminus of the α1 region has elevated B factors in 1UBQ but not in this new ubiquitin structure. Possibly this difference occurs because the close crystal contacts of Glu24 and Asn25 in the new ubiquitin structure reduce conformational motions in that region and lower the observed B factors. It is also possible that crystal contacts also contribute to our ability to identify the flipped state in Asp52-Gly53 in ubiquitin; Asp52 engages in crystal contacts and our structure shows a new hydrogen bond between Glu24(Oδ) and Gly53(NH), both of which presumably help to stabilize the otherwise poorly populated state.
Chemical shift differences between microcrystals and solution
This new structure gives us a new opportunity to consider the origin of differences in chemical shift of the solution versus the microcrystalline state. While this may not be unique to ubiquitin, in that the α Spectrin SH3 crystal structure by Agarwal et al. (Protein Data Bank entry 2NUZ) is in a condition similar to conditions used for solid state NMR.36 Furthermore, the growing body of work on GB1, there is also the availability of X-ray and SSNMR data under consistent conditions.37 In principle, these structures offer rich information, allowing to us correlate SSNMR parameters with a high resolution X-ray structures and thereby advance the development of SSNMR methods and interpretation. In this regard, ubiquitin is an interesting case because of the particularly large volume of NMR work done to date on it.
Figure 6 shows the Cα, CO, and N chemical shift differences between the solid state assignment and the solution assignments. This comparison was previously discussed by Igumenova et al.6 There are two solution assignments, the one from Wand et al.17 [biological magnetic resonance bank (BMRB) 6466] and the other from Cornilescu et al.38 (BMRB 6457). We used the chemical shifts of the former assignment for our comparison because the pH value used in that study (5.7) is somewhat closer to our sample conditions (4.2) than is that of Cornilescu et al. (6.6). The residues that exhibit significant perturbations of the isotropic chemical shifts (i.e., where at least two backbone chemical shift differences fall outside the 89% confidence interval of the best-fit Gaussian curve of differences) are found in the solvent-exposed portions of helix α1 (Glu24, Asn25, and Asp32) and loop α2-β5 (Asn60, Glu64, and Thr66). Many of the residues in these regions have close contacts with neighboring ubiquitin molecules in this new crystal structure. For example, the side chains of loop α2–β5 have crystal contacts with the side chains of the solvent exposed portion of helix α1 in neighboring ubiquitin molecules and intermolecular hydrogen bonds are formed on this surface (Fig. 7). As mentioned above, the space group of the newly reported structure (P3221) is different from 1UBQ (P212121) resulting in different crystal contacts. Notably, the regions with strongly perturbed chemical shifts have good (but not perfect) correlations with crystal contacts in the new ubiquitin structure. For example, residues 24, 25, 32, and 64, which are among the most perturbed, are involved in extensive crystal contacts (large numbers of intermolecular contacts). Asn60 has short distance crystal contacts and form hydrogen bonds with a neighboring ubiquitin molecule [the Asn60(CO) to Gln31(Nɛ2) distance is 3.1 Å)] In contrast, for 1UBQ, none of the perturbed chemical shifts are near or at crystal contacts. Assuming that changes in solid state chemical shifts upon crystallization are often caused by crystal contacts, this analysis is consistent with our assertion that the crystal packing symmetry of the microcrystals used in SSNMR is the same as those seen in this newly reported ubiquitin structure. In order to make sure the difference in pH value is not the main reason cause the chemical shift differences, Igumenova et al. also compared solid state assignments to solution state N assignments at pH 4.539 (a value close to our solid state NMR conditions) and they identified the same site as outlier. We also made the plots of two sets of shift differences (shift differences between solid shifts and solution shift at pH 5.7 and 4.5) with respect to residue numbers, which is shown in Supplementary Figure 1. It shows that the choice of pH for the solution reference value (5.6 vs. 4.5) is not of crucial importance in this case. We conclude that chemical shift differences between microcrystal and solution relate to some other aspect, such as the crystal contacts or presence precipitant.
Figure 6.
