

Abstract
The composition of tumor infiltrating lymphocytes (TIL) is heterogeneous. In addition, the ratio of various subpopulations in the tumor microenvironment is highly dependent on the nature of the host's immune response. Here, we characterize Foxp3-expressing CD8+ T cells in the tumor that demonstrate effector function and accumulate in the context of an effective antitumor response. CD8+Foxp3+ T cells are induced in TIL in regressing tumors of FVB/N mice treated with a GM-CSF secreting HER-2/neu targeted whole cell vaccine. Foxp3 expression in tumor antigen-specific CD8 T cells is restricted to the tumor microenvironment and influenced by cues in the tumor. Interestingly, Foxp3+ and Foxp3− CD8+ T cells have similar IFN-γ production and antigen-specific degranulation after stimulation with RNEU420–429, the immunodominant HER-2/neu (neu) epitope in this model. Adoptive transfer studies, using RNEU(420–429)-specific effector T cells into neu-N mice (a model that results in immune tolerance to neu), confirm that CD8+Foxp3+ T cells are present in tumors only if there is an existing pool of tumor-rejecting effector T cells. CD8+Foxp3+ TILs mark the presence of tumor-rejecting antigen-specific T cells and their accumulation serves as a marker for an effective T cell response.
Keywords: tumor infiltrating T cells, Foxp3, vaccine, immunotherapy
A recent study demonstrated that tumor-specific CD8+ T cell receptor-I (TcR-I) T cells transferred into prostate tumor-bearing mice trafficked to the prostate and acquired suppressive activity when tested in vitro. The report raised concerns that the development of these suppressor cells in the tumor microenvironment may eliminate the potency of T cells primed in the periphery or delivered during adoptive immunotherapy.1 Although a subset of these CD8+ T cells were found to be Foxp3+, the experiments focused on CD8+ (TcR-I) T cells as a whole. In addition, others have described suppressive CD8+CD25+Foxp3+ Treg cell clones derived from bulk prostate TIL lines generated from patients.2 Experimental assays suggested that they are suppressive. Interestingly, transforming growth factor (TGF-β) has been shown to induce Foxp3 expression in CD8+ TcR transgenic T cells and naive CD4+ T cells resulting in the generation of inducible Tregs.3,4 There has been minimal focus on the CD8+Foxp3+ subset of TILs, in part, because the conditions that promote CD8+Foxp3+ T cell induction appear to be restricted to the effector site making isolation of these cells in large numbers challenging.
In our study, we utilize a clinically relevant murine tumor model that exhibits immune tolerance to characterize these elusive tumor-specific CD8+Foxp3+ T cells. HER-2/neu transgenic (neu-N) mice, derived from the FVB/N mouse strain, express rat neu under the control of a mammary-specific promoter, which results in spontaneous mammary tumors.5,6 As opposed to FVB/N mice, neu-N mice develop peripheral tolerance to neu and cannot generate a neu-specific CD8+ T cell response using a neu-specific whole-cell granulocyte macrophage colony stimulating factor (GM-CSF)-secreting vaccine. Our studies led us to the discovery that 9–15% of the CD8+ T cells found in regressing tumors of vaccinated FVB/N mice expressed Foxp3, and that this expression was limited to the tumor-infiltrating lymphocytes. Further characterization of these cells suggested that they are most abundant in the microenvironment of immunogenic tumors. The availability of tumor-specific clonotypic T cells allowed us to further characterize the conditions that enhance or impair the presence of these cells. Although neu specific-CD8+Foxp3+ TILs share some similarity to previously described CD8+ “regulatory” populations in cell surface marker expression and in vitro suppressive capacity, we find that based on analysis of both endogenously generated and adoptively transferred CD8+Foxp3+ tumor antigen-specific TILs, CD8+Foxp3+ TILs mark the presence of tumor-rejecting antigen-specific T cells and accumulation of these T cells serves as a marker for an effective T cell response.
Material and Methods
Mice
FVB/N mice (Taconic) and neu-N mice (Jackson) were purchased. Foxp3gfp knockin mice were provided by Alexander Rudensky, University of Washington.7 FVB/N mice were bred to Foxp3gfp knockin mice resulting in heterozygous, F1 hybrids that express green fluorescent protein (GFP) (F1 FVB.Foxp3gfp). FVB.Foxp3gfp heterozygous mice that were backcrossed nine generations were also generated. Experiments used 6- to 12-week-old mice in protocols approved by the Animal Care and Use Committee of Johns Hopkins. Clone 100 T-cell receptor transgenic mice have been described.8 A majority of CD8+ T cells (>90%) from these mice express the high-avidity, RNEU(420–429)-specific TCR. RNEU(420–429) is the immunodominant major histocompatibility complex (MHC) class I epitope recognized by neu-specific CD8+ T cells.9
Cell lines and media
The GM-CSF-secreting vaccine cell lines, 3T3GM and 3T3neuGM, the NT2.5 neu-expressing tumor line, and the T2Dq line were grown as previously described.5
TIL isolation
Mice were injected with NT2.5 cells into the mammary pad. Seven days later, 3T3neuGM or 3T3GM cells (3 × 106 irradiated [5000 rads]) were injected subcutaneously, equally divided among three limbs 7 days after tumor. At various time points after vaccination, tumors were digested using hyaluronidase (Sigma, St. Louis, MO), collagenase type IV (Invitrogen, Carlsbad, CA) and trypsin (Sigma).
Tumor challenge and adoptive transfers
F1 FVB.Foxp3gfp mice were injected with 5 × 106 NT2.5 cells into the mammary pad. 3T3neuGM cells were given seven days after tumor. For adoptive transfer of CD4 subsets, 105 CD4+Foxp3gfp− or CD4+Foxp3gfp+ sorted splenocytes from tumor challenged mice were transferred 5 days after vaccination. Tumors were excised 14 days after vaccination. For adoptive transfer of clonotypic RNEU(420–429)-specific T cells, splenocytes were purified from Thy 1.2, TCR transgenic mice using Dynal CD8 negative selection beads (Invitrogen) and transferred to Thy 1.1 FVB/N (5 × 105 T cells) or neu-N (1– 6 × 106 T cells) tumor challenged mice. Mice were monitored for tumor growth or TILs were collected 6–12 days after vaccination. Cyclophosphamide (Baxter), 100mg/kg was administered on the day before vaccination.
Peptides
RNEU(420–429) (PDSLRDLSVF) and NP118–126 (RPQASGVYM) peptides were synthesized by the Peptide Synthesis Facility (Johns Hopkins, Baltimore, MD). NP118–126 is an irrelevant H2Dq-binding peptide.10
Antibodies and flow cytometric (FACS) analysis
Anti-Foxp3, glucocorticoid-induced tumor necrosis factor receptor (GITR) and interferon (IFN-γ) were from eBioscience (San Diego, CA). Streptavidin-PE was from Invitrogen (Carlsbad, CA). Other antibodies were from BD Biosciences (San Jose, CA). Data was collected using a BD FACSCalibur and analyzed using CellQuest and FlowJo.
Cells sorting
Cells were stained with anti-CD8 AlexaFluor-488 or APC, Anti-CD4 CyChrome or PerCP, anti-Thy 1.2 PE and anti-CD25 biotin (clone 7D4) or PE. Streptavidin-PE was used as a secondary and cells sorted using a BD FACSAria.
Immunofluorescence
Tissues were imbedded in Tissue-Tek OCT (VWR). Anti-Foxp3 rabbit polyclonal serum was provided by Marc Gavin, Washington University. Alexa Fluor 594 goat anti-rabbit IgG F(ab′)2 fragment and AlexaFluor 488 Streptavidin (Invitrogen, Carlsbad, CA) and anti-CD8 biotin were purchased.
