

Abstract
β2-integrins of neutrophils play a critical role in innate immune defense but they also participate in tissue destruction during autoimmune inflammation. p190RhoGAP, a regulator of Rho-family small GTPases, is required for integrin signal transduction in fibroblasts. Prior studies have also suggested a role for p190RhoGAP in β2 integrin signaling in neutrophils. To directly test that possibility, we have generated a novel targeted mutation completely disrupting the p190RhoGAP-encoding gene in mice. p190RhoGAP deficiency led to perinatal lethality and defective neural development, precluding the analysis of neutrophil functions in adult p190RhoGAP−/− animals. This was overcome by transplantation of fetal liver cells from p190RhoGAP−/− fetuses into lethally irradiated wild type recipients. Neutrophils from such p190RhoGAP−/− bone marrow chimeras developed normally and expressed normal levels of various cell surface receptors. Though p190RhoGAP−/− neutrophils showed moderate reduction of β2 integrin-mediated adherent activation, they showed mostly normal migration in β2-integrin-dependent in vitro and in vivo assays and normal β2 integrin-mediated killing of serum-opsonized S. aureus and E. coli. A neutrophil- and β2 integrin-dependent transgenic model of the effector phase of autoimmune arthritis also proceeded normally in p190RhoGAP−/− bone marrow chimeras. In contrast, all the above responses were completely blocked in CD18−/− neutrophils or CD18−/− bone marrow chimeras. These results suggest that p190RhoGAP likely does not play a major indispensable role in β2 integrin-mediated in vitro and in vivo neutrophil functions or the effector phase of experimental autoimmune arthritis.
Keywords: p190RhoGAP, neutrophils, integrins, signaling, knockout mice
INTRODUCTION
Neutrophils play critical roles in host defense against invading microorganisms but their improper activation can also lead to significant damage to host tissues (1). Neutrophils are equipped with very powerful antimicrobial mechanisms including reactive oxygen species-generating protein complexes, hydrolytic enzymes, other antimicrobial proteins and peptides, as well as highly efficient microbial sensing, chemotactic and phagocytic capabilities. The activation of neutrophils needs to be tightly controlled to ensure that this weaponry is only targeted against the invading microbes but not against the host tissue. Indeed, improper activation of neutrophils leads to severe tissue damage and inflammation, as can be seen in certain autoimmune inflammatory diseases such as autoimmune arthritis (2-4).
β2 (CD11/CD18) integrins play a major role in various functions of neutrophils, as indicated by the inability of CD18-deficient human or murine neutrophils to become activated in an adhesion-dependent manner, migrate to the site of inflammation or to kill bacteria upon a direct encounter with the microbes (5-8). Indeed, it is generally accepted that the severe bacterial infections in human patients lacking the β2 integrin chain CD18 result from disfunction of the neutrophil lineage (5).
Neutrophil β2-integrins trigger complex and yet incompletely understood intracellular signal transduction pathways that are likely triggered by receptor-proximal activation of Hck, Fgr and Lyn, the three members of the Src tyrosine kinase family expressed in these cells (7, 9-12). β2 integrin-mediated activation of Src-family kinases are likely linked to functional responses of the cells through members of the Rho family of small GTPases (13). Of those members, both Rac1 and Rac2 participate in β2 integrin-mediated adhesion and migration of neutrophils (14, 15), Rac2 plays a major role in respiratory burst and granule release (14-22),Cdc42 is required for neutrophil migration through proper regulation of Mac-1 (αMβ2) integrin function (23), whereas RhoA is thought to play an important role in regulation of neutrophil polarization and migration (24, 25). However, the intracellular signals linking receptor-proximal activation of Src-family kinases to regulation of Rho-family members in neutrophils are very poorly understood.
Integrins, Src-family kinases and Rho-family small GTPases are also present in most non-hematopoietic cell types and mediate a spectrum of diverse biological functions including lamellipodia, filopodia and stress fiber formation, as well as cellular migration (26-33). Integrin engagement also leads to a transient suppression of Rho activity which is likely required to relieve cytoskeletal tension and hence allow the necessary shape changes to occur (34-36).
In contrast to cells of hematopoietic origin (e. g. neutrophils), the mechanisms linking integrin-proximal activation of Src-family kinases to regulation of Rho-family small GTPases are better understood in non-hematopoietic cells and is thought to be mediated in large part by p190RhoGAP (also known as glucocorticoid receptor DNA binding factor 1 (Grlf1) or p190-A), a 190 kDa GTPase activating protein (GAP) acting on Rho-family small GTPases. p190RhoGAP is a major substrate of Src-family kinases in fibroblasts (37-41) and in the central nervous system (42) with integrin ligation thought to be a major mechanism of Src-family-mediated phosphorylation of p190RhoGAP in fibroblasts and melanoma cells (34, 43-45). On the other hand, p190RhoGAP possesses GAP activity on Rho-family small GTPases both in cell-free and cell-based in vitro assays (46, 47), providing a possible link between integrin ligation and regulation of the actin cytoskeleton. In agreement with that possibility, overexpression of p190RhoGAP promoted fibroblast spreading and migration (48) and integrin-mediated neurite outgrowth (42, 49), whereas blocking p190RhoGAP function decreased fibroblast adhesion/spreading, polarization and directed migration, and it blocked β1 integrin-mediated invasion of a melanoma cell line (43, 48, 50, 51). Furthermore, genomic deletion of the N-terminal region of p190RhoGAP-encoding gene in mice led to perinatal lethality and neural defects (51), likely due to defective integrin-mediated axon outgrowth (42). Embryonic fibroblasts carrying that p190RhoGAP mutation also showed defective migration in a Transwell system and defective migration and polarization in an in vitro wound-healing assay (52). Taken together, p190RhoGAP appears to be required for diverse integrin-mediated functions in non-hematopoietic cells, most likely by linking Src-family kinases to Rho-family small GTPases and the actin cytoskeleton.
The above studies on non-hematopoietic cells raise the possibility that p190RhoGAP plays a similar role in cells of hematopoietic origin. That possibility is supported by the presence and stimulus-dependent translocation to the plasma membrane of p190RhoGAP in neutrophils (53-56), its phosphorylation by Src-family kinases in macrophages (57) and its co-localization with the αVβ3 integrin and Src-family kinases in the podosomes of osteoclasts (58). Furthermore, recombinant p190RhoGAP increases the GTPase activity of Rac1 and Rac2 and limits the superoxide production in a cell-free NADPH oxidase assay system (53), raising the possibility that p190RhoGAP may regulate the NADPH oxidase. Finally, crosslinking of β2-integrins of human neutrophils induces the translocation of p190RhoGAP to the membrane fraction, and increases its GAP activity on RhoA and its association with p120RasGAP (55), all of which are blocked by an inhibitor of Src-family kinases (55). Collectively, those studies raise the possibility that p190RhoGAP participates in the regulation of integrin-mediated functional responses of myeloid lineage cells such as neutrophils.
As mentioned above, a genetically modified mouse strain carrying a hypomorphic allele expressing a truncated version of p190RhoGAP has been generated before (51). However, no mouse strain with a complete genetic deficiency of p190RhoGAP has yet been described.
The above studies prompted us to generate a novel mouse strain completely lacking p190RhoGAP and to use those mice to test the role of p190RhoGAP in integrin-mediated neutrophil functions, as well as the development of a neutrophil-mediated, integrin-dependent in vivo model of autoimmune inflammatory arthritis. Contrary to our expectations, p190RhoGAP did not prove to play a major indispensable role in integrin-mediated neutrophil functions and the effector phase of experimental autoimmune arthritis.
MATERIALS AND METHODS
Generation of the p190RhoGAP null mutation
Our strategy aimed at the complete disruption of the p190RhoGAP-encoding gene was to delete the first 1.5 kb of exon 1, eliminate the translation initiation site and to drive the expression of a β-Galactosidase-Neomycin-resistance (β-Geo) selection/reporter fusion protein by the endogenous p190RhoGAP promoter. To this end, a targeting vector consisting of a 1.4 kb BamHI-EcoRI 5′ genomic fragment (containing sequences upstream of exon 1 of the p190RhoGAP-encoding gene), a promoter-less β-Geo selection/reporter cassette in the sense orientation, a 6.5 kb XbaI 3′ genomic fragment (containing the 3′ part of exon 1 and the 5′ part of intron 1), as well as a 3′ negative selection cassette encoding the diphtheria toxin A subunit driven by the PGK promoter (PGK-DTA) has been constructed. The targeting vector was linearized and electroporated into D3 ES cells, followed by selection in G418. Correctly targeted ES cell clones were detected by Southern blot analysis of HindIII digested genomic DNA, using a 5′ external probe detecting a 3.8 kb wild-type or a 3.2 kb mutant allele. 34 out of 47 ES cell clones were heterozygous for p190RhoGAP, indicating a very high (72%) targeting efficiency likely because the expression of the β-Geo fusion protein was dependent on the promoter activity of the p190RhoGAP-encoding gene in ES cells. Heterozygous ES cells were injected into 3.5-day old C57BL/6 blastocysts and the generated chimeric mice were bred to C56BL/6 mice to obtain germline transmission of the mutation which was confirmed by Southern blot analysis of the offspring’s tail DNA. The primary sequence of the mutant allele was confirmed by PCR amplification and sequencing. The mutant allele is being submitted to the Mouse Genome Informatics database as the Grlf1tm2JSet mutation.
Animals
The above-described null mutation of the p190RhoGAP-encoding gene (Grlf1tm2JSet, referred to as p190RhoGAP−) was maintained in heterozygous form by breeding p190RhoGAP+/− carriers with C57BL/6 mice. Offspring were genotyped by allele-specific PCR reaction using 5′-ACA GCA AAG GAC AAG TAT GAG T-3′ wild type-specific and 5′-GAG TCG AAT CAA GCT GAT C-3′ mutant-specific forward primers along with the 5′-CCA TCA AGT CAA AGG GAA TC-3′ common reverse primer, giving 410 and 251 bp products for the wild type and mutant alleles, respectively. Mice carrying the KRN T-cell-receptor transgene (59) were obtained from Diane Mathis and Christophe Benoist (Harvard Medical School, Boston, MA) and were maintained and used as described (60). Complete CD18-deficient (Itgb2tm2Bay/tm2Bay, referred to as CD18−/−) mice (6) were obtained from Arthur Beaudet (Baylor College of Medicine, Houston, TX). All mice were backcrossed to the C57BL/6 genetic background for 8 or more generations. Wild type control C57BL/6 mice were purchased from the Hungarian National Institute of Oncology. NOD mice, as well as a congenic strain carrying the CD45.1 allele on the C57BL/6 genetic background (B6.SJL-Ptprca) were purchased from the Jackson Laboratory. Mice were kept in individually sterile ventilated cages (Tecniplast) in a conventional facility. All animal experiments were approved by the Semmelweis University Animal Experimentation Review Board.