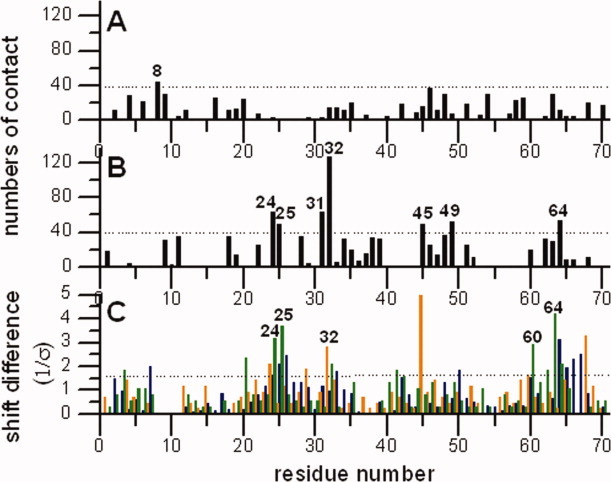
Plots of the numbers of intermolecular contacts (numbers of intermolecular C, N, and O atoms pairs, which have distance shorter than 5Å) with residues numbers in the 1UBQ (A) and the new ubiquitin structure (B) the numbers of intermolecular contacts were calculated by a home-written program, ACCEPT-NMR (described elsewhere). (C) The Cα (orange), CO (green), and N (blue) chemical shift differences between solid state assignments by Igumenova et al. and solution assignments by Wand et al. The shift differences were fitted by a Gaussian function and plotted the shift differences relative to the standard derivation of the mean σ. The shift differences falling outside the confidence interval of 89% on the best fitting Gaussian curve (dashed line, 1.6 σ) are considered to be outliers. Most of the residues having larger Cα, CO, and N perturbed shifts (label the residues have greater perturbed shifts at least two backbone chemical shifts are perturbed) also have higher numbers of intermolecular contacts in our new ubiquitin structure, but not in the 1UBQ structure.
Figure 7.
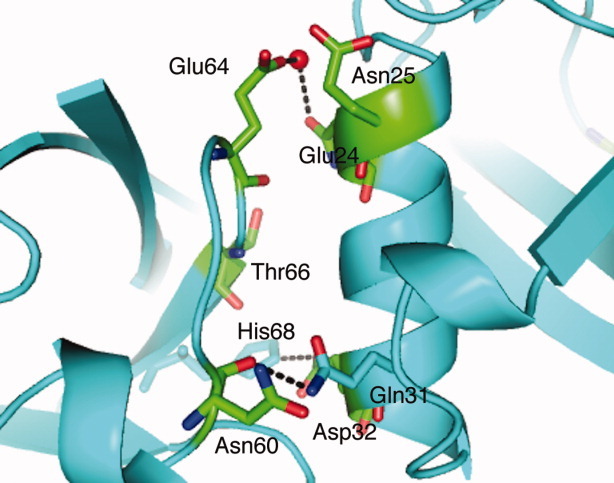
The correlation between crystal contacts of our new ubiquitin crystal structure and the perturbation of the solid state chemical shift (shown in green). Residues that have perturbed chemical shifts, including, solvent exposed part of helix 1 (Glu24, Asn25, and Asp32), have close contacts with loop α2-β5 (Asn60 and Glu64) and some of them form intermolecular hydrogen bonds.
In contrast to the analysis of Cα, CO, and N shifts discussed above, we could not find significant correlations between many of the perturbations in the Cβ chemical shifts and crystal contacts. Using recent developed tools, some chemical shifts can be predicted based on three-dimensional structures. We used a chemical shift predication tool called SPARTA40 (version 2007.04.016) that predicts backbone isotropic chemical shifts based on torsion angle ϕ/ψ/χ1 information from crystal structures. Based on this tool, our SSNMR derived chemical shifts were equally compatible with the conformation of ubiquitin in 1UBQ structure as with our new ubiquitin crystal structure. This is probably because relatively few backbone torsion angles differ when comparing our new ubiquitin structure and 1UBQ. The regions that have greater predicted shift differences (loop β1–β2) are also the regions of great plasticity, and the SSNMR chemical shift data are missing due to this mobility and unfortunately can not be compared. In other words, the local conformational changes between the two crystalline forms or between solution and crystalline structures appear to be poorly predicted by statistically based predictive tools for the moment.