Intracellular cytokine staining
Plates were coated with anti-CD3 (50 μl of 1 μg/ml in PBS) or PBS. T cell/TIL samples were added to each well (2 × 105 cells). For antigen-specific activation, T2-Dq cells were pulsed for 2–3 hr with RNEU(420–429) or NP118–126 (each at 10 μg/ml). T2-Dq (1 × 105) cells and T cells/TIL were added (2 × 105). GolgiStop (BDBiosciences) was added at 0.67 μl/ml. After 6 hr, cells were stained, fixed and permeabilized using PermFix Buffer (BDBiosciences).
CD107 assay
Intracellular cytokine staining (ICS) was performed with anti-CD107a/b FITC (final concentration of each antibody 2.5 μg/ml) or Rat IgG isotype control FITC (5 μg/ml). The percent CD107a/b+ was determined by comparison with CD107a/b FITC-staining in unstimulated samples and the isotype control FITC-staining in stimulated samples.
In vitro suppression
Sorted CD4+Foxp3gfp−CD25− responder splenocytes were isolated from F1 FVB.Foxp3gfp mice. TILs were isolated from tumors of F1 FVB.Foxp3gfp mice. Accessory cells were T cell depleted splenocytes (3,000 Rads). T cell depletion was accomplished with anti-Thy1.2 ascites followed by rabbit complement (Cedarlane Laboratories, Burlington, NC). Soluble anti-CD3 at 5 μg/ml (Clone 145-2C11, BD Biosciences) was added. Each well contained 104 responder cells, 5 × 104 irradiated accessory cells, with or without 5 ug/ml anti-CD3. A total of 104 TILs or splenic Tregs were added to one set of wells (1:1 Responder:Suppressor ratio). After 48 hr, tritium (Ambient) was placed into each well at 5 μl/ml for 16 hr.
In vitro culture
For experiments using F1 mice, cells were sorted and stimulated with anti-CD3/CD28 dynabeads (Dynal Biotech) at a 1:1 ratio for 1 week. IL-2 (R&D Systems) was added at 20 units/ml and TGF-β (R&D Systems) at 2–5 ng/ml.
Statistics
Statistical significance of the time course data was determined by two-way analysis of variance testing and Bonferroni post test. Student's t test was applied to compare two treatment groups using GraphPad Prism software (San Diego, CA).
Results
CD8+Foxp3+ TIL increase in a temporal fashion and are found most abundantly in the tumor microenvironment of regressing tumors
In an effort to study the differences in TIL populations between FVB/N and neu-N mice, we isolated TILs from mice vaccinated with either 3T3 cells genetically modified to express both neu and GM-CSF (3T3neuGM) or mock-vaccination (3T3GM) at various time points. On day 7, 5 × 106 NT2.5 cells were injected into the mammary pad of FVB/N or neu-N mice. On day 0, 3T3neuGM or 3T3GM cells were administered. TILs were collected on days 6, 10 and 15. All tumors in 3T3neuGM-treated FVB/N mice started regressing between 7 and 10 days after treatment and were completely eradicated by day 28. Tumors progressed in all other groups (unpublished data). There were a negligible number of T cells in the tumors of neu-N mice. Analysis of the regressing tumors in FVB/N mice demonstrated a large percentage of Foxp3-expressing CD8+ T cells in the tumor, but these were not found in the spleen or tumor draining lymph nodes (Fig. 1a). In contrast, although CD4+Foxp3+ T cells are increased in the tumor microenvironment (20–36% of CD4+ TILs), they also make up 9–10% of the splenic and lymph node-derived CD4+ T cells. By absolute numbers, total CD8+ T cells and CD8+Foxp3+ T cells were found most abundantly in the regressing tumors of FVB/N mice treated with 3T3neuGM (Fig. 1b, left and right panel). 3T3neuGM and 3T3GM-treated neu-N mice and 3T3GM-treated FVB/N mice had a negligible number of CD8+Foxp3+ TILs. In addition, these cells increased over time in the regressing tumors (p = 0.03). Although CD8+Foxp3+ TILs were detected in mock-vaccinated FVB/N mice by percentage, these were fewer in absolute number (Fig. 1b, middle and right panel). The presence of CD8+Foxp3+ T cells appears to be restricted to the immunogenic setting, because they are always detected in the regressing tumors in FVB/N mice but not in the progressing tumors isolated from neu-N mice.
Figure 1.
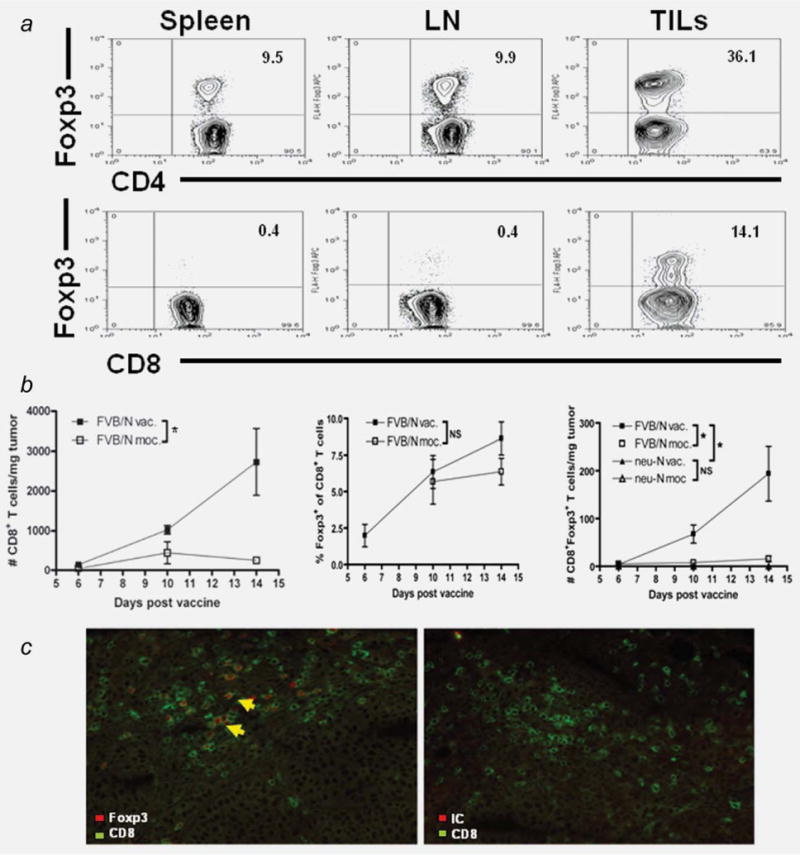
CD8+Foxp3+ T cells comprise a significant number of tumor infiltrating T cells in tumors of FVB/N mice treated with 3T3neuGM. (a), FVB/N mice were tumor challenged and vaccinated as described in Material and Methods. Splenocytes, tumor draining lymph nodes and TILs were harvested 14 days after vaccination. Top row (CD4+ gate), Bottom row (CD8+ gate). Isotype control staining was never >0.1% in the populations analyzed. (b), TILs were harvested from FVB/N or neu-N mice on the designated days after vaccination with 3T3neuGM or 3T3GM. Left panel, absolute number of CD8+ TILs. Middle panel, percent of CD8+Foxp3+ TILs. Right panel, absolute number of CD8+Foxp3+ TILs. The number of T cells per mg of tumor was calculated based on absolute number of viable TIL, the percentages of each T cell subset determined by flow cytometry, and the tumor weight. All tumors in 3T3neuGM-treated FVB/N mice start regressing between days 7–10 after treatment. Tumors progress in all other groups (unpublished data). The graphs are the compiled data of more than six independently conducted experiments. Error bars = standard error of the mean. NS = p > 0.05. *p ≤ 0.05. c, Immunofluorescence using (left) Green = CD8staining, Red = Foxp3 staining; (right) Green = CD8 staining, Red = control rabbit serum staining. Sections are from tumor harvested from a vaccinated FVB/N mouse. Anti-Foxp3 showed specific staining of tumor infiltrating CD8+ T cells. Image using fluorescent microscope set up to image FITC (Alexa Fluor 488) and Texas Red (Alexa Fluor 594). [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Foxp3 expression was confirmed by quantitative RT-PCR in sorted cells with high CD25 expression and by immunofluorescence staining of explanted tumors. Using RT-PCR, Foxp3 expression was detected in the CD8+CD25+ T cells and not detected in the CD8+CD25− T cells (unpublished data). Using immunofluorescence, CD8 surface expression colocalized with intracellular Foxp3 staining in a portion of the T cells (Fig. 1c).