Generation of bone marrow chimeras
Bone marrow chimeras with p190RhoGAP−/− hematopoietic system were generated using a fetal liver transplantation approach using E15.5-E18.5 fetuses from timed matings of p190RhoGAP+/− carriers as described (61). p190RhoGAP−/− fetuses were identified by accelerated immunoblot analysis of fetal brain tissue lysates using an anti-p190RhoGAP antibody and occasionally by the presence of the (less penetrant) neural closure defects. The genotypes of the fetuses were confirmed and further analyzed after the transplantation by immunoblotting for β-galactosidase (which is expressed from the p190RhoGAP− allele) and, occasionally, by allele-specific PCR reaction from genomic DNA samples. Unfractionated fetal liver cell suspensions were injected intravenously into lethally irradiated (11.5 Gy) recipients carrying the CD45.1 allele on the C57BL/6 genetic background (approx. 8 recipients per fetal liver). An equal number of control chimeras were also generated using p190RhoGAP-expressing (p190RhoGAP+/+ or p190RhoGAP+/−) sibling fetuses and will be referred to as wild type chimeras. CD18−/− and control wild type bone marrow chimeras were generated by using bone marrow cells from adult donors. Mixed bone marrow chimeras were generated by using a mixture of different donor cells (see below).
Repopulation of the hematopoietic compartment by donor-derived cells was tested 4-6 weeks after transplantation by flow cytometry (see below) and is expressed as the percentage of CD45.2-positive cells in the Gr1-positive granulocyte gate. Bone marrow chimeras were usually used 5-10 weeks after the transplantation.
Neutrophil isolation
Human neutrophils were isolated from venous blood of healthy volunteers by dextran sedimentation followed by Ficoll-Paque PLUS (GE Healthcare) gradient centrifugation and hypotonic lysis of remaining red blood cells as described (62). For further purification of neutrophils, the cells were subjected to positive magnetic separation using human CD16 Microbeads (Miltenyi Biotec) following the manucfacturers’ instructions (63). The purity of the preparation was tested by flow cytometry (see below), as well as by the differential analysis of 1000 leukocytes from each preparation on May-Grünwald-Giemsa stained Cytospin slides.
Unless otherwise stated, mouse neutrophils were isolated from the bone marrow of the femurs and tibias by hypotonic lysis followed by Percoll (GE Healthcare) gradient centrifugation at room temperature using sterile and endotoxin-free reagents as described (64). Neutrophil assays were performed at 37 °C in Hank’s balanced salt solution (Invitrogen) supplemented with 20 mM HEPES, pH 7.4.
For in vitro Zigmond chamber experiments, murine neutrophils were isolated by Percoll gradient centrifugation (65), then cultivated for 24 hours in RPMI 1640 medium (Biochrom) supplemented with 20% conditioned medium from WEHI-3B cells (German Collection of Microorganisms and Cell Cultures).
Flow cytometry
Human neutrophils were labeled with unconjugated anti-CD16 (3G8), followed by FITC-labeled goat anti-mouse antibodies. Isolated murine neutrophils, peripheral blood samples or peritoneal lavage fluids were stained with PE-conjugated anti-Gr1 (RB6-8C5), FITC-conjugated anti-CD45.2 (clone 104) or biotinylated anti-CD11b (M1/70) antibodies, or unconjugated antibodies against CD18 (C71/16), CD11a (M17/4), FcγRII/III (2.4G2) or FcγRIV (9E9; obtained from Jeffrey Ravetch, Rockefeller University, New York, NY) (66). KRN transgene-positive mice were identified as described (60). Unconjugated and biotinylated antibodies were visualized with FITC-conjugated mouse anti-rat IgG, streptavidin-Cy3 (Jackson Immunoresearch, West Grove, PA) or streptavidin-FITC. Unless otherwise stated, all flow cytometry reagents were purchased from BD Biosciences. All staining was performed in the presence of 5% FCS (Invitrogen). Samples were fixed in FACS Lysing Solution (BD Biosciences) and analyzed on a BD Biosciences FACSCalibur using the CellQuest software. Neutrophils were identified based on Gr1-positive staining and their forward/side scatter characteristics.
In vitro functional assays
For integrin-dependent activation, murine neutrophils were plated on a surface coated with 150 μg/ml human fibrinogen (Calbiochem) and stimulated by 50 ng/ml murine TNF (Peprotech), 1 μg/ml Pam3CSK4 (EMC Microcollections) or 10 ng/ml murine GM-CSF (Peprotech) as described (7, 67). For immune complex-mediated activation, the cells were plated on immobilized HSA–anti-HSA immune complexes (both from Sigma) without any additional stimulus as described (68). Adhesion-independent activation of neutrophils was performed in Mg2+-free media essentially as described (7, 62, 64, 67), using 100 nM PMA (Sigma), 3 μM fMLP (Sigma), 50 ng/ml TNF or 10 ng/ml GM-CSF. Where indicated, cells were primed with 50 ng/ml TNF for 25 min or pretreated with 10 μM cytochalasin B (Sigma) for 10 min prior to cell activation. Where necessary, the reaction was stopped after 10 (signaling studies and CB+fMLP-induced degranulation) or 30 min (other degranulation experiments and integrin-dependent spreading responses; TNF or GM-CSF induced integrin-upregulation).
Superoxide release was determined by a real-time cytochrome c (Sigma) reduction test as described (69), using a Labsystems Multiskan Ascent multiplate reader in dual wavelength (550 and 540 nm) kinetic measurement mode. To simplify the presentation, unstimulated control values were subtracted from those of stimulated samples. Exocytosis of gelatinase was determined by in-gel gelatinase zymography as described (67, 68). Cell spreading was assessed after formalin fixation, using a Leica DMI 6000B inverted microscope with a 20× phase contrast objective, connected to a Leica DFC480 CCD camera.
Measurement of bacterial survival
Bacterial killing experiments were performed essentially as described (70). Briefly, S. aureus or E. coli were opsonized with normal mouse serum, followed by incubation with wild type, p190RhoGAP−/− or CD18−/− neutrophils for 30 minutes at a neutrophil:bacteria ratio of 1:100 (S. aureus) or 1:10 (E. coli). Samples were taken at the indicated time points, treated with saponin to lyse the neutrophils, diluted in LB media in 96-well plates, and their absorbance at 650 nm (which corresponds to bacterial growth) was followed by a Labsystems Multiskan Ascent multiplate reader overnight with constant agitation. The assay was calibrated using samples containing known numbers of live bacteria.
Biochemical and signaling studies
Unstimulated wild type and p190RhoGAP−/− neutrophils were lysed in SDS-PAGE sample buffer containing 1% Triton X-100-based lysis buffer supplemented with 0.1% SDS and 0.5% sodium deoxycholate (RIPA) that was preheated to 96 °C (7, 64), run on SDS-PAGE and immunoblotted with antibodies against p190RhoGAP (Clone 30 from BD Biosciencies or Clone D2D6 from Millipore), followed by peroxidase-labeled anti-mouse secondary antibodies (GE Healthcare).
For p38 MAP kinase assays, neutrophils were stimulated in Mg2+-free media in suspension and lysed in RIPA. The RIPA-soluble fractions were run on SDS-PAGE and immunoblotted with phospho-specific (#9211 from Cell Signaling) or non-phospho-specific (Santa Cruz) antibodies against the p38 MAP kinase.
In vitro migration
Transwell migration assays were performed essentially as described (7, 67). Briefly, Transwell inserts with polycarbonate filters of 3 μm pore size (Corning) were precoated with human fibrinogen, filled with neutrophil suspensions and inserted into media containing the indicated concentrations of fMLP. After 60 min, the number of neutrophils in the lower compartment was assessed by an acid phosphatase assay (7).
The migration of individual neutrophils was followed using a Zigmond chamber assay. 1.5 × 106 PMN were suspended in HBSS supplemented with 1% gelatine, 0.25% BSA, 0.1% glucose, 20 mM Hepes, 1.2 mM Ca2+ and 1 mM Mg2+, seeded onto coverslips (Carl Roth) coated with 50 μg/ml murine fibrinogen (Sigma) and incubated for 20 minutes at 37°C. The coverslips were attached to a Zigmond chamber as described (71) and the two grooves were filled with medium with or without 10 μM fMLP. After incubation for 10 minutes at 37°C, time-lapse video microscopy was performed using a Zeiss 200M microscope equipped with a Plan-Apochromat 20×/0.75 NA objective. Off-line analysis of migration tracks was carried out using ImageJ 1.43f (NIH) and the manual tracking plugin (written by Fabrice P. Cordeliès, Institute Curie, Orsay, France). The diagrams were plotted using the chemotaxis and migration tool 1.01 provided by Ibidi. For the generation of the rose diagram, an angular sector with an interior angle of 90 degrees was rotated by 1 degree steps, the cells that ended up in that angle were counted and the counts were grouped in 10-degree intervals with the radius of each wedge indicating the accumulated cell number.
In vivo migration
A competitive migration assay during sterile peritonitis in mixed bone marrow chimeras was performed as described (7, 60). Briefly, CD45.2-expressing p190RhoGAP−/− and CD45.1-expressing wild type bone marrow cells were mixed at varying ratios (10–80% CD45.2-expressing cells) and injected into lethally irradiated CD45.1-expressing recipient mice. Control chimeras were generated using C57BL/6 or CD18−/− mice as CD45.2-expressing donors along with CD45.1-expressing wild type donor cells. 5-8 weeks after transplantation, mixed bone marrow chimeras were injected intraperitoneally with 2 ml 3% thioglycollate broth (Heipha Diagnostics). Blood was taken directly before, as well as 2 and 4 hours after the injection, and the peritoneal cavity was lavaged at 4 hours. The relative percentage of CD45.1 and CD45.2-expressing neutrophils in the peripheral blood and peritoneal lavage samples was determined by flow cytometry. Relative migration of neutrophils was calculated as described (60).
K/B×N serum transfer arthritis
Mice carrying the KRN T-cell receptor transgene (59) on the C57BL/6 genetic background were mated with NOD mice. KRN transgene-positive (K/B×N) and transgene-negative (B×N) offsprings were identified as described (60, 61) and their sera were taken and pooled separately.
Arthritis was induced by a single intraperitoneal injection of 400 μl arthritogenic (K/B×N) or control (B×N) serum, followed by daily assessment of arthritis development. Scoring of visible clinical signs of arthritis, measurement of the ankle thickness and the assessment of the articular function was performed and analyzed as described (60, 61).
Presentation of data and statistical analysis
Error bars in representative experiments correspond to SD of duplicate or triplicate readings. When showing summary of multiple independent experiments, error bars represent SEM from the indicated number of experiments. For statistical analysis, control values were subtracted from stimulated ones wherever possible, and the resulting data were analyzed by Student’s paired two-population or unpaired 2-tailed two-population t-test. p values below 0.05 were considered statistically significant.
RESULTS
p190RhoGAP is present in human and murine neutrophils
To document the expression of p190RhoGAP in neutrophils, whole cell lysates of cells obtained using a standard human neutrophil isolation protocol (discontinuous Ficoll gradient centrifugation) were subjected to immunoblotting with two different monoclonal antibodies against p190RhoGAP. As shown in Fig 1A, both the “Clone 30” antibody (which binds to the central region of the molecule) and the D2D6 antibody (which recognizes the N-terminal region) revealed a single protein band at 190 kDa, indicating the presence of p190RhoGAP.