Conclusions
A new ubiquitin crystal structure was determined which corresponds to the form used in many SSNMR studies. The structure is folded in a form similar to previously reported ubiquitin structures, validating the prior SSNMR work on this system. Perturbed solid state chemical shifts when compared with solution shifts can be mostly explained by the presence of crystal contacts in this new ubiquitin crystal. One difference between the new structure and the prior monomeric crystal structure is that the Asp52/Gly53 peptide plane is flipped out to engage in intermolecular contacts, as it does in deubiquitinating enzyme complexes. This work clearly demonstrates that the flip is possible in the apomolecule.
Materials and Methods
Protein expression and purification
BL21 (DE3) cells were transformed with a p′AED vector (T7 expression system) in which ubiquitin gene was subcloned41 and grown in 1L Luria Broth (LB) medium at 37°C with ampicillin (1 mg/L). One liter of culture was grown in a 2 L flask at 37°C with 250 rpm shaking. When the optical density (OD600) of the culture reached 0.7, protein expression was induced by adding 1 ml of 1 mM isopropyl β-D-1-thiogalactopyranoside (IPTG) into each liter of medium. After 8 h of expression, the cells were harvested by centrifugation at 5000×g for 30 min. Approximately 1 to 1.5 g cell pellet was mixed with 20 ml glacial acetic acid. The cell lysate was centrifuged at 4000xg for 1 h and the supernatant neutralized to pH ˜ 5 with 5 M KOH. This solution was then dialyzed twice against deionized water and a third time against 50 mM ammonium acetate, pH 4.5. A 5 ml SP Sepharose™ Fast Flow (Amersham Pharmacia Biotech AB) resin column (per liter of culture) was equilibrated in 100 ml of 50 mM ammonium acetate, pH 4.5. The cell lysate was loaded onto the SP sepharose resin column, and then washed with 100 ml of equilibration buffer at a flow rate of 1 ml/min. The protein was then eluted with 150 ml of elution buffer (50 mM ammonium acetate, pH 5.5) at a flow rate of 1 ml/min and collected in 5 ml fractions. The protein concentrations of the fractions were determined using optical absorption at 280 nm. The plasmid was re-sequenced and the results were in perfect agreement with a prior coding modification for the wide type sequence.41 The resulting protein was characterized by mass spectrometry.
Protein crystallization
Commercial ubiquitin (Sigma-Aldrich, St Louis, MO) and ubiquitin prepared as described above were used to prepare the microcrystalline samples. Ubiquitin crystals were formed at 4°C by the hanging-drop vapor diffusion method: 2 μl of a 10 mg/ml protein solution was mixed with 2 μl of the reservoir solution (50-56 % MPD and 8-18 % (v/v) glycerol solution in 27mM sodium citrate (pH 4.0-4.2) buffer). Long, thin (0.5mm × 0.01mm × 0.01mm) crystals were obtained after approximately 4 days (Fig. 8).
Figure 8.

Ubiquitin crystals viewed under 20x magnification. The largest crystal is approximately 0.5 mm × 0.01 mm × 0.01 mm.
Structure determination and refinement
X-ray diffraction data were collected to 1.8 Å resolution at the X29A beamline of the National Synchrotron Light Source (NSLS). Diffraction images were processed with the HKL package.42 The structure was solved by molecular replacement with the program COMO43 using the structure of human ubiquitin12 as the search model. The model was refined with the program CNS44 and manually re-built using the program O.45
Acknowledgments
Special thanks to Dr. Clay Bracken for kindly providing ubiquitin plasmid, Mical Nobel for some crystallization conditions screening and Ivan V. Sergeyev for crystal contact calculation program ACCEPT-NMR.