CD8+Foxp3+ TILs have an activated effector phenotype
To further characterize the CD8+Foxp3+ T cells, TIL samples were stained for T cell activation markers and Treg-associated markers (Fig. 2a). TILs express activation markers regardless of their Foxp3 expression. No differences between the Foxp3+ and Foxp3− T cell subsets were detected when stained for CD3(positive), CD44(CD44hi), CD69(positive) and CD62L(CD62Llo) expression. CD8+Foxp3+ T cells expressed higher levels of the Treg markers CD25, GITR and cytotoxic T-lymphocyte antigen 4 (CTLA4) when compared to CD8+Foxp3− T cells. These markers while often associated with Treg populations are also markers of T cell activation. A similar trend is seen upon staining the CD4+Foxp3+ T cells.
Figure 2.
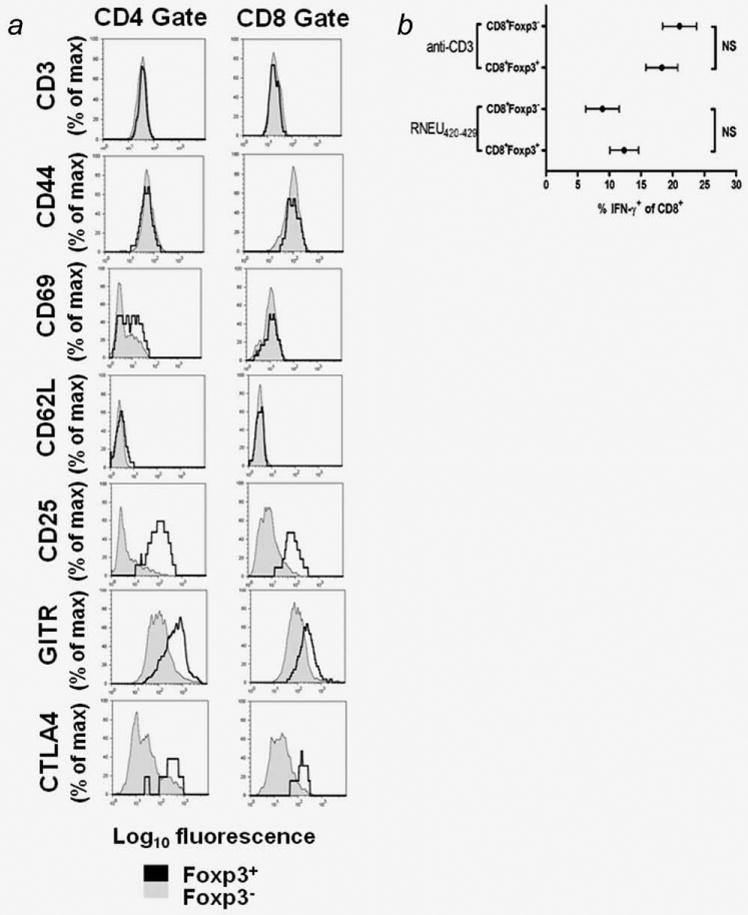
CD8+Foxp3+TILs express surface markers and cytokines associated with effector function. a, Cells were analyzed by flow cytometry. Shaded histograms = Foxp3− subsets. Open histograms = Foxp3+ subsets. Foxp3+ cells showed higher expression of CD25, GITR and CTLA4 then Foxp3− cells. b, Foxp3 expression and IFN-γ secretion were determined by ICS following in vitro stimulation with plate-bound anti-CD3 or RNEU(420–429)-pulsed APCs. The percent IFN-γ+ was determined by subtracting the percent IFN-γ+ in the unstimulated sample (or irrelevant peptide) from the percent IFN-γ+ in the stimulated sample (or RNEU(420–429) peptide). The graphs are the compiled data of more than six independently conducted experiments. Error bars = standard error of the mean, NS = p > 0.05.
Identification of cells that expressed both CD8 and Foxp3 led us to question whether these cells would secrete cytokines that were classically associated with effector function. Similar percentages of IFN-γ producing CD8+Foxp3+ and CD8+Foxp3− T cells were observed as assessed by ICS after anti-CD3 stimulation (p = 0.78) (Fig. 2b). We next explored antigen specificity. A hallmark of the CD8+ T cell response in the FVB/N mice is a strong response to the RNEU(420–429) peptide.9 TILs were stimulated with antigen presenting cells pulsed with RNEU(420–429). The Foxp3 status of cells made no difference in the percentage of CD8+ T cells that secreted IFN-γ (p > 0.05) (Fig. 2b). The IFN-γ expression pattern of CD8+Foxp3+ TILs is consistent with an effector phenotype.
Because it is difficult to obtain a sufficient number of TILs for an in vitro lysis analysis, we used CD107a/b staining as a surrogate for degranulation and lytic ability.11 TILs were stimulated for 5 hr with plate-bound anti-CD3 (Fig. 3a) or RNEU(420–429)-pulsed APCs (Fig. 3b) in the presence of anti-CD107a/b antibodies. Significant numbers of both CD8+Foxp3+ and CD8+Foxp3− T cells showed CD107a/b staining on stimulation with plate-bound anti-CD3 or RNEU(420–429)-pulsed APCs (Fig. 3c). Again, no difference was observed in the CD8+Foxp3+ versus the CD8+Foxp3− T cell populations (p > 0.05). Thus, the CD8+Foxp3+ T cells appear to have similar function to CD8+Foxp3− T cells.
Figure 3.