Figure 1. p190RhoGAP expression in neutrophils.
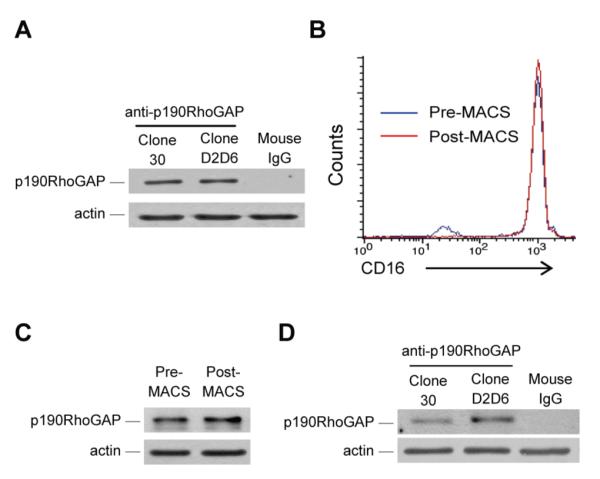
A, Immunoblot analysis of p190RhoGAP expression in lysates of human neutrophils (prepared by a standard procedure) with the indicated antibodies. B, Flow cytometric analysis of CD16 expression in neutrophil preparations before (pre-MACS) and after (post-MACS) a second-step magnetic purification of CD16-positive cells. C, Immunoblot analysis of pre-MACS and post-MACS neutrophil lysates for p190RhoGAP expression using the “Clone 30” antibody. D, Immunoblot analysis of p190RhoGAP expression in murine neutrophil lysates with the indicated antibodies. Immunoblotting for actin in panels A, C and D serve as loading controls. Each panel represents 4-8 independent experiments with similar results.
The above isolation protocol allows a good separation of neutrophils but the resulting cell suspension still contains some other leukocytes (primarily eosinophils). To obtain a higher purity of our neutrophil preparations, they were subjected to a second isolation step, i. e. the positive magnetic cell separation (MACS) of CD16-expressing cells (eosinophils of healthy subjects and most mononuclear cells do not express CD16). As shown in Fig 1B, our standard neutrophil peparation contained a small CD16-negative population (in addition to a dominant CD16-positive population) which was completely eliminated by the magnetic separation step. However, since some mononuclear cells may still express CD16 and one group suggested that eosinophils from allergic patients may also express this receptor (72), we further assessed the purity of the cell suspensions by microscopic leukocyte differential analysis on Cytospin preparations. While our standard neutrophil preparation contained 96.6 ± 1.6% (n = 3) neutrophils with the remaining cells being mostly eosinophils, cells obtained after the MACS procedure contained 99.1 ± 0.5% neutrophils, 0.2 ± 0.2% eosinophils, 0.2 ± 0.3% lymphocytes, 0.03 ± 0.06% monocytes and 0.4 ± 0.1% other leukocytes that could not be clearly identified morphologically (n = 3). This distribution indicated that the cell suspension obtained after the magnetic isolation of CD16-positive cells is a very pure neutrophil population. Importantly, removal of CD16-negative cells did not affect the p190RhoGAP immunoreactivity (Fig 1C), indicating that p190RhoGAP is indeed expressed in human neutrophils.
To test whether p190RhoGAP is also expressed in murine neutrophils and whether our antibodies are able to recognize murine p190RhoGAP, we also tested the presence of the protein in mouse neutrophils. As shown in Fig 1D, both the “Clone 30” and the D2D6 anti-p190RhoGAP antibodies recognized a 190 kDa protein band in lysates of mouse bone marrow-derived neutrophils.
Taken together, these experiments indicate that p190RhoGAP is indeed expressed in the neutrophil lineage in both humans and mice.
Generation of a novel genomic null mutation of p190RhoGAP in mice
A targeted mutation of the p190RhoGAP-encoding Grlf1 gene (Grlf1tm1JSet mutation, referred to as the p190RhoGAPhypo allele) was generated previously by replacing the 5′ 1.5 kb region of exon 1 (containing the normal translation initiation site) with a PGK-Neo cassette in the reverse orientation (Fig 2A) (51). Unfortunately, that mutation proved to generate a hypomorphic allele due to the presence of a truncated p190RhoGAP protein (51) supposedly expressed from a cryptic downstream internal translation initiation site (Fig 2A). This was likely made possible by the lack of upstream translation initiation and termination sites in the mutant transcript, allowing the “walking” of the ribosome to the cryptic translation initiation site.
Figure 2. Generation of a novel p190RhoGAP-deficient mouse strain.

A, General genomic organization of the p190RhoGAP locus and the resulting protein expression in case of the wild type (p190RhoGAP+), the previously generated hypomorphic mutant (p190RhoGAPhypo), as well as the novel complete null mutant (p190RhoGAP−) allele. B, The strategy used to target the p190RhoGAP locus to obtain the p190RhoGAP− allele. Restriction sites of EcoRI (“E”), HindIII (“H”), XbaI (“X”) and BamHI (“B”) are marked. The red bar shows the position of the probe used for Southern blot analysis. PGK-DTA, negative selection cassette encoding the diphtheria toxin A-chain under the control of the phosphoglycerate kinase promoter. C, Southern blot analysis of HindIII fragments of genomic DNA isolated from different ES cell clones of the indicated p190RhoGAP genotypes. D, Immunoblot analysis of ES cell clones of the indicated p190RhoGAP genotypes and of p190RhoGAPhypo/hypo murine embryonic fibroblasts using an anti-p190RhoGAP (“Clone 30”) and a p120RasGAP (loading control) antibody. E, Analysis of the genotype of late gestation fetuses obtained from a p190RhoGAP+/− × p190RhoGAP+/− mating by allele-specific PCR reactions from fetal liver DNA or by immunoblotting with antibodies against the indicated proteins from brain cell lysates (immunoblotting for actin serves as loading control). F, Percentage of E15.5-E18.5 fetuses of the indicated genotype. G, Macroscopic phenotype of a p190RhoGAP−/− fetus with severe neural closure defect.
We have utilized a different targeting strategy to generate a novel mouse strain with a complete null mutation of the p190RhoGAP-encoding gene (Grlf1tm2JSet mutation). In the new targeting vector, the same 1.5 kb region of exon 1 was replaced by a promoterless selection/reporter cassette encoding a β-Geo fusion protein in the sense orientation (Figs 2A-B), ensuring that a fully functional upstream open reading frame prevents the use of the downstream cryptic internal translation initiation site. An additional advantage of this strategy was that β-Geo was expressed from the endogenous p190RhoGAP promoter, strongly increasing the targeting efficiency (since the targeting vector had to be inserted into a gene active in ES cells, such as that encoding p190RhoGAP), as well as allowing the later analysis of p190RhoGAP promoter activity by testing β-galactosidase expression. The detailed targeting strategy is depicted in Fig 2B. The primary sequence of the mutant allele can be found in the GenBank database (www.ncbi.nlm.nih.gov/genbank) under accession number HM365221. The resulting Grlf1tm2JSet mutation will be referred to as the p190RhoGAP− allele.
The targeting vector was electroporated in ES cells. Southern blot analysis of G418-resistant mutant ES cell lines revealed the expected 3.2 kb band corresponding to correct gene targeting. Immunoblotting of p190RhoGAP+/− ES cells using the “Clone 30” (Fig 2D) and the A5D12 (51) (not shown) antibodies (both recognizing the central region of p190RhoGAP) confirmed the lack of a truncated version of the protein whereas the truncated protein was clearly present in p190RhoGAPhypo/hypo murine embryonic fibroblasts. Confirmed p190RhoGAP+/− ES cells were injected into wild type blastocysts to obtain p190RhoGAP+/− founder mice.
Initial characterization of the p190RhoGAP− mutation
While p190RhoGAP+/− mice were born at the expected ratios and were viable and fertile, they did not give rise to live p190RhoGAP−/− offspring beyond the age of one week (not shown), indicating that (similar to the p190RhoGAPhypo/hypo mutation (51)) the p190RhoGAP−/− mutation is a lethal mutation. We next analysed p190RhoGAP−/− fetuses obtained from timed mating of heterozygous p190RhoGAP+/− carriers. The genotype of the fetuses were determined by allele-specific PCR reactions from genomic DNA, as well as by immunoblotting for p190RhoGAP and β-galactosidase in fetal brain cell lysates (Fig 2E). 18% (compared to the expected 25%) of 153 live late-gestation fetuses were of p190RhoGAP−/− genotype (Fig 2F) and about one third (9 out of 27) of the p190RhoGAP−/− fetuses (but none of the p190RhoGAP+/+ or p190RhoGAP+/− ones) showed exencephaly and spina bifida (Fig 2G). The severity of this defect ranged from a hardly visible phenotype to a very obvious exteriorization of both the brain and the lower spinal cord as shown in Fig 2G. Homozygous p190RhoGAP−/− mice also exhibited defective kidney development which was present in all animals tested but ranged from moderate dysplasia of one kidney to complete agenesis of both kidneys (not shown).
Taken together, the complete genetic deficiency of p190RhoGAP leads to perinatal lethality, moderately decreased survival in embryonic life, a partially penetrant neural closure defect and defective kidney development. Except for the kidney developmental defect (which was not assessed before), those phenotypes are similar to the ones previously seen in the p190RhoGAPhypo/hypo mutants (51).
Generation of p190RhoGAP-deficient neutrophils by fetal liver transplantation
The perinatal lethality p190RhoGAP−/− mice precluded the analysis of p190RhoGAP−/− neutrophils from adult mice. This was overcome by generating chimeric mice with p190RhoGAP−/− hematopoietic compartment in an otherwise normal environment. To this end, lethally irradiated recipients carrying the CD45.1 allele were injected intravenously with fetal liver cells of p190RhoGAP−/− or wild type control fetuses (both donor strains carry the CD45.2 allele). Flow cytometric analysis of peripheral blood samples 4-6 weeks after transplantation (Fig 3A) revealed that 99.7 ± 0.2% and 99.8 ± 0.2% (n = 70) of circulating neutrophils of wild type and p190RhoGAP−/−chimeras, respectively, were of donor origin. Immunoblot analyses confirmed that neutrophils isolated from p190RhoGAP−/− bone marrow chimeras did indeed lack the p190RhoGAP protein (Fig 3B).
Figure 3. Generation of p190RhoGAP−/− neutrophils by fetal liver transplantation.

A, Flow cytometric analysis of the donor-specific CD45.2 allele on peripheral blood neutrophils from a recipient mouse, a mouse of the donor genetic background (C57BL/6) and a p190RhoGAP−/− bone marrow chimera. B, Analysis of p190RhoGAP expression in neutrophils isolated from the bone marrow of wild type and p190RhoGAP−/− chimeras using the “Clone 30” monoclonal antibody (staining for actin serves as a loading control). C, Flow cytometric analysis of the expression of the Gr-1 granulocyte marker and of Fcγ-receptors on neutrophils isolated from the bone marrow of wild type and p190RhoGAP−/− chimeras. D, Flow cytometric analysis of the expression of various β2 integrin subunits on neutrophils isolated from the bone marrow of wild type, CD18−/− and p190RhoGAP−/− chimeras. Staining with an isotype control antibody in C and D shows fluorescence intensity due to nonspecific antibody binding. Each panel is representative of 3 or more independent experiments with similar results.