References
- 1.Cavanagh J, Fairbrother WJ, Palmer AG, Rance M, Skelton NJ. Protein NMR spectroscopy : principles and practice. Amsterdam, Boston: Academic Press; 2007. [Google Scholar]
- 2.Igumenova TI, McDermott AE, Zilm KW, Martin RW, Paulson EK, Wand AJ. Assignments of carbon NMR resonances for microcrystalline ubiquitin. J Am Chem Soc. 2004;126:6720–6727. doi: 10.1021/ja030547o. [DOI] [PubMed] [Google Scholar]
- 3.Seidel K, Lange A, Becker S, Hughes CE, Heise H, Baldus M. Protein solid-state NMR resonance assignments from (13C, 13C) correlation spectroscopy. Phys Chem Chem Phys. 2004;6:5090–5093. [Google Scholar]
- 4.Schubert M, Manolikas T, Rogowski M, Meier BH. Solid-state NMR spectroscopy of 10% 13C labeled ubiquitspectral simplification and stereospecific assignment of isopropyl groups. J Biomol NMR. 2006;35:167–173. doi: 10.1007/s10858-006-9025-x. [DOI] [PubMed] [Google Scholar]
- 5.Morcombe CR, Paulson EK, Gaponenko V, Byrd RA, Zilm KW. 1H-15N correlation spectroscopy of nanocrystalline proteins. J Biomol NMR. 2005;31:217–230. doi: 10.1007/s10858-005-0527-8. [DOI] [PubMed] [Google Scholar]
- 6.Igumenova TI, Wand AJ, McDermott AE. Assignment of the backbone resonances for microcrystalline ubiquitin. J Am Chem Soc. 2004;126:5323–5331. doi: 10.1021/ja030546w. [DOI] [PubMed] [Google Scholar]
- 7.Zech SG, Wand AJ, McDermott AE. Protein structure determination by high-resolution solid-state NMR spectroscopy: application to microcrystalline ubiquitin. J Am Chem Soc. 2005;127:8618–8626. doi: 10.1021/ja0503128. [DOI] [PubMed] [Google Scholar]
- 8.Seidel K, Etzkorn M, Heise H, Becker S, Baldus M. High-resolution solid-state NMR studies on uniformly 13C,15N -labeled ubiquitin. Chembiochem. 2005;6:1638–1647. doi: 10.1002/cbic.200500085. [DOI] [PubMed] [Google Scholar]
- 9.Manolikas T, Herrmann T, Meier BH. Protein structure determination from 13C spin-diffusion solid-state NMR spectroscopy. J Am Chem Soc. 2008;130:3959–3966. doi: 10.1021/ja078039s. [DOI] [PubMed] [Google Scholar]
- 10.Lorieau JL, McDermott AE. Conformational flexibility of a microcrystalline globular proteorder parameters by solid-state NMR spectroscopy. J Am Chem Soc. 2006;128:11505–11512. doi: 10.1021/ja062443u. [DOI] [PubMed] [Google Scholar]
- 11.Schneider R, Seidel K, Etzkorn M, Lange A, Becker S, Baldus M. Probing molecular motion by double-quantum (13C,13C) solid-state nmr spectroscopy: application to ubiquitin. J Am Chem Soc. 2010;132:223–233. doi: 10.1021/ja906283h. [DOI] [PubMed] [Google Scholar]
- 12.Vijaykumar S, Bugg CE, Cook WJ. Structure of ubiquitin refined at 1.8 Å resolution. J Mol Biol. 1987;194:531–544. doi: 10.1016/0022-2836(87)90679-6. [DOI] [PubMed] [Google Scholar]
- 13.Ramage R, Green J, Muir TW, Ogunjobi OM, Love S, Shaw K. Synthetic, structural and biological studies of the ubiquitin system—the total chemical synthsis of ubiquitin. Biochem J. 1994;299:151–158. doi: 10.1042/bj2990151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.DeLano WL, Lam JW. PyMOL: A communications tool for computational models. Abstr Pap Am Chem Soc. 2005;230:U1371–U1372. [Google Scholar]
- 15.Wilkinson KD, Mayer AN. Alcohol-induced conformational changes of ubiquitin. Arch Biochem Biophys. 1986;250:390–399. doi: 10.1016/0003-9861(86)90741-1. [DOI] [PubMed] [Google Scholar]
- 16.Lange OF, Lakomek NA, Fares C, Schroder GF, Walter KFA, Becker S, Meiler J, Grubmuller H, Griesinger C, de Groot BL. Recognition dynamics up to microseconds revealed from an RDC-derived ubiquitin ensemble in solution. Science. 2008;320:1471–1475. doi: 10.1126/science.1157092. [DOI] [PubMed] [Google Scholar]
