

Abstract
Presenilins are ubiquitous, intramembrane proteins that function in Alzheimer's disease (AD) as the catalytic component of the γ-secretase complex. Familial AD mutations in presenilin are known to exacerbate lysosomal pathology. Hence, we sought to elucidate the function endogenous, wild-type presenilins play in autophagy-mediated protein degradation. We report the finding that genetic deletion or knockdown of presenilins alters many autophagy-related proteins demonstrating a buildup of autophagosomes, indicative of dysfunction in the system. Presenilin-deficient cells inefficiently clear long-lived proteins and fail to build up autophagosomes when challenged with lysosomal inhibitors. Our studies further show that γ-secretase inhibitors do not adversely impact autophagy, indicating that the role of presenilins in autophagy is independent of γ-secretase activity. Based on our findings, we conclude that endogenous, wild-type presenilins are necessary for proper protein degradation through the autophagosome–lysosome system by functioning at the lysosomal level. The role of presenilins in autophagy has many implications for its function in neurological diseases such as AD.
Introduction
The presenilins are ubiquitous, polytopic proteins (Spasic et al., 2006) localized to many intracellular compartments, including the endoplasmic reticulum, Golgi complex, lysosomes, and autophagosomes (Kovacs et al., 1996; Pasternak et al., 2003; Yu et al., 2004). Mutated presenilins underlie the majority of familial Alzheimer's disease (FAD) cases (Mann et al., 1996; Borchelt et al., 1997; Gómez-Isla et al., 1999), because they are the catalytic component of γ-secretase, cleaving amyloid precursor protein (APP) to form β-amyloid (Aβ) (Xia et al., 1998; Palacino et al., 2000; Lanz et al., 2003; Page et al., 2008). In addition to APP, many γ-secretase substrates have now been identified, suggesting that γ-secretase and presenilins are important for many cellular processes (Parks and Curtis, 2007; Selkoe and Wolfe, 2007).
The portfolio of presenilin functions extends beyond their catalytic role in γ-secretase. Presenilins play a major role in calcium homeostasis (LaFerla, 2002). In addition, γ-secretase-independent presenilins function in phosphoinositide-3 kinase/Akt (PI3K) signaling (Baki et al., 2008). Beyond these roles, possible presenilin functions are immense, because at least 40 interacting partners have been identified (Parks and Curtis, 2007; Wakabayashi et al., 2009). Consequently, the presenilins are situated to directly or indirectly impact many cellular pathways.
Mounting evidence is pointing to a role for presenilins in autophagy. The three major types of autophagy in higher eukaryotes are macroautophagy, chaperone-mediated autophagy, and microautophagy. Macroautophagy, hereafter referred to as autophagy, involves the sequestration of cellular contents by an autophagosome and subsequent fusion of the autophagosome to a lysosome for degradation of its contents. Autophagy is critical for maintaining cytosolic amino acid pool, degrading organelles, long-lived proteins, and aggregates, and protecting against neurodegeneration (Mortimore and Pösö, 1987; Ravikumar et al., 2002; Onodera and Ohsumi, 2005; Berger et al., 2006; Hara et al., 2006; Komatsu et al., 2006; Yang et al., 2006; Sarkar et al., 2009).
FAD-linked presenilin mutations lead to increased lysosomal pathology in both mouse models and humans (Cataldo et al., 2004). In addition, presenilin-1 (PS1) knock-out neurons build up telecephalin in autophagosomes (Esselens et al., 2004) and α- and β-synuclein in degradative organelles (Wilson et al., 2004), suggesting that certain proteins are not efficiently cleared through autophagy in the absence of presenilin-1. Also, impairment in autophagy was found to increase presenilin-1 expression and alter γ-secretase activity (Ohta et al., 2010). Finally, recently published data demonstrated that autophagy and lysosomal proteolysis require presenilin-1 (Lee et al., 2010). In this study, presenilin-1 knock-out blastocysts showed decreased long-lived protein proteolysis and a buildup of autophagosomes. Additionally, they observed that cells with low or absent presenilin-1 had decreased acidification of lysosomes attributable to dysfunctional glycosylation and targeting of the v-ATPase VOa1 subunit.