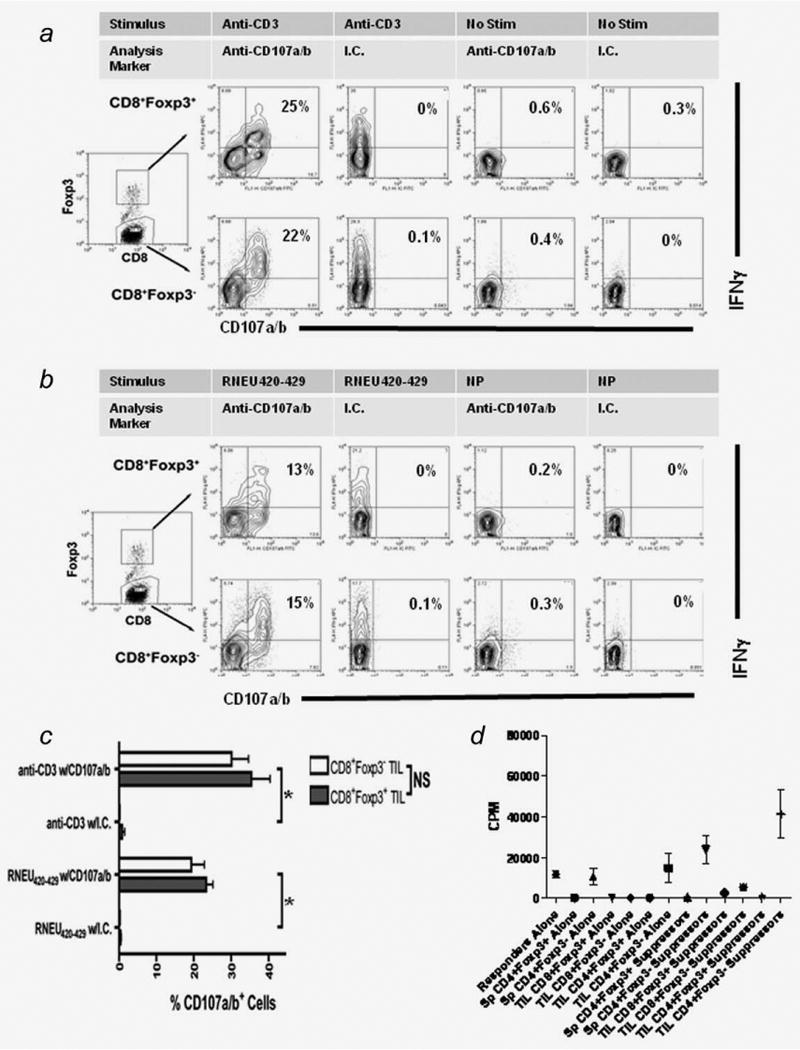
CD8+Foxp3+ possess both lytic ability and suppressive capacity in vitro. a, Top row = gated on CD8+Foxp3+ TILs. Bottom row = gated on CD8+Foxp3− TILs. Foxp3 expression, IFN-γ secretion and CD107 staining were determined after stimulation with plate-bound anti-CD3. b, Top row = gated on CD8+Foxp3+ TILs. Bottom row = gated on CD8+Foxp3− TILs. Foxp3 expression, IFN-γ secretion and CD107 staining were determined after stimulation with RNEU(420–429)-pulsed APC. Isotype control staining of CD107+IFNγ+ ceslls was always less than 0.5%. c, Compiled data from the CD107 assays reveals that no differences in lytic activity exist between the CD8+Foxp3+ and CD8+Foxp3− TIL populations (p > 0.05). The plots are representative of the data from three independently conducted experiments. NS = p > 0.05. d, TILs were harvested from F1 FVB.Foxp3gfp mice. Cells were sorted based on their CD4, CD8, Foxp3gfp and CD25 status. Responders and suppressors were plated in a 1:1 ratio. Responder cells were CD4+Foxp3gfp−CD25− splenocytes. The various suppressor populations are noted in the figure (Sp = spleen, TIL = tumor infiltrating lymphocytes). The stimulus was soluble anti-CD3.
Although the CD8 effector function of these cells was unexpected, the question of their suppressive capacity still needed to be addressed. To obtain a purified population of CD8+Foxp3+ cells that could be used in an ex-vivo study, FVB/N mice were bred to Foxp3gfp knockin mice.7 Splenocytes and TILs from neu-vaccinated, tumor bearing F1 FVB.Foxp3gfp mice were sorted. The different TIL T cell subsets were CD8+Foxp3gfp+CD25hi, CD8+Foxp3gfp−CD25lo, CD4+Foxp3gfp+CD25hi and CD4+Foxp3gfp−CD25lo. The responder cells were CD4+Foxp3gfp−CD25lo F1 splenocytes. Because the Foxp3 gene is on the the X chromosome, only one-half of Foxp3+ cells in heterozygous female F1 mice express GFP because of random X chromosome inactivation. Therefore, there is possible contamination of Foxp3+ (GFP negative) cells in the populations labeled as Foxp3−. GFP+ populations were sorted to >95% purity, and responders were stimulated using soluble anti-CD3 and accessory cells. Cells were plated in a 1:1 responder:suppressor ratio. Suppression of proliferation was seen in all three Foxp3-expressing populations including the CD8+Foxp3+ TILs (Fig. 3d). These results are consistent with the finding that Foxp3-expressing cell populations have suppressive capacity in vitro.2–4 This finding is also in line with the report that CD8+ TcR-I prostate-infiltrating TILs have suppressive capacity in vitro.1 Given technical limitations in the capacity to collect sufficient CD8+Foxp3+ TILs for in vivo suppression assays, we were only able to show a trend toward the suppression of vaccine-induced neu-specific T cells (Supporting Information Fig. S1). Therefore, these cells are not conclusively “regulatory” but they share surface phenotypic expression and in vitro suppression capacity with other regulatory cells.
TGF-β plays a role in both the maintenance and induction of Foxp3 in CD8+ T cells
To test whether or not the TIL-derived CD8+Foxp3+ T cells required signals found in the tumor microenvironment to maintain Foxp3 expression, we used TILs generated from the F1 FVB.Foxp3gfp mice and sorted the T cell populations so that the initial cells plated in culture were all Foxp3+. The CD8+Foxp3gfp+ and CD4+Foxp3gfp+ TILs and CD4+Foxp3gfp+ splenocytes were stimulated in vitro for 1 week using anti-CD3/CD28 beads supplemented with IL-2. The purity of a representative CD8+Foxp3gfp+ population before the in vitro stimulation is provided in Supporting Information Fig. S2. A majority of splenic Tregs (93.6%) maintained Foxp3 expression, whereas a small percentage of the CD4+Foxp3gfp+ TILs (6.9%) and even smaller percentage of CD8+Foxp3gfp+ TILs (0.3%) maintained Foxp3 expression (Fig. 4a). Many of the tumor-induced Foxp3+ cells T cells revert to Foxp3− negative cells when provided anti-CD3/CD28 and IL-2, which are stimuli known to sustain splenic Tregs in culture.
Figure 4.
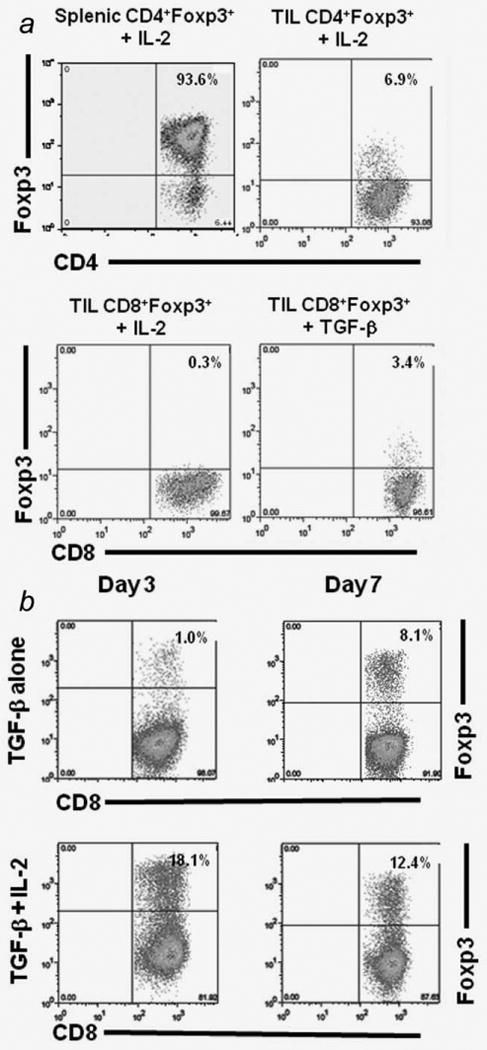
Foxp3 expression in CD8+ T cells is dependent on the cytokine milieu. a, TILs were cultured with anti-CD3/CD28 beads with either IL-2 or TGF-β for 1 week. The percentages of each T cell subset was determined by flow cytometry. b, Naïve RNEU(420–429) peptide-specific T cells were stimulated with anti-CD3/CD28 beads and 5ng/ml of TGF-β alone or TGF-β + 20units/ml of IL-2.