Our bone marrow neutrophil isolation protocol yielded comparable numbers of wild type and p190RhoGAP−/− neutrophils per mouse (not shown). p190RhoGAP−/− neutrophils expressed normal levels of the Gr1 granulocyte differentiation marker (Fig 3C) and the general leukocyte marker CD45 (Fig 3A). Surface staining with antibodies recignizing a shared FcγRII/FcγRIII epitope or the FcγRIV molecule did not reveal any difference between the two genotypes (Fig 3C). The p190RhoGAP−/− mutation did not affect expression of the β2 integrin chain CD18 or the α-chains of LFA-1 (CD11a) or Mac-1 (CD11b) either, while all those proteins were missing from the surface of CD18−/− neutrophils (Fig 3D). Taken together, genetic deficiency of p190RhoGAP did not affect neutrophil maturation or the expression of major cell surface integrins or Fcγ-receptors.
Integrin-mediated adherent activation of p190RhoGAP−/− neutrophils
Given the role of p190RhoGAP in integrin signaling in non-hematopoietic cell types (42, 43, 48, 50-52) and its proposed role in β2 integrin signaling in neutrophils (55), we next tested various integrin-mediated functional responses of p190RhoGAP−/− neutrophils.
β2 integrin-mediated neutrophil activation (73, 74) was tested by monitoring the superoxide release of cells plated on a fibrinogen-coated surface in the presence of TNF. While CD18−/− neutrophils, in agreement with previous findings (7), were completely defective in this assay, p190RhoGAP−/− neutrophils showed a mostly normal response (Fig 4A) with no difference between the wild type and p190RhoGAP−/− cells during the first 30 mins (p = 0.20; n = 23) but a moderate but statistically significant decrease in the p190RhoGAP−/− response by the end of the 60-min assay period (p = 0.004; n = 23). Analysis of superoxide release at suboptimal (5 or 2 ng/ml) TNF concentration did not reveal a considerable difference between wild type and p190RhoGAP−/− neutrophils either (Fig 4B), indicating that the lack of an effect of the p190RhoGAP−/− mutation was not due to supramaximal stimulation of the cells.
Figure 4. Integrin-mediated adherent activation of p190RhoGAP−/− neutrophils.

A-B, Superoxide release of wild type, CD18−/− or p190RhoGAP−/− neutrophils stimulated with recombinant murine TNF on a fibrinogen (Fbg) coated surface. Panel A shows the kinetic analysis of superoxide release at 50 ng/ml TNF concentration in one representative experiment. Error bars represent SD values of triplicate readings. Panel B shows average and SEM of superoxide relase data at the indicated time points from the indicated number of independent experiments at 50, 5 and 2 ng/ml TNF-concentrations, respectively. C-D, Spreading of neutrophils stimulated with 50 ng/ml TNF on a Fbg surface during a 30-min incubation. Panel C shows phase-contrast photomicrographs from a representative experiment. Panel D shows average and SEM of the percentage of spread cells from 5 independent experiments. E-H, Superoxide release of Fbg-adherent neutrophils stimulated with 1 μg/ml Pam3CSK4 (E-F) or 10 ng/ml GM-CSF (G-H). Panels E and G show representative kinetic curves. Error bars represent SD values of triplicate readings. Panels F and H show the average and SEM of superoxide relase data at the 60 min timepoint from 5 and 8 independent experiments, respectively. *, statistically significant difference (p<0.05) between the indicated groups.
The above activation system also leads to spreading of the cells over the adhesion surface which is entirely dependent on CD18 (not shown). As shown in Fig 4C, TNF-treated p190RhoGAP−/− neutrophils were able to spread over fibrinogen, though the percentage of fully spread cells appeared to be partially decreased (p = 0.04; n = 5). Hence, p190RhoGAP appears to make a partial contribution to neutrophil spreading.
Analysis of adherent activation in the presence of other proinflammatory agonists revealed that superoxide release of fibrinogen-adherent neutrophils triggered by the TLR2 ligand Pam3CSK4 was slightly decreased in p190RhoGAP−/− neutrophils by the 60-min time point (p = 0.046; n = 5). Superoxide release triggered by GM-CSF under similar conditions was also moderately reduced in the mutant cells; however, this did not reach statistical significance (p = 0.06; n = 8). Therefore, p190RhoGAP−/− likely makes a moderate contribution to adhesion-dependent activation of neutrophils triggered by soluble agonists other than TNF. This is in contrast with the complete defect of those responses in CD18−/− neutrophils (60).
Taken together, while p190RhoGAP−/− neutrophils were moderately less responsive in various CD18-dependent adherent activation assays, none of those responses were dramatically affected by the genetic deficiency of p190RhoGAP.
In vitro migration of p190RhoGAP-deficient neutrophils
The critical role of β2 integrins in in vitro and in vivo migration of human and mouse neutrophils (7, 75) prompted us to examine the role of p190RhoGAP in neutrophil migration.
In agreement with our previous report (7), CD18−/− neutrophils were completely defective in migrating towards increasing concentrations of the bacterial tripeptide fMLP in an in vitro Transwell assay. However, p190RhoGAP−/− neutrophils were able to migrate towards 1, 3 or 10 μM fMLP (Fig 5A and data not shown) and they even showed a tendency to slightly increased migration (Figs 5A-B) which, however, did not reach statistical significance (p = 0.09 ; n = 8).
Figure 5. In vitro and in vivo migration of p190RhoGAP−/− neutrophils.

A-B, Migration of wild type, CD18−/− and p190RhoGAP−/− neutrophils towards fMLP through fibrinogen-coated Transwell membranes of 3 μm pore size. Panel A shows the mean and SD values of duplicate readings from one representative experiment at the indicated concentrations of fMLP while panel B shows the average and SEM of 8 (wild type and p190RhoGAP−/−) or 3 (CD18−/−) independent experiments at 10 μM fMLP. C-E, Migration of wild type and p190RhoGAP−/− neutrophils towards a gradient of 10 μM fMLP on a fibrinogen-coated surface in a Zigmond chamber. C-D, Single cell tracks (C) and rose diagrams (D) of the disctribution of migration angles of wild type and p190RhoGAP−/− neutrophils migrating towards an fMLP gradient for 12 min. Results from a single representative experiment with 43 wild type and 39 p190RhoGAP−/− cells are shown. E, Quantitative analysis of directionality, migration velocity, accumulated distance and Euclidean distance of neutrophils of the indicated genotypes. Mean ± SEM values obtained from 11 chambers with wild type neutrophils from 5 different mice and 10 chambers with p190RhoGAP−/− cells from 4 different mice are shown. F-G, Competitive migration of CD45.2-expressing wild type, CD18−/− and p190RhoGAP−/− neutrophils vs. CD45.1-expressing (otherwise wild type) neutrophils during thioglycollate-induced sterile peritonitis in mixed bone marrow chimeras. F, Percentage of CD45.2-expressing wild type, p190RhoGAP−/− cells in the blood and the peritoneal lavage fluid of each individual mouse. The thin diagonal line marks points of identical percentage of CD45.2 cells in the blood and the peritoneum. Each data point represent one mouse from one representative experiment. Error bars represent SD from three blood samples taken at different time points from the same mouse. G, Relative migratory capacity of CD45.2-expressing wild type, CD18−/− or p190RhoGAP−/− neutrophils relative to the CD45.1-expressing cells. Mean and SEM of data obtained from 25 (wild type and p190RhoGAP−/−) or 4 (CD18−/−) mixed bone marrow chimeras tested in three independent experiments. *, statistically significant difference (p<0.05) between the indicated groups.
The directed chemotactic migration of individual neutrophils was tested by time-lapse video microscopy using a Zigmond chemotactic chamber with 10 μM fMLP added to one side of the chamber. We have previously shown that this assay is completely dependent on the presence of CD18 on the neutrophil surface (65). As shown in the individual migration tracks (Fig 5C) and the rose diagrams indicating the distribution of migration angles (Fig 5D), p190RhoGAP deficiency did not affect the migration of neutrophils along the fMLP gradient. This was also confirmed by the quantitative analysis of the directionality of cell migration (p = 0.30), the total accumulated distance (p = 0.60), Euclidean distance (p = 0.68) and migration velocity (p = 0.55) (Fig 5E) (n = 11 and 10 chambers with wild type and p190RhoGAP−/− neutrophils, respectively, throughout).
In vivo migration of p190RhoGAP-deficient neutrophils
We also tested the migration of neutrophils to the site of inflammation by a competitive in vivo migration assay (7, 60) where the tissue accumulation of neutrophils of two different genotypes are directly compared within the same individual mouse. To this end, mixed bone marrow chimeras carrying two genetically different hematopoietic cell populations (CD45.1-expressing wild type along with CD45.2-expressing wild type, p190RhoGAP−/− or CD18−/−) were generated and tested in a thioglycollate-induced sterile peritonitis model. No difference between the percentage of CD45.2-positive neutrophils in the blood and the peritoneum could be observed when both the CD45.1 and CD45.2 expressing cells were of otherwise wild type origin (Fig 5F). In contrast, when CD45.1-expressing wild type and CD45.2-expressing CD18−/− neutrophils were present, the percentage of the CD45.2-expressing (i. e. CD18−/−) neutrophils in the peritoneum was significantly lower than that in the bloodstream (Fig 5F), indicating a cell-autonomous requirement for CD18 during neutrophil migration into the inflamed peritoneum (7). Importantly, when CD45.2-expressing p190RhoGAP−/− cells were present along with CD45.1-expressing wild type cells, the percentage of p190RhoGAP−/− neutrophils in the peritoneum was similar to, or even slightly higher than, that in the blood, indicating that p190RhoGAP−/− neutrophils do not have a cell-autonomous migration defect. Statistical analysis of data from three independent experiments did not reveal a statistically significant difference between the relative migratory capacity of CD45.2-expressing wild type and p190RhoGAP−/− neutrophils (p = 0.13; n = 3). However, the difference between the two genotypes became highly significant when the result from each mouse (even if from the same experiment) was considered an independent data point (p = 2.7 × 10−6; n = 25; Fig 5G).
Taken together, p190RhoGAP is not required for CD18-mediated in vitro or in vivo migration of neutrophils and the absence of p190RhoGAP may even slightly promote neutrophil migration under certain conditions.
Normal bacterial killing by p190RhoGAP-deficient neutrophils
β2 integrins appear to participate in killing of invading microorganisms through recognition of complement C3b fragments by the αMβ2 integrin Mac-1 (6, 8, 76). This has prompted us to test the role of p190RhoGAP in bacterial killing by neutrophils.