- 17.Wand AJ, Urbauer JL, McEvoy RP, Bieber RJ. Internal dynamics of human ubiquitin revealed by 13C-relaxation studies of randomly fractionally labeled protein. Biochemistry. 1996;35:6116–6125. doi: 10.1021/bi9530144. [DOI] [PubMed] [Google Scholar]
- 18.Chang SL, Tjandra N. Temperature dependence of protein backbone motion from carbonyl 13C and amide 15N NMR relaxation. J Magn Reson. 2005;174:43–53. doi: 10.1016/j.jmr.2005.01.008. [DOI] [PubMed] [Google Scholar]
- 19.Peschard P, Kozlov G, Lin T, Mirza A, Berghuis AM, Lipkowitz S, Park M, Gehring K. Structural basis for ubiquitin-mediated dimerization and activation of the ubiquitin protein ligase Cbl-b. Mol Cell. 2007;27:474–485. doi: 10.1016/j.molcel.2007.06.023. [DOI] [PubMed] [Google Scholar]
- 20.Kozlov G, Nguyen L, Lin T, De Crescenzo G, Park M, Gehring K. Structural basis of ubiquitin recognition by the ubiquitin-associated (UBA) domain of the ubiquitin ligase EDD. J Biol Chem. 2007;282:35787–35795. doi: 10.1074/jbc.M705655200. [DOI] [PubMed] [Google Scholar]
- 21.Teo H, Veprintsev DB, Williams RL. Structural insights into endosomal sorting complex required for transport (ESCRT-I) recognition of ubiquitinated proteins. J Biol Chem. 2004;279:28689–28696. doi: 10.1074/jbc.M400023200. [DOI] [PubMed] [Google Scholar]
- 22.Misaghi S, Galardy PJ, Meester WJN, Ovaa H, Ploegh HL, Gaudet R. Structure of the ubiquitin hydrolase UCH-L3 complexed with a suicide substrate. J Biol Chem. 2005;280:1512–1520. doi: 10.1074/jbc.M410770200. [DOI] [PubMed] [Google Scholar]
- 23.Reyes-Turcu FE, Horton JR, Mullally JE, Heroux A, Cheng XD, Wilkinson KD. The ubiquitin binding domain ZnFUBP recognizes the C-terminal diglycine motif of unanchored ubiquitin. Cell. 2006;124:1197–1208. doi: 10.1016/j.cell.2006.02.038. [DOI] [PubMed] [Google Scholar]
- 24.Renatus M, Parrado SG, D'Arcy A, Eidhoff U, Gerhartz B, Hassiepen U, Pierrat B, Riedl R, Vinzenz D, Worpenberg S, Kroemer M. Structural basis of ubiquitin recognition by the deubiquitinating protease USP2. Structure. 2006;14:1293–1302. doi: 10.1016/j.str.2006.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hu M, Li PW, Li MY, Li WY, Yao TT, Wu JW, Gu W, Cohen RE, Shi YG. Crystal structure of a UBP-family deubiquitinating enzyme in isolation and in complex with ubiquitin aldehyde. Cell. 2002;111:1041–1054. doi: 10.1016/s0092-8674(02)01199-6. [DOI] [PubMed] [Google Scholar]
- 26.Samara NL, Datta AB, Berndsen CE, Zhang XB, Yao TT, Cohen RE, Wolberger C. Structural insights into the assembly and function of the SAGA deubiquitinating module. Science. 2010;328:1025–1029. doi: 10.1126/science.1190049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sato Y, Yoshikawa A, Yamagata A, Mimura H, Yamashita M, Ookata K, Nureki O, Iwai K, Komada M, Fukai S. Structural basis for specific cleavage of Lys 63-linked polyubiquitin chains. Nature. 2008;455:358–362. doi: 10.1038/nature07254. [DOI] [PubMed] [Google Scholar]
- 28.Sgourakis NG, Patel MM, Garcia AE, Makhatadze GI, McCallum SA. Conformational dynamics and structural plasticity play critical roles in the ubiquitin recognition of a UIM domain. J Mol Biol. 2010;396:1128–1144. doi: 10.1016/j.jmb.2009.12.