Foxp3 gene expression can be induced in CD4+CD25− T cells by stimulation with TGF-β and anti-CD3/anti-CD28.12,13 TGF-β plays a role in enhancer related activation of Foxp3 expression.14 Given that TGF-β is known to be expressed in a variety of tumor microenvironments and its role in the induction and maintenance of Foxp3 expression has been well documented, we tested whether or not TGF-β could maintain Foxp3 expression in the CD8+ TILs. Although IL-2 alone is not sufficient to maintain Foxp3 expression in culture, TGF-β administration did maintain a small percentage (3.4%) of CD8+Foxp3+ TILs (Fig. 4a). This data shows that, similar to recent findings in CD4+Foxp3+ T cells, transcriptional control of Foxp3 in CD8+ T cells may be influenced by the cytokine milieu.
To further assess the effects of TGF-β on inducing Foxp3 expression in CD8+ T cell populations, we analyzed TGF-β regulation of Foxp3 expression using Clone 100 TCR transgenic T cells. The majority of CD8+ T cells (>90%) from these mice express the high-avidity, RNEU(420–429)-specific TCR. Specifically, we stimulated naïve RNEU(420–429) peptide-specific T cells under conditions known to express inducible CD4+Foxp3+ T cells (anti-CD3/CD28 + TGF-β). We found that these conditions also induced Foxp3 expression in the CD8+ RNEU(420–429) peptide-specific T cells (Fig. 4b). However, this induction is more pronounced only at later time points, which correlates well with the data that CD8+Foxp3+ TILs increase over time in regressing tumors. The addition of IL-2 and TGF-β to cultures promoted robust Foxp3 expression and abrogated the difference in the time points.
Foxp3 expression can be induced in vivo in high avidity tumor antigen-specific CD8+ T cells
We next considered whether Foxp3 could be induced in high avidity tumor antigen-specific T cells in an in vivo setting. To address this question, FVB/N (Thy 1.1+) mice received a tumor challenge on day 7, vaccination on day 0 and adoptive transfer of the TCR transgenic (Thy 1.2+) RNEU-specific CD8+ T cells on day +1, and TIL were collected and analyzed on days 6 and 12. The results showed that a proportion of the adoptively transferred CD8+ T cells became Foxp3+ within the TILs (Fig. 5). These cells were confirmed to be Foxp3 negative before transfer. In the three groups tested (no vaccination, 3T3GM mock vaccine or 3T3neuGM), a similar percent of Thy 1.2+ TILs became Foxp3+ by day 12. Interestingly, when TILs were collected at the day 6 time point, the CD8+ TILs in the 3T3neuGM treated mice had a lower percentage and absolute number of Foxp3+ cells than at the day 12 time point (Fig. 5, column a and b, respectively). A reasonable explanation for this finding is that when the RNEU-specific T cells are transferred into mice that have been vaccinated, the vaccine primed dendritic cells activate peripheral T cells. At the early time point (day 6) when these activated T cells enter the tumor microenvironment they are not as susceptible to the stimuli in the microenvironment and do not express Foxp3. At the later time point (day 12), the tumors have almost completely regressed, and the increased percentage of Foxp3-expressing T cells may be explained by their longer exposure to the signals in the tumor microenvironment. Importantly, the absolute number of CD8+Foxp3+ TILs is greater in the 3T3neuGM treated mice at day 12. Thus, this experimental system allowed us to confirm expression of Foxp3 in CD8+ tumor-specific T cells that are derived from high avidity effector CD8+ T cells that normally do not express Foxp3.
Figure 5.
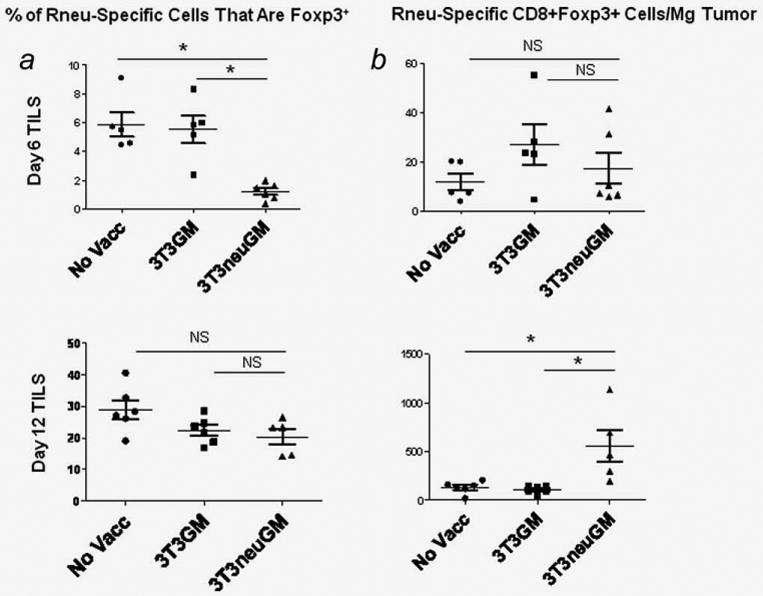
Foxp3 expression is induced in adoptively transferred tumor antigen-specific CD8+ effector T cells. FVB/N mice were challenged with NT2.5 on day −7, vaccinated with 3T3GM, 3T3neuGM, or not vaccinated on day 0, CD8+ clonotypic RNEU(420–429)-specific T cells from Thy1.2+ mice were adoptively transferred on day 1, and TILs were collected on day 6 and day 12. CD8+ clonotypic RNEU(420–429)-specific T cells were isolated from spleens of TCR transgenic mice using CD8 negative selection and documented to be Foxp3−. At the earlier time point (top row), the percentage of Foxp3+ CD8 cells is less in the mice vaccinated with 3T3neuGM. The percentage is equivalent at the later time point (bottom row). Column a: Foxp3 expression of the Thy1.2+ cells was determined by intracellular staining. The percentages of each T cell subset was determined by flow cytometry. Column b: Foxp3 expressing Thy1.2+ T cells were normalized to tumor weight. At the early time point (day 6) when these activated T cells enter the tumor microenvironment they are not as susceptible to the stimuli in the microenvironment and do not express Foxp3. At the later time point (day 12), the tumors have almost completely regressed, and the increased percentage of Foxp3-expressing T cells may be explained by their longer exposure to the tumor microenvironment. The absolute number of CD8+Foxp3+ TILs is greater in the 3T3neuGM treated mice at the later time point. Data are representative of two independent experiments. NS = p > 0.05. *p < 0.05.