As shown in Fig 6A, incubation of wild type neutrophils with serum-opsonized S. aureus led to a gradual killing of approx. 90% of the bacteria by the end of the 30-min incubation period. As expected (8), CD18−/− neutrophils showed a strongly reduced capacity to kill those microbes. Importantly, p190RhoGAP−/− neutrophils were as effective in killing S. aureus bacteria as their wild type counterparts (Fig 6B; p = 0.83; n = 3). Analysis of killing of serum-opsonized E. coli bacteria by neutrophils also revealed a significant contribution of CD18 to that response (Fig 6C). However, p190RhoGAP−/− neutrophils did not show any defect in killing E. coli either (Fig 6D; p = 0.69; n = 3). Collectively, these results indicate that p190RhoGAP does not play a major role in β2 integrin-mediated killing of Gram-positive or Gram-negative bacteria.
Figure 6. Normal killing of Gram-positive and Gram-negative bacteria by p190RhoGAP−/− neutrophils.
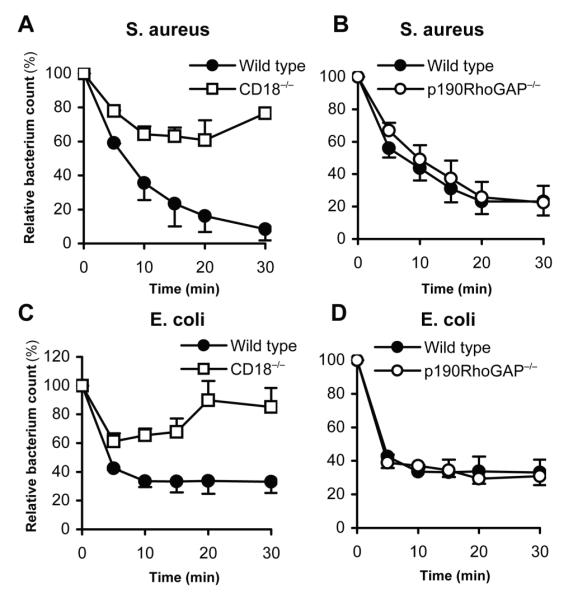
In vitro killing of S. aureus (A-B) or E. coli (C-D) by CD18−/− (A, C) or p190RhoGAP−/− (B, D) neutrophils. Bacterial counts at the indicated time points are expressed relative to those at the 0 time point. Mean and SEM values from two (panel A) or three (panels B-D) independent experiments are shown.
Integrin-independent functional responses of neutrophils
Neutrophils also perform a number of integrin-independent tasks. Though prior studies have mainly suggested the role of p190RhoGAP in integrin signal transduction, we also aimed to test its possible role in integrin-independent neutrophil functions.
As shown in Fig 7A, the Fcγ-receptor-dependent (68) respiratory burst of neutrophils triggered by immobilized IgG immune complexes proceeded normally in p190RhoGAP−/− cells. Neutrophils can also be activated by the bacterial tripeptide fMLP after priming by a pro-inflammatory agonist such as TNF or when pretreated with the cytoskeleton-disrupting agent cytochalasin B (CB). As shown in Figs 7B-C, the genetic deficiency of p190RhoGAP did not affect those responses either. The mutant cells also responded normally to the non-physiological PKC agonist PMA (Fig 7D). p190RhoGAP was not required for the exocytosis of gelatinase granules triggered by the TNF or GM-CSF cytokines or the fMLP peptide (the latter one in the presence of CB) either (Fig 7E). p190RhoGAP−/− neutrophils also showed normal activation of the p38 MAP kinase pathway by TNF or Pam3CSK4 (Fig 7F). Finally, upregulation of CD11b in response to activation by TNF (Fig 7G) or GM-CSF (Fig 7H) proceeded normally in p190RhoGAP−/− neutrophils.
Figure 7. p190RhoGAP is not required for integrin-independent neutrophil functions.

A-D, Superoxide release of wild type and p190RhoGAP−/− neutrophils plated on immobilized IgG immune complexes (A), stimulated with 3 μM fMLP (B-C) or activated by 100 nM PMA (D). Neutrophils were preincubated in the presence or absence of 50 ng/ml murine TNF (B) or in the presence of 10 μM cytochalasin B (CB; panel C). E, Exocytosis of gelatinase determined by gelatinase zymography from neutrophils stimulated with 50 ng/ml murine TNF, 3 μM fMLP or 10 ng/ml murine GM-CSF. F, Western blot analysis of p38 MAP kinase phosphorylation in neutrophils stimulated with 50 ng/ml TNF or 1 μg/ml Pam3CSK4 (Pam3). G-H, CD11b expression of neutrophils stimulated with 50 ng/ml TNF (G) or 10 ng/ml GM-CSF (H). Each panel is representative of 3 or more independent experiments with similar results. Unstimulated control values were subtracted in A-D. Error bars in A-D represent SD of triplicate readings.
Taken together, diverse integrin-independent neutrophil functions mediated by Fcγ-receptors, G-protein-coupled receptors, Toll-like receptors or cytokine receptors proceeded normally in the absence of p190RhoGAP.
K/B×N serum transfer arthritis in p190RhoGAP−/− bone marrow chimeras
Improper activation of neutrophils also participates in inflammation and tissue damage during autoimmune inflammatory diseases (1, 2, 77, 78). Since some of those diseases are mediated by β2-integrins (79), we tested the possible role of p190RhoGAP in autoimmune inflammation using the K/B×N serum transfer arthritis model.
As shown in Fig 8A, arthritogenic K/B×N serum triggered a significant swelling and visible inflammation of the hind paws of wild type bone marrow chimeras whereas CD18−/− bone marrow chimeras were protected from the disease, as expected (79). Importantly, p190RhoGAP−/− bone marrow chimeras showed a qualitatively similar disease to wild type chimeras (Fig 8A). This was also confirmed by clinical scoring of the animals (Fig 8B) which showed nearly complete protection of CD18−/− chimeras but normal disease course in p190RhoGAP−/− chimeras (p = 0.31; n = 11 on day 8). Measurement of the ankle thickness also showed protection of CD18−/− chimeras whereas the ankle thickness of the p190RhoGAP−/− bone marrow chimeras were increased similar to wild type chimeras (p = 0.097; n = 11 on day 8).
Figure 8. K/B×N serum transfer arthritis in p190RhoGAP−/− bone marrow chimeras.

A-C, Wild type, CD18−/− or p190RhoGAP−/− bone marrow chimeras were injected with 400 μl arthritogenic (K/B×N) or non-arthritic control serum and arthritis development in the hind limbs was followed. A, photographs of the hind limbs taken 8 days after serum injection. B-C, Time course of hind limb clinical scores (B) and ankle thickness (C) during the two weeks after serum injection. D, The articular function of the chimeras was tested by measuring the ability of the chimeras to hold on to a custom-made wire grid. The experiment was repeated 15 times on each mouse between days 6 and 10 after serum injection. The results were combined to “holding on curves” showing the percentage of the mice still holding on to the grid after a given time period. Data in B-D show average and SEM from 11 mice per group in case of wild type and p190RhoGAP−/− chimeras, and 6 mice per group in case of CD18−/− chimeras.
Autoimmune arthritis also leads to arthritis-induced loss of articular function. This was assessed by testing the ability of the mice to hold on to the lower side of a custom-made wire grid (60, 61). This experiment was performed several times on each chimeras during the plateau phase of the disease and the results were converted to “holding-on” curves similar to Kaplan-Meier survival curves (Fig 8D). While control serum-treated mice were mostly able to hold on to the grid for the entire 20 s assay period, wild type chimeras injected with arthritogenic K/B×N serum fell off the grid within the first few seconds, indicating an arthritis-induced loss of articular function (Fig 8D). CD18−/− bone marrow chimeras were mostly protected from this response whereas p190RhoGAP−/− chimeras injected with arthritogenic K/B×N serum fell off the grid in a similar course as wild type bone marrow chimeras (p= 0.34; n = 11 at the 20 s time point) (Fig 8D).
Taken together, p190RhoGAP is not required for development of macroscopic signs of arthritis or of arthritis-induced loss of articular function in the K/B×N serum transfer model.
DISCUSSION
Prior studies of non-hematopoietic cell types (42, 43, 48, 50-52) indicated that p190RhoGAP is a critical component of integrin signal transduction in those cells and correlative data in neutrophils and other phagocytic lineage cells (53-58, 80) suggested that this is also true for neutrophils. We addressed that issue more directly by the generation and analysis of a novel mouse strain with a complete genetic defect of p190RhoGAP expression (Fig 2). Contrary to our expectations, our detailed studies revealed that p190RhoGAP is largely dispensable for a number of β2 integrin-dependent neutrophil functions (Figs 4-6), as well as a neutrophil- and β2 integrin-mediated in vivo autoimmune arthritis model (Fig 8), indicating that p190RhoGAP does not play a major indispensable role in integrin signal transduction in neutrophils.
We were quite surprised by our initial results and feared a possible technical problem behind them. Therefore, we first attempted to confirm that p190RhoGAP is indeed expressed in the neutrophil lineage. Despite our significant efforts, we have not been able to demonstrate p190RhoGAP immunoreactivity above background levels by immunofluorescent staining (not shown), possibly due to the tendency of neutrophils to bind IgG in a non-specific manner. As an alternative approach, we took a second magnetic separation step to increase the purity of our cell suspensions (Fig 1B) and showed that p190RhoGAP is indeed expressed in those highly purified neutrophils (Fig 1C). To confirm that our assays indeed require integrins, we performed parallel analyses of CD18−/− neutrophils (Figs 4-6 and 8). We also extended our studies to a number of conceptually different in vitro (Figs 4-6) and in vivo (Fig 8) functional responses. All those studies confirmed that p190RhoGAP is largely dispensable for CD18-mediated neutrophil functions in vitro and arthritis development in vivo. Finally, the use of a novel null mutation of the p190RhoGAP-encoding gene (Fig 2) excluded the possibility of functional compensation by a truncated version of the protein. These results argue against a technical problem behind the lack of a major p190RhoGAP−/− neutrophil phenotype.
There are a number of possible explanations for our findings. First, Rac1/2, Cdc42 and RhoA may play opposite effects in certain neutrophil functions (as exemplified by prior studies in phagocytic lineage cells (24, 81, 82)) leading to the lack of a major net effect of the genetic deficiency of p190RhoGAP. In addition, a number of other RhoGAPs (including Bcr and p50RhoGAP) are also present in neutrophils (56, 83) and may regulate Rho-family small GTPases in p190RhoGAP−/− cells. It is also possible that p190RhoGAP plays a unique role in some specific aspect(s) of neutrophil activation that have not been revealed by the experimental approaches used in this study.