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hammes GG, Chang YC, Oas TG. Conformational selection or induced fit: a flux description of reaction mechanism. Proc Natl Acad Sci U S A. 2009;106:13737–13741. doi: 10.1073/pnas.0907195106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Boehr DD, Nussinov R, Wright PE. The role of dynamic conformational ensembles in biomolecular recognition. Nat Chem Biol. 2009;5:789–796. doi: 10.1038/nchembio.232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ramanathan A, Agarwal PK. Computational identification of slow conformational fluctuations in proteins. J Phys Chem B. 2009;113:16669–16680. doi: 10.1021/jp9077213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Massi F, Grey MJ, Palmer AG. Microsecond timescale backbone conformational dynamics in ubiquitin studied with NMR R-1p relaxation experiments. Protein Sci. 2005;14:735–742. doi: 10.1110/ps.041139505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lienin SF, Bremi T, Brutscher B, Bruschweiler R, Ernst RR. Anisotropic intramolecular backbone dynamics of ubiquitin characterized by NMR relaxation and MD computer simulation. J Am Chem Soc. 1998;120:9870–9879. [Google Scholar]
- 34.Majumdar A, Ghose R. Probing slow backbone dynamics in proteins using TROSY-based experiments to detect cross-correlated time-modulation of isotropic chemical shifts. J Biomol NMR. 2004;28:213–227. doi: 10.1023/B:JNMR.0000013705.98136.99. [DOI] [PubMed] [Google Scholar]
- 35.Lorieau J. 2005. p. 372. Protein dynamics:solid state NMR and computational studies, Ph.D. dissertation: Department of Chemistry, Columbia University, New York, pp.
- 36.Pauli J, Baldus M, van Rossum B, de Groot H, Oschkinat H. Backbone and side-chain 13C and 15N signal assignments of the alpha-spectrin SH3 domain by magic angle spinning solid-state NMR at 17.6 tesla. Chembiochem. 2001;2:272–281. doi: 10.1002/1439-7633(20010401)2:4<272::AID-CBIC272>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 37.Schmidt HLF, Sperling LJ, Gao YG, Wylie BJ, Boettcher JM, Wilson SR, Rienstra CA. Crystal polymorphism of protein GB1 examined by solid-state NMR spectroscopy and X-ray diffraction. J Phys Chem B. 2007;111:14362–14369. doi: 10.1021/jp075531p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cornilescu G, Marquardt JL, Ottiger M, Bax A. Validation of protein structure from anisotropic carbonyl chemical shifts in a dilute liquid crystalline phase. J Am Chem Soc. 1998;120:6836–6837. [Google Scholar]
- 39.Varadan R, Walker O, Pickart C, Fushman D. Structural properties of polyubiquitin chains in solution. J Mol Biol. 2002;324:637–647. doi: 10.1016/s0022-2836(02)01198-1. [DOI] [PubMed] [Google Scholar]
- 40.Shen Y, Bax A. Protein backbone chemical shifts predicted from searching a database for torsion angle and sequence homology. J Biomol NMR. 2007;38:289–302. doi: 10.1007/s10858-007-9166-6. [DOI] [PubMed] [Google Scholar]
- 41.Lazar GA, Desjarlais JR, Handel TM. De novo design of the hydrophobic core of ubiquitin. Protein Sci. 1997;6:1167–1178. doi: 10.1002/pro.5560060605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. San Diego: Academic Press Inc; 1997. pp. 307–326. Macromolecular crystallography, Part A.: pp. [DOI] [PubMed] [Google Scholar]
- 43.Jogl G, Tao X, Xu YW, Tong L. COMO: a program for combined molecular replacement. Acta Crystallogr Sect D-Biol Crystallogr. 2001;57:1127–1134. doi: 10.1107/s0907444901006783. [DOI] [PubMed] [Google Scholar]
- 44.Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr Sect D-Biol Crystallogr. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 45.Jones TA, Zou JY, Cowan SW, Kjeldgaard M. Improved methods for building protein models in electron-density maps and the location of errors in these models. Acta Crystallogr Sect A. 1991;47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]