Manipulating conditions to promote an effective immune response promotes the expansion of CD8+Foxp3+ TILs
The above data suggest that this CD8+Foxp3+ TIL population is induced during the evolution of a productive immune response (vaccinated FVB/N mice). This observation led us to consider whether or not conditions that could enhance a tumor-specific T cell response could increase the percentage of CD8+Foxp3+ TILs. We applied our repeated observation in vitro that CD4+Foxp3− cells enhance T cell proliferation to determine whether this observation held true in vivo. F1 FVB.Foxp3gfp mice were given tumor and vaccination as in the prior experiments. Five days after vaccination with 3T3neuGM, either CD4+Foxp3gfp+ or CD4+Foxp3gfp− splenocytes from tumor challenged syngeneic mice were adoptively transferred into the F1 FVB.Foxp3gfp mice from which TILs would be collected on day 14 after vaccination. Providing additional CD4+Foxp3+ Tregs did not alter the percentage of CD8+Foxp3+ TILs. However, providing CD4+FoxP3− T cells increased the percentage of CD8+Foxp3+ TILs (Fig. 6a). Thus, manipulating conditions thought to promote an effective immune response also promotes the expansion of CD8+Foxp3+ TILs. A possible explanation for this finding is that the CD4+ T cells stimulate expansion of the pool of tumor-specific T cells as well as add stimulatory cytokines such as IL-2 to the TGF-β expression already present in the tumor's microenvironment.
Figure 6.
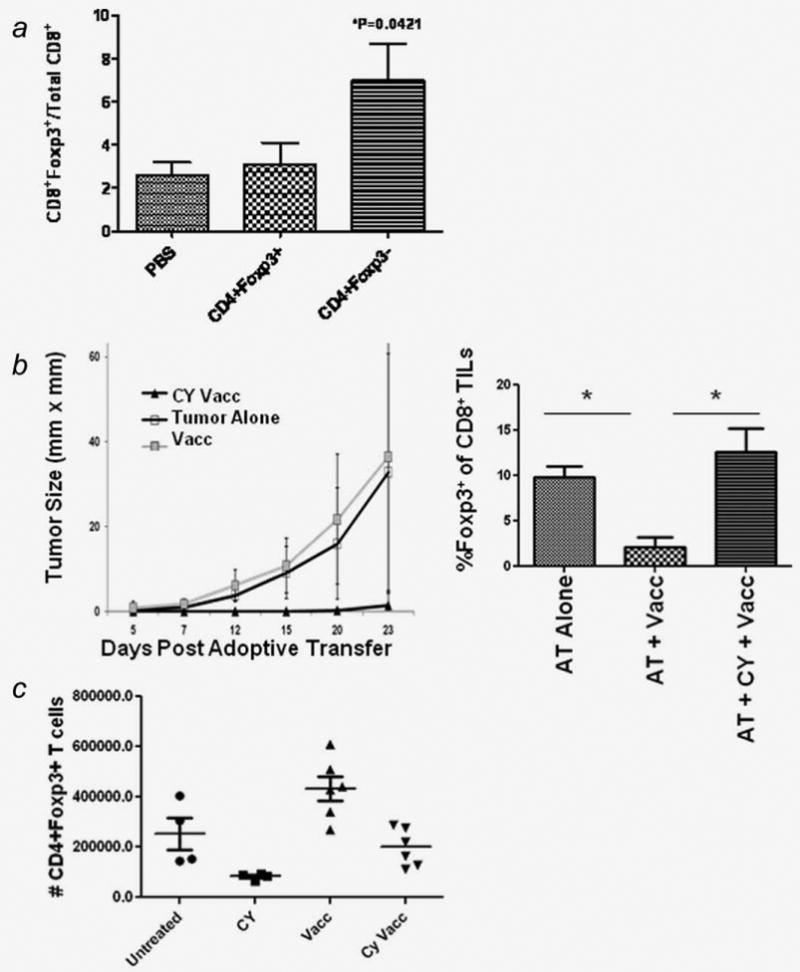
Manipulating conditions to promote an effective immune response promotes the expansion of CD8+Foxp3+ TILs. a, adoptive transfer of CD4+Foxp3gfp− populations increases the percentage of CD8+Foxp3+ TILs. F1 FVB.Foxp3gfp mice were challenged with NT2.5 on day -7, vaccinated with 3T3neuGM on day 0, 1 × 105 CD4+ T cell subsets were adoptively transferred on day 5, and TILs were collected on day 14. The adoptively transferred T cells were obtained from spleens of tumor challenged F1 FVB.Foxp3gfpmice and sorted based on CD4 and Foxp3gfp expression. The percentages of each T cell subset was determined by flow cytometry. Data are representative of two independent experiments. *p < 0.05. b, Neu-N mice were challenged with NT2.5 cells on day 7, given Cyclophosphamide on day 1, and vaccinated with 3T3neuGM on day 0. CD8+ RNEU(420–429)-specific T cells from Thy1.2+ mice were adoptively transferred on day 1. Tumors were monitored for growth after adoptive transfer (AT) of 6 × 106 T cells (left panel). TILs were collected on day 10 after AT of 1 × 106 T cells (right panel). Foxp3 expressing Thy1.2+ T cells were determined by flow cytometry. Data are representative of two independent experiments. *p < 0.05. c, Neu-N mice were challenged with NT2.5 cells on day 7, given Cyclophosphamide on day 1, and vaccinated with 3T3neuGM on day 0. Vaccine draining lymph nodes were harvested on day 2 and analyzed for expression of CD4 and Foxp3. The absolute number of CD4+Foxp3+ T cells was based on the total lymphocyte count and the percentage of CD4+Foxp3+ T cells determined by flow cytometry. These results were confirmed using F1 Neu-N.Foxp3gfp mice (unpublished data).
One factor that may contribute to the lack of induction of Foxp3 expression in tumor-antigen specific T cells in a tolerant host is the lack of a significant pool of CD8+ antigen-specific T cells presumably due to deletion of these cells. It is therefore possible that neu-N mice fail to demonstrate CD8+Foxp3+ TIL due to lack of endogenous RNEU-specific T cells or due to immune regulatory mechanisms that inhibit these T cells. By adoptively transferring clonotypic RNEU(420–429) specific T cells into neu-N mice under T cell activating conditions, we were able to test whether the provision of these tumor-specific T cells would be sufficient to generate CD8+Foxp3+ TILs. The high avidity RNEU-specific T cells were therefore transferred into neu-N mice under three separate conditions. T cells were transferred into tumor-bearing mice that were either: unvaccinated, a condition in which the RNEU(420–429) specific T cells are not specifically being primed in vaccine draining lymph nodes; vaccinated with 3T3neuGM, a condition that actively tolerizes the adoptively transferred RNEU-specific T cells; or vaccinated with 3T3neuGM in sequence with Treg cell modifying doses of cyclophosphamide (CY) the day before vaccination, a condition which enhances the anti-tumor activity of the RNEU-specific T cells. We have previously shown that CY depletes cycling Tregs allowing the generation and activation of endogenous high avidity RNEU(420–429) specific T cells.15 In this study, we found that CY given before vaccination is necessary to promote successful tumor clearance even with the adoptive transfer of large numbers of T cells, up to 6 × 106 high avidity RNEU(420–429) specific T cells (Fig. 6b, left panel). CD8+Foxp3+ TILs were induced in the tumors of both the AT alone group and the group given AT + CY + 3T3neuGM but not the AT + 3T3neuGM group (Fig. 6b, right panel). This observation suggests that CD8+Foxp3+ TILs are only found when tumor antigen-specific T cells are present and not subjected to enhanced tolerizing conditions. We could not enhance CD8+Foxp3+ TILs from the group of mice in which the T cells were primed with vaccine only, a situation that induces active tolerance. Work from others have demonstrated that GM-CSF whole cell vaccine strategies increase the absolute number of Tregs compared to unvaccinated mice.16 As further support of this finding, lymph nodes from tumor bearing neu-N mice vaccinated with 3T3neuGM have increased numbers of CD4+Foxp3+ T cells and the addition of CY reduces this number (Fig. 6c). Therefore, vaccination alone in a tolerogenic system results in a condition in which RNEU(420–429) specific T cells are actively tolerized in the presence of Tregs, and does not result in the induction of CD8+Foxp3+ TILs. However, the numbers of CD8+Foxp3+ TILs can be amplified within the tumor when techniques that inhibit vaccine-induced Treg expansion are utilized (AT + CY + 3T3neuGM).