An additional possibility may be that p190-B, another member of the p190RhoGAP family (84, 85), may functionally compensate for the lack of p190RhoGAP in neutrophils. However, the phenotypes of p190-B-deficient mice are quite distinct from those of p190RhoGAP−/− animals (51, 86-89). p190-B also fails to be phosphorylated by Src-family kinases or to associate with p120RhoGAP (89), two features that are considered to be critical for the participation of p190RhoGAP in integrin signal transduction events. Furthermore, p190RhoGAP-deficient murine embryonic fibroblasts show a migration defect (52) despite the expression of the p190-B isoforms in those cells ((86, 90) and data not shown). Critical C-terminal phosphorylation sites required for the participation of p190RhoGAP in polarized cell migration are also missing from p190-B (52) and p190-B also lacks phosphorylation sites thought to be important for PKC-mediated regulation of p190RhoGAP (91). Those results suggest that p190-B is likely not able to compensate for the lack of p190RhoGAP in integrin-mediated functional responses. However, the possibility of functional compensation could only be formally excluded by the analysis of double mutant cells genetically deficient of both p190RhoGAP-family members. Unfortunately, the combined genetic deficiency of both p190RhoGAP and p190-B leads to very early embryonic lethality (likely before E9.5) (89) precluding the generation of bone marrow chimeras from the double mutant fetal liver cells.
It should be noted that, despite the lack of a major or general phenotype in p190RhoGAP−/− neutrophils, we did see some moderate differences in those cells in certain assays. Those included slightly decreased superoxide release and spreading during integrin-mediated activation (Fig 4), as well as a slight increase of neutrophil migration under certain in vitro or in vivo conditions (Fig 5). Therefore, p190RhoGAP may play a partial, modulatory role in integrin signal transduction in neutrophils, and it should be emphasized again that more detailed analyses (e. g. sophisticated microscopic techniques) may also reveal additional differences between the fine behavior of wild type and p190RhoGAP−/− neutrophils in the future.
Though our study was primarily prompted by prior reports showing a major positive role for p190RhoGAP in integrin signal transduction, p190RhoGAP has also been proposed to be a negative regulator of the NADPH oxidase, e. g. by decreasing the level of GTP-bound Rac in the oxidase complex (53). Since the genetic deficiency of p190RhoGAP did not affect adhesion-independent respiratory burst of neutrophils (Figs 7A-D) and it caused a moderate decrease in adhesion-mediated superoxide production (Figs 4A-B, 4E-H), we conclude that p190RhoGAP is not a major negative regulator of the NADPH oxidase.
We have also generated a novel p190RhoGAP-deficient mouse strain during the course of this study (Fig 2) which, in contrast to the previously generated hypomorphic p190RhoGAP-mutant strain (51), does not express any truncated version of the protein (Fig 2D). Analyses so far have revealed very similar phenotypes of p190RhoGAP−/− and p190RhoGAPhypo/hypo mice (such as perinatal lethality, limited embryonic survival and incompletely penetrant neural tube closure defects). Further analyses will be required to reveal whether there is any possible differences between the two strains, which would indicate an independent functional role of the C-terminal portion of p190RhoGAP.
Taken together, our results obtained using a novel knockout mouse strain indicate that p190RhoGAP does not play a major indispensable role in integrin signaling of neutrophils. Those findings should trigger the re-evaluation of the current view of integrin signaling in neutrophils and the identification of the components of the p190RhoGAP-independent pathway linking engagement of β2 integrins to reorganization of the actin cytoskeleton.
ACKNOWLEDGMENTS
We thank Arthur Beaudet for the CD18−/− mice; Diane Mathis and Christoph Benoist for the KRN transgenic strain; Jeff Ravetch for the FcγRIV mAb; Edina Simon, Mónika Duleba, Marcell Szabó, Gábor Petheő Mátyás Mayer and Katy Niedung for help with experiments; Erzsébet Ligeti and Zoltán Jakus for inspiring suggestions and careful reading of the manuscript; and József Mandl and Anna Sebestyén for access to equipment.
This work was supported by the the European Research Council (Starting Independent Investigator Award No. 206283 to A. M.), the Hungarian Office for Research and Technology (Anyos Jedlik Award No. NKFP-A1-0069/2006 to A. M.), the Wellcome Trust (International Senior Research Fellowship No. 087782 to A. M.) and the Deutsche Forschungsgemeinschaft (Wa 1048/2-3 to B. W.).
Footnotes
Publisher's Disclaimer: “This is an author-produced version of a manuscript accepted for publication in The Journal of Immunology (The JI). The American Association of Immunologists, Inc. (AAI), publisher of The JI, holds the copyright to this manuscript. This manuscript has not yet been copyedited or subjected to editorial proofreading by The JI; hence it may differ from the final version published in The JI (online and in print). AAI (The JI) is not liable for errors or omissions in this author-produced version of the manuscript or in any version derived from it by the United States National Institutes of Health or any other third party. The final, citable version of record can be found at www.jimmunol.org.”
DISCLOSURES The authors declare that they have no competing financial interests.
REFERENCES
- 1.Nathan C. Neutrophils and immunity: challenges and opportunities. Nat Rev Immunol. 2006;6:173–182. doi: 10.1038/nri1785. [DOI] [PubMed] [Google Scholar]
- 2.Wipke BT, Allen PM. Essential role of neutrophils in the initiation and progression of a murine model of rheumatoid arthritis. J Immunol. 2001;167:1601–1608. doi: 10.4049/jimmunol.167.3.1601. [DOI] [PubMed] [Google Scholar]
- 3.Jonsson H, Allen P, Peng SL. Inflammatory arthritis requires Foxo3a to prevent Fas ligand-induced neutrophil apoptosis. Nat Med. 2005;11:666–671. doi: 10.1038/nm1248. [DOI] [PubMed] [Google Scholar]
- 4.Tanaka D, Kagari T, Doi H, Shimozato T. Essential role of neutrophils in anti-type II collagen antibody and lipopolysaccharide-induced arthritis. Immunology. 2006;119:195–202. doi: 10.1111/j.1365-2567.2006.02424.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bunting M, Harris ES, McIntyre TM, Prescott SM, Zimmerman GA. Leukocyte adhesion deficiency syndromes: adhesion and tethering defects involving β2 integrins and selectin ligands. Curr Opin Hematol. 2002;9:30–35. doi: 10.1097/00062752-200201000-00006. [DOI] [PubMed] [Google Scholar]
- 6.Scharffetter-Kochanek K, Lu H, Norman K, van Nood N, Munoz F, Grabbe S, McArthur M, Lorenzo I, Kaplan S, Ley K, Smith CW, Montgomery CA, Rich S, Beaudet AL. Spontaneous skin ulceration and defective T cell function in CD18 null mice. J Exp Med. 1998;188:119–131. doi: 10.1084/jem.188.1.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mócsai A, Zhou M, Meng F, Tybulewicz VL, Lowell CA. Syk is required for integrin signaling in neutrophils. Immunity. 2002;16:547–558. doi: 10.1016/s1074-7613(02)00303-5. [DOI] [PubMed] [Google Scholar]
- 8.Ferrante A, Martin AJ, Bates EJ, Goh DH, Harvey DP, Parsons D, Rathjen DA, Russ G, Dayer JM. Killing of Staphylococcus aureus by tumor necrosis factor-α-activated neutrophils. The role of serum opsonins, integrin receptors, respiratory burst, and degranulation. J Immunol. 1993;151:4821–4828. [PubMed] [Google Scholar]
- 9.Berton G, Mócsai A, Lowell CA. Src and Syk kinases: key regulators of phagocytic cell activation. Trends Immunol. 2005;26:208–214. doi: 10.1016/j.it.2005.02.002. [DOI] [PubMed] [Google Scholar]
- 10.Lowell CA, Fumagalli L, Berton G. Deficiency of Src family kinases p59/61hck and p58c-fgr results in defective adhesion-dependent neutrophil functions. J Cell Biol. 1996;133:895–910. doi: 10.1083/jcb.133.4.895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mócsai A, Ligeti E, Lowell CA, Berton G. Adhesion-dependent degranulation of neutrophils requires the Src family kinases Fgr and Hck. J Immunol. 1999;162:1120–1126. [PubMed] [Google Scholar]
- 12.Pereira S, Zhou M, Mócsai A, Lowell C. Resting murine neutrophils express functional α4 integrins that signal through Src family kinases. J Immunol. 2001;166:4115–4123. doi: 10.4049/jimmunol.166.6.4115. [DOI] [PubMed] [Google Scholar]
- 13.Bokoch GM. Regulation of innate immunity by Rho GTPases. Trends Cell Biol. 2005;15:163–171. doi: 10.1016/j.tcb.2005.01.002. [DOI] [PubMed] [Google Scholar]
- 14.Gu Y, Filippi MD, Cancelas JA, Siefring JE, Williams EP, Jasti AC, Harris CE, Lee AW, Prabhakar R, Atkinson SJ, Kwiatkowski DJ, Williams DA. Hematopoietic cell regulation by Rac1 and Rac2 guanosine triphosphatases. Science. 2003;302:445–449. doi: 10.1126/science.1088485. [DOI] [PubMed] [Google Scholar]
- 15.Sun CX, Downey GP, Zhu F, Koh AL, Thang H, Glogauer M. Rac1 is the small GTPase responsible for regulating the neutrophil chemotaxis compass. Blood. 2004;104:3758–3765. doi: 10.1182/blood-2004-03-0781. [DOI] [PubMed] [Google Scholar]
- 16.Roberts AW, Kim C, Zhen L, Lowe JB, Kapur R, Petryniak B, Spaetti A, Pollock JD, Borneo JB, Bradford GB, Atkinson SJ, Dinauer MC, Williams DA. Deficiency of the hematopoietic cell-specific Rho family GTPase Rac2 is characterized by abnormalities in neutrophil function and host defense. Immunity. 1999;10:183–196. doi: 10.1016/s1074-7613(00)80019-9. [DOI] [PubMed] [Google Scholar]