Discussion
In this report, we describe four findings that provide new insights into effector T cell populations within the tumor's microenvironment. First, a subpopulation of T cells defined as CD8+Foxp3+ T cells, are induced at the tumor site of regressing tumors but not in immune tolerant conditions. Second, despite having an effector phenotype with some features of other regulatory cells, CD8+Foxp3+ T cells can be associated with a desired anti-tumor immune response. Third, Foxp3 expression in TIL subsets is dependent on ongoing signals from the microenvironment. Fourth, only antigen-specific T cells that have not been actively tolerized can acquire effector function and Foxp3 expression.
For therapeutic vaccine strategies to achieve optimal benefit, it is essential to understand the elements necessary for adequate priming and expansion of tumor-specific T cells. It is also critical to understand the effects of vaccination strategies on T cell populations both at peripheral sites and within the tumor's microenvironment, and the complex interplay that occurs in the microenvironment where effectors must infiltrate and kill their targets. Our studies show for the first time that CD8+Foxp3+ T cells are localized to the TIL of regressing tumors and mark the presence of potentially functional tumor-specific T cells. In addition, induction of these T cells in TIL may serve as a marker for an effective T cell response. In both preclinical and clinical studies, vaccines have been able to induce robust T cell responses in the peripheral blood or lymph nodes. However, there has been a dissociation between systemic and intratumoral responses.16 Few studies have correlated vaccine-induced tumor regressions with tumor-specific T cell populations within the tumor's microenvironment. Yet, T cells must maintain their active state within the tumor's environment to serve as a true biomarker of tumor regression.
Some studies suggest that CD4+CD25+Foxp3+ Tregs in tumors contribute to immune tolerance and are a correlative marker of poorer patient outcomes in breast, ovarian and pancreatic cancers.17–20 Other studies suggest that it is Teff/Treg ratios that correlate with effective anti-tumor responses.21,22 In Hodgkin's lymphoma, Tregs in the tumor microenvironment are actually associated with improved clinical outcomes.23 In our model, the ratio of Tregs to effectors does not necessarily correlate with an active immune response as suggested by similar ratios of CD4Tregs/CD4effs in TILs isolated from both the FVB/N and neu-N mice. In fact, by absolute numbers, the trend shows that CD4+ Tregs were more abundant in the actively regressing tumors (Supporting Information Fig. S3). Foxp3 expression in the tumor microenvironment may serve as an activation marker in T cells. Another hypothesis is that they serve to dampen an anti-tumor response as it resolves. Interestingly, the transfer of Tregs into Apcmin/+ results in regression of intestinal tumors.24 This is primarily due to the anti-inflammatory role of the Treg in this model. In our model, the best correlative marker of an effective anti-tumor response is the presence of CD8+Foxp3+ TILs.
The effector phenotype of a population with regulatory features is somewhat surprising. Activation markers such as CD25, GITR and CTLA4 are expressed on the majority of TILs as would be expected in ongoing immune responses. The use of Foxp3 as a Treg lineage marker has made it possible to distinguish Tregs from effectors cells. Studies currently show that Foxp3+ cells of any origin are suppressive. Even human CD8+ T cells that express Foxp3 transiently in vitro as a result of activation have suppressive capacity. CD8+Foxp3+ TILs also have potent in vitro suppressive capacity. Therefore, our report does not contradict prior studies on reportedly regulatory CD8+ T cell populations. Our data on vaccine-induced endogenously generated and adoptively transferred transgenic tumor-specific T cells which shows a correlation with an anti-tumor response stresses the importance that induction of Foxp3 expression has to be understood within the context of the host's immune response.
The interactions among the diverse components of the tumor microenvironment are becoming increasingly complex. Any one of the elements of the environment, from the diverse populations to the evolving cytokine milieu, could be critical in the induction or maintenance of the Foxp3 phenotype. Although most studies focus on the CD4 lineage, TGF-β induced Foxp3 induction and maintenance is not a novel concept. Priming and expansion of tumor-specific T cells under activating conditions appears to increase the pool of T cells that will subsequently enter the tumor microenvironment. Intact signaling through the T cell receptor and TGF-β/smad-3 pathway are likely necessary for Foxp3 induction. Because Foxp3 expression suggests active signaling through the TCR, CD8+Foxp3+ T cells signify a population of cells in which TCR signaling is not impaired. It is possible that these cells could be recruited to function as anti-tumor effectors. A pure population of clonotypic CD8+ effector T cells with tumor lysis capability can convert a proportion of active T cells into Foxp3+ T cells. We therefore propose a model in which effector CD8+ T cells initially traffic into the tumor's microenvironment and continue to expand to meet the demands of the antigen load within a T cell activating cytokine mileu. As the tumor begins to regress and the antigen load diminishes, changes in the cytokine mileu mediated by cytokines such as TGF-beta induce upregulation of Foxp3 expression within a subpopulation of the effector CD8+ T cells. Given that there is some suggestion that these cells acquire some suppressive capacity, a hypothesis generated from these findings is that these T cells then control the effector arm of the response, acting as a feedback mechanism to quickly turn off the activation status of the larger effector T cell population.
In conclusion, our findings identify a novel CD8+ T cell population within the tumor microenvironment that demonstrates tumor antigen specificity and effector function. Their Foxp3 expression depends on the cytokine mileu. We also show for the first time that the CD8+Foxp3+ TILs can serve as a potential marker for an effective anti-tumor immune response. The presence of Foxp3+ T cells as a marker of an active immune response may appear contradictory. However, these cells are present only in a host environment that is supportive of a functioning effector T cell population and appear to be antigen-specific. Thus, these findings provide new insights into the nature of Foxp3+ cells in the microenvironment and suggest caution in interpreting any analysis which involves the prognostic or predictive implications of the presence of Foxp3+ subsets.
Supplementary Material
Footnotes
Additional Supporting Information may be found in the online version of this article.
Through a licensing agreement by Johns Hopkins University to Bio Sante, Johns Hopkins University has the potential to receive royalties in the future.