- 17.Glogauer M, Marchal CC, Zhu F, Worku A, Clausen BE, Foerster I, Marks P, Downey GP, Dinauer M, Kwiatkowski DJ. Rac1 deletion in mouse neutrophils has selective effects on neutrophil functions. J Immunol. 2003;170:5652–5657. doi: 10.4049/jimmunol.170.11.5652. [DOI] [PubMed] [Google Scholar]
- 18.Kim C, Dinauer MC. Rac2 is an essential regulator of neutrophil nicotinamide adenine dinucleotide phosphate oxidase activation in response to specific signaling pathways. J Immunol. 2001;166:1223–1232. doi: 10.4049/jimmunol.166.2.1223. [DOI] [PubMed] [Google Scholar]
- 19.Filippi MD, Szczur K, Harris CE, Berclaz PY. Rho GTPase Rac1 is critical for neutrophil migration into the lung. Blood. 2007;109:1257–1264. doi: 10.1182/blood-2006-04-017731. [DOI] [PubMed] [Google Scholar]
- 20.Abdel-Latif D, Steward M, Macdonald DL, Francis GA, Dinauer MC, Lacy P. Rac2 is critical for neutrophil primary granule exocytosis. Blood. 2004;104:832–839. doi: 10.1182/blood-2003-07-2624. [DOI] [PubMed] [Google Scholar]
- 21.Ambruso DR, Knall C, Abell AN, Panepinto J, Kurkchubasche A, Thurman G, Gonzalez-Aller C, Hiester A, deBoer M, Harbeck RJ, Oyer R, Johnson GL, Roos D. Human neutrophil immunodeficiency syndrome is associated with an inhibitory Rac2 mutation. Proc Natl Acad Sci U S A. 2000;97:4654–4659. doi: 10.1073/pnas.080074897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Williams DA, Tao W, Yang F, Kim C, Gu Y, Mansfield P, Levine JE, Petryniak B, Derrow CW, Harris C, Jia B, Zheng Y, Ambruso DR, Lowe JB, Atkinson SJ, Dinauer MC, Boxer L. Dominant negative mutation of the hematopoietic-specific Rho GTPase, Rac2, is associated with a human phagocyte immunodeficiency. Blood. 2000;96:1646–1654. [PubMed] [Google Scholar]
- 23.Szczur K, Zheng Y, Filippi MD. The small Rho GTPase Cdc42 regulates neutrophil polarity via CD11b integrin signaling. Blood. 2009;114:4527–4537. doi: 10.1182/blood-2008-12-195164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xu J, Wang F, Van Keymeulen A, Herzmark P, Straight A, Kelly K, Takuwa Y, Sugimoto N, Mitchison T, Bourne HR. Divergent signals and cytoskeletal assemblies regulate self-organizing polarity in neutrophils. Cell. 2003;114:201–214. doi: 10.1016/s0092-8674(03)00555-5. [DOI] [PubMed] [Google Scholar]
- 25.Pestonjamasp KN, Forster C, Sun C, Gardiner EM, Bohl B, Weiner O, Bokoch GM, Glogauer M. Rac1 links leading edge and uropod events through Rho and myosin activation during chemotaxis. Blood. 2006;108:2814–2820. doi: 10.1182/blood-2006-01-010363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bouvard D, Brakebusch C, Gustafsson E, Aszodi A, Bengtsson T, Berna A, Fassler R. Functional consequences of integrin gene mutations in mice. Circ Res. 2001;89:211–223. doi: 10.1161/hh1501.094874. [DOI] [PubMed] [Google Scholar]
- 27.Mitra SK, Schlaepfer DD. Integrin-regulated FAK-Src signaling in normal and cancer cells. Curr Opin Cell Biol. 2006;18:516–523. doi: 10.1016/j.ceb.2006.08.011. [DOI] [PubMed] [Google Scholar]
- 28.Shattil SJ. Integrins and Src: dynamic duo of adhesion signaling. Trends Cell Biol. 2005;15:399–403. doi: 10.1016/j.tcb.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 29.Huveneers S, Danen EH. Adhesion signaling - crosstalk between integrins, Src and Rho. J Cell Sci. 2009;122:1059–1069. doi: 10.1242/jcs.039446. [DOI] [PubMed] [Google Scholar]
- 30.Playford MP, Schaller MD. The interplay between Src and integrins in normal and tumor biology. Oncogene. 2004;23:7928–7946. doi: 10.1038/sj.onc.1208080. [DOI] [PubMed] [Google Scholar]
- 31.Miranti CK, Brugge JS. Sensing the environment: a historical perspective on integrin signal transduction. Nat Cell Biol. 2002;4:E83–90. doi: 10.1038/ncb0402-e83. [DOI] [PubMed] [Google Scholar]
- 32.DeMali KA, Wennerberg K, Burridge K. Integrin signaling to the actin cytoskeleton. Curr Opin Cell Biol. 2003;15:572–582. doi: 10.1016/s0955-0674(03)00109-1. [DOI] [PubMed] [Google Scholar]
- 33.Nobes CD, Hall A. Rho, rac, and cdc42 GTPases regulate the assembly of multimolecular focal complexes associated with actin stress fibers, lamellipodia, and filopodia. Cell. 1995;81:53–62. doi: 10.1016/0092-8674(95)90370-4. [DOI] [PubMed] [Google Scholar]
- 34.Arthur WT, Petch LA, Burridge K. Integrin engagement suppresses RhoA activity via a c-Src-dependent mechanism. Curr Biol. 2000;10:719–722. doi: 10.1016/s0960-9822(00)00537-6. [DOI] [PubMed] [Google Scholar]
- 35.Ren XD, Kiosses WB, Sieg DJ, Otey CA, Schlaepfer DD, Schwartz MA. Focal adhesion kinase suppresses Rho activity to promote focal adhesion turnover. J Cell Sci. 2000;113:3673–3678. doi: 10.1242/jcs.113.20.3673. [DOI] [PubMed] [Google Scholar]
- 36.Ren XD, Kiosses WB, Schwartz MA. Regulation of the small GTP-binding protein Rho by cell adhesion and the cytoskeleton. EMBO J. 1999;18:578–585. doi: 10.1093/emboj/18.3.578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ellis C, Moran M, McCormick F, Pawson T. Phosphorylation of GAP and GAP-associated proteins by transforming and mitogenic tyrosine kinases. Nature. 1990;343:377–381. doi: 10.1038/343377a0. [DOI] [PubMed] [Google Scholar]
- 38.Moran MF, Polakis P, McCormick F, Pawson T, Ellis C. Protein-tyrosine kinases regulate the phosphorylation, protein interactions, subcellular distribution, and activity of p21ras GTPase-activating protein. Mol Cell Biol. 1991;11:1804–1812. doi: 10.1128/mcb.11.4.1804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Settleman J, Narasimhan V, Foster LC, Weinberg RA. Molecular cloning of cDNAs encoding the GAP-associated protein p190: implications for a signaling pathway from ras to the nucleus. Cell. 1992;69:539–549. doi: 10.1016/0092-8674(92)90454-k. [DOI] [PubMed] [Google Scholar]
- 40.Chang JH, Gill S, Settleman J, Parsons SJ. c-Src regulates the simultaneous rearrangement of actin cytoskeleton, p190RhoGAP, and p120RasGAP following epidermal growth factor stimulation. J Cell Biol. 1995;130:355–368. doi: 10.1083/jcb.130.2.355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chang JH, Wilson LK, Moyers JS, Zhang K, Parsons SJ. Increased levels of p21ras-GTP and enhanced DNA synthesis accompany elevated tyrosyl phosphorylation of GAP-associated proteins, p190 and p62, in c-src overexpressors. Oncogene. 1993;8:959–967. [PubMed] [Google Scholar]
- 42.Brouns MR, Matheson SF, Settleman J. p190 RhoGAP is the principal Src substrate in brain and regulates axon outgrowth, guidance and fasciculation. Nat Cell Biol. 2001;3:361–367. doi: 10.1038/35070042. [DOI] [PubMed] [Google Scholar]
- 43.Nakahara H, Mueller SC, Nomizu M, Yamada Y, Yeh Y, Chen WT. Activation of β1 integrin signaling stimulates tyrosine phosphorylation of p190RhoGAP and membrane-protrusive activities at invadopodia. J Biol Chem. 1998;273:9–12. doi: 10.1074/jbc.273.1.9. [DOI] [PubMed] [Google Scholar]
- 44.Hernandez SE, Settleman J, Koleske AJ. Adhesion-dependent regulation of p190RhoGAP in the developing brain by the Abl-related gene tyrosine kinase. Curr Biol. 2004;14:691–696. doi: 10.1016/j.cub.2004.03.062. [DOI] [PubMed] [Google Scholar]
- 45.Bass MD, Morgan MR, Roach KA, Settleman J, Goryachev AB, Humphries MJ. p190RhoGAP is the convergence point of adhesion signals from α5β1 integrin and syndecan-4. J Cell Biol. 2008;181:1013–1026. doi: 10.1083/jcb.200711129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Settleman J, Albright CF, Foster LC, Weinberg RA. Association between GTPase activators for Rho and Ras families. Nature. 1992;359:153–154. doi: 10.1038/359153a0. [DOI] [PubMed] [Google Scholar]
- 47.Ridley AJ, Self AJ, Kasmi F, Paterson HF, Hall A, Marshall CJ, Ellis C. rho family GTPase activating proteins p190, bcr and rhoGAP show distinct specificities in vitro and in vivo. EMBO J. 1993;12:5151–5160. doi: 10.1002/j.1460-2075.1993.tb06210.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Arthur WT, Burridge K. RhoA inactivation by p190RhoGAP regulates cell spreading and migration by promoting membrane protrusion and polarity. Mol Biol Cell. 2001;12:2711–2720. doi: 10.1091/mbc.12.9.2711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liang X, Draghi NA, Resh MD. Signaling from integrins to Fyn to Rho family GTPases regulates morphologic differentiation of oligodendrocytes. J Neurosci. 2004;24:7140–7149. doi: 10.1523/JNEUROSCI.5319-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.McGlade J, Brunkhorst B, Anderson D, Mbamalu G, Settleman J, Dedhar S, Rozakis-Adcock M, Chen LB, Pawson T. The N-terminal region of GAP regulates cytoskeletal structure and cell adhesion. EMBO J. 1993;12:3073–3081. doi: 10.1002/j.1460-2075.1993.tb05976.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Brouns MR, Matheson SF, Hu KQ, Delalle I, Caviness VS, Silver J, Bronson RT, Settleman J. The adhesion signaling molecule p190 RhoGAP is required for morphogenetic processes in neural development. Development. 2000;127:4891–4903. doi: 10.1242/dev.127.22.4891. [DOI] [PubMed] [Google Scholar]
- 52.Jiang W, Betson M, Mulloy R, Foster R, Levay M, Ligeti E, Settleman J. p190A RhoGAP is a glycogen synthase kinase-3-β substrate required for polarized cell migration. J Biol Chem. 2008;283:20978–20988. doi: 10.1074/jbc.M802588200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Heyworth PG, Knaus UG, Settleman J, Curnutte JT, Bokoch GM. Regulation of NADPH oxidase activity by Rac GTPase activating protein(s) Mol Biol Cell. 1993;4:1217–1223. doi: 10.1091/mbc.4.11.1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dusi S, Donini M, Wientjes F, Rossi F. Translocation of p190rho guanosine triphosphatase-activating protein from cytosol to the membrane in human neutrophils stimulated with different agonists. Biochem Biophys Res Commun. 1996;219:859–862. doi: 10.1006/bbrc.1996.0323. [DOI] [PubMed] [Google Scholar]
- 55.Dib K, Melander F, Andersson T. Role of p190RhoGAP in β2 integrin regulation of RhoA in human neutrophils. J Immunol. 2001;166:6311–6322. doi: 10.4049/jimmunol.166.10.6311. [DOI] [PubMed] [Google Scholar]