References
- 1.Shafer-Weaver KA, Anderson MJ, Stagliano K, Malyguine A, Greenberg NM, Hurwitz AA. Cutting edge: tumor-specific CD8+ T cells infiltrating prostatic tumors are induced to become suppressor cells. J Immunol. 2009;183:4848–52. doi: 10.4049/jimmunol.0900848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kiniwa Y, Miyahara Y, Wang HY, Peng W, Peng G, Wheeler TM, Thompson TC, Old LJ, Wang RF. CD8+ Foxp3+ regulatory T cells mediate immunosuppression in prostate cancer. Clin Cancer Res. 2007;13:6947–58. doi: 10.1158/1078-0432.CCR-07-0842. [DOI] [PubMed] [Google Scholar]
- 3.Kapp JA, Honjo K, Kapp LM, Xu X, Cozier A, Bucy RP. TcR transgenic CD8+ T cells activated in the presence of TGFβ express FoxP3 and mediate linked suppression of primary immune responses and cardiac allograft rejection. Int Immunol. 2006;18:1549–62. doi: 10.1093/intimm/dxl088. [DOI] [PubMed] [Google Scholar]
- 4.Chen W, Jin W, Hardegen N, Lei KJ, Li L, Marinos N, McCrady G, Wahl SM. Conversion of peripheral CD4+CD25− naive T cells to CD4+CD25+ regulatory T cells by TGF-β induction of transcription factor Foxp3. J Exp Med. 2003;198:1875–86. doi: 10.1084/jem.20030152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Reilly RT, Gottlieb MB, Ercolini AM, Machiels JP, Kane CE, Okoye FI, Muller WJ, Dixon KH, Jaffee EM. HER-2/neu is a tumor rejection target in tolerized HER-2/neu transgenic mice. Cancer Res. 2000;60:3569–76. [PubMed] [Google Scholar]
- 6.Guy CT, Webster MA, Schaller M, Parsons TJ, Cardiff RD, Muller WJ. Expression of the neu protooncogene in the mammary epithelium of transgenic mice induces metastatic disease. Proc Natl Acad Sci USA. 1992;89:10578–82. doi: 10.1073/pnas.89.22.10578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol. 2003;4:330–6. doi: 10.1038/ni904. [DOI] [PubMed] [Google Scholar]
- 8.Manning EA, Ullman JG, Leatherman JM, Asquith JM, Hansen TR, Armstrong TD, Hicklin DJ, Jaffee EM, Emens LA. A vascular endothelial growth factor receptor-2 inhibitor enhances antitumor immunity through an immune-based mechanism. Clin Cancer Res. 2007;13:3951–9. doi: 10.1158/1078-0432.CCR-07-0374. [DOI] [PubMed] [Google Scholar]
- 9.Ercolini AM, Machiels JP, Chen YC, Slansky JE, Giedlen M, Reilly RT, Jaffee EM. Identification and characterization of the immunodominant rat HER-2/neu MHC class I epitope presented by spontaneous mammary tumors from HER-2/neu-transgenic mice. J Immunol. 2003;170:4273–80. doi: 10.4049/jimmunol.170.8.4273. [DOI] [PubMed] [Google Scholar]
- 10.Lee DR, Rubocki RJ, Lie WR, Hansen TH. The murine MHC class I genes, H-2Dq and H-2Lq, are strikingly homologous to each other, H-2Ld, and two genes reported to encode tumor-specific antigens. J Exp Med. 1988;168:1719–39. doi: 10.1084/jem.168.5.1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Betts MR, Brenchley JM, Price DA, De Rosa SC, Douek DC, Roederer M, Koup RA. Sensitive and viable identification of antigen-specific CD8+ T cells by a flow cytometric assay for degranulation. J Immunol Methods. 2003;281:65–78. doi: 10.1016/s0022-1759(03)00265-5. [DOI] [PubMed] [Google Scholar]
- 12.Chen W, Jin W, Hardegen N, Lei KJ, Li L, Marinos N, McGrady G, Wahl SM. Conversion of peripheral CD4+CD25-naive T cells to CD4+CD25+ regulatory T cells by TGF-beta induction of transcription factor Foxp3. J Exp Med. 2003;198:1875–86. doi: 10.1084/jem.20030152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fantini MC, Becker C, Monteleone G, Pallone F, Galle PR, Neurath MF. Cutting edge: TGF-beta induces a regulatory phenotype in CD4+CD25- T cells through Foxp3 induction and down-regulation of Smad7. J Immunol. 2004;172:5149–53. doi: 10.4049/jimmunol.172.9.5149. [DOI] [PubMed] [Google Scholar]
- 14.Tone Y, Furuuchi K, Kojima Y, Tykocinski ML, Greene MI, Tone M. Smad3 and NFAT cooperate to induce Foxp3 expression through its enhancer. Nat Immunol. 2008;9:194–202. doi: 10.1038/ni1549. [DOI] [PubMed] [Google Scholar]
- 15.Ercolini AM, Ladle BH, Manning EA, Pfannenstiel LW, Armstrong TD, Machiels JP, Bieler JG, Emens LA, Reilly RT, Jaffee EM. Recruitment of latent pools of high-avidity CD8(+) T cells to the antitumor immune response. J Exp Med. 2005;201:1591–602. doi: 10.1084/jem.20042167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Quezada SA, Peggs KS, Simpson TR, Shen Y, Littman DR, Allison JP. Limited tumor infiltration by activated T effector cells restricts the therapeutic activity of regulatory T cell depletion against established melanoma. J Exp Med. 2008;205:2125–38. doi: 10.1084/jem.20080099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bates GJ, Fox SB, Han C, Leek RD, Garcia JF, Harris AL, Banham AH. Quantification of regulatory T cells enables the identification of high-risk breast cancer patients and those at risk of late relapse. J Clin Oncol. 2006;24:5373–80. doi: 10.1200/JCO.2006.05.9584. [DOI] [PubMed] [Google Scholar]
- 18.Curiel TJ, Coukos G, Zou L, Alvarez X, Cheng P, Mottram P, Evdemon-Hogan M, Conejo-Garcia JR, Zhang L, Burow M, Zhu Y, Wei S, et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med. 2004;10:942–9. doi: 10.1038/nm1093. [DOI] [PubMed] [Google Scholar]
- 19.Liyanage UK, Moore TT, Joo HG, Tanaka Y, Herrmann V, Doherty G, Drebin JA, Strasberg SM, Eberlein TJ, Goedegebuure PS, Linehan DC. Prevalence of regulatory T cells is increased in peripheral blood and tumor microenvironment of patients with pancreas or breast adenocarcinoma. J Immunol. 2002;169:2756–61. doi: 10.4049/jimmunol.169.5.2756. [DOI] [PubMed] [Google Scholar]
- 20.Liyanage UK, Moore TT, Joo HG, Tanaka Y, Herrmann V, Doherty G, Drebin JA, Strasberg SM, Eberlein TJ, Goedegebuure PS, Linehan DC. Prevalence of regulatory T cells is increased in peripheral blood and tumor microenvironment of patients with pancreas or breast adenocarcinoma. J Immunol. 2002;169:2756–61. doi: 10.4049/jimmunol.169.5.2756. [DOI] [PubMed] [Google Scholar]
- 21.Quezada SA, Peggs KS, Curran MA, Allison JP. CTLA4 blockade and GM-CSF combination immunotherapy alters the intratumor balance of effector and regulatory T cells. J Clin Invest. 2006;116:1935–45. doi: 10.1172/JCI27745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gao Q, Qiu SJ, Fan J, Zhou J, Wang XY, Xiao YS, Xu Y, Li YW, Tang ZY. Intratumoral balance of regulatory and cytotoxic T cells is associated with prognosis of hepatocellular carcinoma after resection. J Clin Oncol. 2007;25:2586–93. doi: 10.1200/JCO.2006.09.4565. [DOI] [PubMed] [Google Scholar]
- 23.Alvaro T, Lejeune M, Salvado MT, Bosch R, Garcia JF, Jaen J, Banham AH, Roncador G, Montalban C, Piris MA. Outcome in hodgkin's lymphoma can be predicted from the presence of accompanying cytotoxic and regulatory T cells. Clin Cancer Res. 2005;11:1467–73. doi: 10.1158/1078-0432.CCR-04-1869. [DOI] [PubMed] [Google Scholar]
- 24.Erdman SE, Sohn JJ, Rao VP, Nambiar PR, Ge Z, Fox JG, Schauer DB. CD4+CD25+ regulatory lymphocytes induce regression of intestinal tumors in ApcMin/+ mice. Cancer Res. 2005;65:3998–4004. doi: 10.1158/0008-5472.CAN-04-3104. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.