- 56.Geiszt M, Dagher MC, Molnar G, Havasi A, Faure J, Paclet MH, Morel F, Ligeti E. Characterization of membrane-localized and cytosolic Rac-GTPase-activating proteins in human neutrophil granulocytes: contribution to the regulation of NADPH oxidase. Biochem J. 2001;355:851–858. doi: 10.1042/bj3550851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Continolo S, Baruzzi A, Majeed M, Caveggion E, Fumagalli L, Lowell CA, Berton G. The proto-oncogene Fgr regulates cell migration and this requires its plasma membrane localization. Exp Cell Res. 2005;302:253–269. doi: 10.1016/j.yexcr.2004.09.005. [DOI] [PubMed] [Google Scholar]
- 58.Saltel F, Chabadel A, Bonnelye E, Jurdic P. Actin cytoskeletal organisation in osteoclasts: a model to decipher transmigration and matrix degradation. Eur J Cell Biol. 2008;87:459–468. doi: 10.1016/j.ejcb.2008.01.001. [DOI] [PubMed] [Google Scholar]
- 59.Kouskoff V, Korganow AS, Duchatelle V, Degott C, Benoist C, Mathis D. Organ-specific disease provoked by systemic autoimmunity. Cell. 1996;87:811–822. doi: 10.1016/s0092-8674(00)81989-3. [DOI] [PubMed] [Google Scholar]
- 60.Jakus Z, Simon E, Frommhold D, Sperandio M, Mócsai A. Critical role of phospholipase Cγ2 in integrin and Fc receptor-mediated neutrophil functions and the effector phase of autoimmune arthritis. J Exp Med. 2009;206:577–593. doi: 10.1084/jem.20081859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Jakus Z, Simon E, Balázs B, Mócsai A. Genetic deficiency of Syk protects mice from autoantibody-induced arthritis. Arthritis Rheum. 2010 doi: 10.1002/art.27438. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mócsai A, Jakus Z, Vántus T, Berton G, Lowell CA, Ligeti E. Kinase pathways in chemoattractant-induced degranulation of neutrophils: The role of p38 mitogen-activated protein kinase activated by Src family kinases. J Immunol. 2000;164:4321–4331. doi: 10.4049/jimmunol.164.8.4321. [DOI] [PubMed] [Google Scholar]
- 63.Petheo GL, Maturana A, Spat A, Demaurex N. Interactions between electron and proton currents in excised patches from human eosinophils. J Gen Physiol. 2003;122:713–726. doi: 10.1085/jgp.200308891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mócsai A, Zhang H, Jakus Z, Kitaura J, Kawakami T, Lowell CA. G-protein-coupled receptor signaling in Syk-deficient neutrophils and mast cells. Blood. 2003;101:4155–4163. doi: 10.1182/blood-2002-07-2346. [DOI] [PubMed] [Google Scholar]
- 65.Schymeinsky J, Sindrilaru A, Frommhold D, Sperandio M, Gerstl R, Then C, Mócsai A, Scharffetter-Kochanek K, Walzog B. The Vav binding site of the non-receptor tyrosine kinase Syk at Tyr 348 is critical for β2 integrin (CD11/CD18)-mediated neutrophil migration. Blood. 2006;108:3919–3927. doi: 10.1182/blood-2005-12-030387. [DOI] [PubMed] [Google Scholar]
- 66.Nimmerjahn F, Bruhns P, Horiuchi K, Ravetch JV. FcγRIV: A novel FcR with distinct IgG subclass specificity. Immunity. 2005;23:41–51. doi: 10.1016/j.immuni.2005.05.010. [DOI] [PubMed] [Google Scholar]
- 67.Mócsai A, Abram CL, Jakus Z, Hu Y, Lanier LL, Lowell CA. Integrin signaling in neutrophils and macrophages uses adaptors containing immunoreceptor tyrosine-based activation motifs. Nat Immunol. 2006;7:1326–1333. doi: 10.1038/ni1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Jakus Z, Németh T, Verbeek JS, Mócsai A. Critical but overlapping role of FcγRIII and FcγRIV in activation of murine neutrophils by immobilized immune complexes. J Immunol. 2008;180:618–629. doi: 10.4049/jimmunol.180.1.618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Jakus Z, Berton G, Ligeti E, Lowell CA, Mócsai A. Responses of neutrophils to anti-integrin antibodies depends on costimulation through low affinity FcγRs: Full activation requires both integrin and nonintegrin signals. J Immunol. 2004;173:2068–2077. doi: 10.4049/jimmunol.173.3.2068. [DOI] [PubMed] [Google Scholar]
- 70.Rada BK, Geiszt M, Kaldi K, Timar C, Ligeti E. Dual role of phagocytic NADPH oxidase in bacterial killing. Blood. 2004;104:2947–2953. doi: 10.1182/blood-2004-03-1005. [DOI] [PubMed] [Google Scholar]
- 71.Zigmond SH. Ability of polymorphonuclear leukocytes to orient in gradients of chemotactic factors. J Cell Biol. 1977;75:606–616. doi: 10.1083/jcb.75.2.606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Davoine F, Lavigne S, Chakir J, Ferland C, Boulay ME, Laviolette M. Expression of FcγRIII (CD16) on human peripheral blood eosinophils increases in allergic conditions. J Allergy Clin Immunol. 2002;109:463–469. doi: 10.1067/mai.2002.121952. [DOI] [PubMed] [Google Scholar]
- 73.Nathan CF. Neutrophil activation on biological surfaces. Massive secretion of hydrogen peroxide in response to products of macrophages and lymphocytes. J Clin Invest. 1987;80:1550–1560. doi: 10.1172/JCI113241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Nathan C, Srimal S, Farber C, Sanchez E, Kabbash L, Asch A, Gailit J, Wright SD. Cytokine-induced respiratory burst of human neutrophils: Dependence on extracellular matrix proteins and CD11/CD18 integrins. J Cell Biol. 1989;109:1341–1349. doi: 10.1083/jcb.109.3.1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lekstrom-Himes JA, Gallin JI. Immunodeficiency diseases caused by defects in phagocytes. N Engl J Med. 2000;343:1703–1714. doi: 10.1056/NEJM200012073432307. [DOI] [PubMed] [Google Scholar]
- 76.Prince JE, Brayton CF, Fossett MC, Durand JA, Kaplan SL, Smith CW, Ballantyne CM. The differential roles of LFA-1 and Mac-1 in host defense against systemic infection with Streptococcus pneumoniae. J Immunol. 2001;166:7362–7369. doi: 10.4049/jimmunol.166.12.7362. [DOI] [PubMed] [Google Scholar]
- 77.Eyles JL, Roberts AW, Metcalf D, Wicks IP. Granulocyte colony-stimulating factor and neutrophils - Forgotten mediators of inflammatory disease. Nat Clin Pract Rheumatol. 2006;2:500–510. doi: 10.1038/ncprheum0291. [DOI] [PubMed] [Google Scholar]
- 78.Looney MR, Su X, Van Ziffle JA, Lowell CA, Matthay MA. Neutrophils and their Fcγ receptors are essential in a mouse model of transfusion-related acute lung injury. J Clin Invest. 2006;116:1615–1623. doi: 10.1172/JCI27238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Watts GM, Beurskens FJ, Martin-Padura I, Ballantyne CM, Klickstein LB, Brenner MB, Lee DM. Manifestations of inflammatory arthritis are critically dependent on LFA-1. J Immunol. 2005;174:3668–3675. doi: 10.4049/jimmunol.174.6.3668. [DOI] [PubMed] [Google Scholar]
- 80.Kim JS, Kim JG, Moon MY, Jeon CY, Won HY, Kim HJ, Jeon YJ, Seo JY, Kim JI, Kim J, Lee JY, Kim PH, Park JB. Transforming growth factor-β1 regulates macrophage migration via RhoA. Blood. 2006;108:1821–1829. doi: 10.1182/blood-2005-10-009191. [DOI] [PubMed] [Google Scholar]
- 81.Diebold BA, Fowler B, Lu J, Dinauer MC, Bokoch GM. Antagonistic cross-talk between Rac and Cdc42 GTPases regulates generation of reactive oxygen species. J Biol Chem. 2004;279:28136–28142. doi: 10.1074/jbc.M313891200. [DOI] [PubMed] [Google Scholar]
- 82.Ory S, Munari-Silem Y, Fort P, Jurdic P. Rho and Rac exert antagonistic functions on spreading of macrophage-derived multinucleated cells and are not required for actin fiber formation. J Cell Sci. 2000;113:1177–1188. doi: 10.1242/jcs.113.7.1177. [DOI] [PubMed] [Google Scholar]
- 83.Voncken JW, van Schaick H, Kaartinen V, Deemer K, Coates T, Landing B, Pattengale P, Dorseuil O, Bokoch GM, Groffen J, O. D, Bokoch GM, Groffen J, Heisterkamp N. Increased neutrophil respiratory burst in bcr-null mutants. Cell. 1995;80:719–728. doi: 10.1016/0092-8674(95)90350-x. [DOI] [PubMed] [Google Scholar]
- 84.Burbelo PD, Miyamoto S, Utani A, Brill S, Yamada KM, Hall A, Yamada Y. p190-B, a new member of the Rho GAP family, and Rho are induced to cluster after integrin cross-linking. J Biol Chem. 1995;270:30919–30926. doi: 10.1074/jbc.270.52.30919. [DOI] [PubMed] [Google Scholar]
- 85.Burbelo PD, Finegold AA, Kozak CA, Yamada Y, Takami H. Cloning, genomic organization and chromosomal assignment of the mouse p190-B gene. Biochim Biophys Acta. 1998;1443:203–210. doi: 10.1016/s0167-4781(98)00207-3. [DOI] [PubMed] [Google Scholar]
- 86.Sordella R, Classon M, Hu KQ, Matheson SF, Brouns MR, Fine B, Zhang L, Takami H, Yamada Y, Settleman J. Modulation of CREB activity by the Rho GTPase regulates cell and organism size during mouse embryonic development. Dev Cell. 2002;2:553–565. doi: 10.1016/s1534-5807(02)00162-4. [DOI] [PubMed] [Google Scholar]
- 87.Chakravarty G, Hadsell D, Buitrago W, Settleman J, Rosen JM. p190-B RhoGAP regulates mammary ductal morphogenesis. Mol Endocrinol. 2003;17:1054–1065. doi: 10.1210/me.2002-0428. [DOI] [PubMed] [Google Scholar]
- 88.Sordella R, Jiang W, Chen GC, Curto M, Settleman J. Modulation of Rho GTPase signaling regulates a switch between adipogenesis and myogenesis. Cell. 2003;113:147–158. doi: 10.1016/s0092-8674(03)00271-x. [DOI] [PubMed] [Google Scholar]
- 89.Matheson SF, Hu KQ, Brouns MR, Sordella R, VanderHeide JD, Settleman J. Distinct but overlapping functions for the closely related p190 RhoGAPs in neural development. Dev Neurosci. 2006;28:538–550. doi: 10.1159/000095116. [DOI] [PubMed] [Google Scholar]
- 90.Vincent S, Settleman J. Inhibition of RhoGAP activity is sufficient for the induction of Rho-mediated actin reorganization. Eur J Cell Biol. 1999;78:539–548. doi: 10.1016/S0171-9335(99)80019-3. [DOI] [PubMed] [Google Scholar]
- 91.Lévay M, Settleman J, Ligeti E. Regulation of the substrate preference of p190RhoGAP by protein kinase C-mediated phosphorylation of a phospholipid binding site. Biochemistry. 2009;48:8615–8623. doi: 10.1021/bi900667y. [DOI] [PMC free article] [PubMed] [Google Scholar]