Our data provide evidence for a role for both PS1 and presenilin-2 (PS2) in autophagy through a γ-secretase-independent mechanism. Although we find no reduction in LysotrackerRed staining as in the study by Lee et al. (2010), our results do indicate that presenilin loss leads to dysfunction at the lysosome.
Materials and Methods
Cell culture.
Mouse embryonic fibroblasts (MEFs) and N2A neuroblastomas were maintained with DMEM (Invitrogen), 1% penicillin/streptomycin (Invitrogen), and 10% FBS (Invitrogen). SHSY5Y neuroblastomas were maintained with DMEM/F-12 (Invitrogen), 10% FBS, 1% nonessential amino acid solution (Invitrogen), and 1% penicillin/streptomycin.
Protein immunoblotting.
Protein extracts were prepared when cells were 85–95% confluent using M-PER protein extraction reagent (Pierce) with Complete-mini protease inhibitor cocktail tablets (Roche). Protein concentrations were determined by the Bradford method. Equal amounts of protein (12–20 μg) were separated by SDS/PAGE on a 4–12 or 10% Bis/Tris or 3–8% Tris-acetate gels (Invitrogen), transferred to polyvinylidene difluoride membranes, blocked for 1 h in 5% (v/v) nonfat milk in Tris-buffered saline, pH 7.5, supplemented with 0.2% Tween 20, and incubated overnight at 4°C with the appropriate primary antibody. Antibodies used in this study include light chain 3 (LC3) (MBL International), mammalian target of rapamycin (mTOR) (Sigma), phosphorylated mTOR (S2448; Cell Signaling Technology), beclin1 (Santa Cruz Biotechnology and Novus), ultraviolet irradiation resistance-associated gene (UVRAG) (Abgent), p150 (Abnova), Vps34 (Invitrogen), Atg12 (Novus), LAMP2a (Invitrogen), Ubiquitin (DAKO), CT20 (Calbiochem), and Actin (Sigma). Membranes were washed five times and then incubated with HRP-conjugated secondary antibodies at room temperature. Quantitative densiometric analyses were performed on digitized images of immunoblots with NIH ImageJ. The background from each blot was subtracted from the raw data for each protein band and was then normalized to the protein loading control β-actin.
EFGP–LC3 and LysotrackerRed quantification.
Enhanced green fluorescent protein (EGFP)–LC3 (Plasmid 11546; Addgene) was transiently transfected using Lipofectamine2000 (Invitrogen). At the time of media replacement, complete medium alone, with rapamycin (0.2 μm in DMSO; Calbiochem), or DMSO was used. LysotrackerRed (75 nm; Invitrogen) was loaded into cells for 30 min before fixation. After treatment, cells were fixed using 4% paraformaldehyde. The Leica DM2500 (Leica Microsystems) confocal microscope was used to obtain the images.
Quantifications for EGFP–LC3 and LysotrackerRed were performed using manual counts of puncta per cell on NIH ImageJ. The identity of the cell type was blinded to the experimenter, and the same puncta criteria were used throughout. Cells expressing high levels of EGFP–LC3 were excluded to avoid counting aggregates of GFP and not autophagosomes.
RNA interference.
Transient transfections of small interfering RNA (siRNA) into SHSY5Y neuroblastomas were performed using the Amaxa nucleofector system (Kit V; Lonza). Two micromolar of each siRNA [scramble, 5′-AAATGTGTGTACGTCTCCTCC-3′; PSEN1, validated Mission siRNA (SASI_Hs01_00043630; Sigma-Aldrich); and PSEN2, validated Mission siRNA (SASI_Hs01_00033516; Sigma-Aldrich)] was used, and knockdown was allowed to proceed for 40 h after which protein extracts were made for immunoblotting. For endoribonuclease-prepared small interfering RNA (esiRNA), 2 μm of each Mission esiRNA [esiRNA EGFP, catalog #EHUEGFP; esiRNA PSEN1, catalog #EHU073361; esiRNA PSEN1, catalog #EHU070541 (Sigma-Aldrich)] was used, and knockdown was allowed to proceed for 48 h after which protein extracts were made for immunoblotting.
Stable knockdown of beclin1 in wild-type mouse embryonic fibroblasts were performed using short hairpin RNA (shRNA) lentiviral delivery. Lentiviral shRNA beclin1 constructs were purchased from Open Biosystems. shRNAs were cotransfected into 293FT cells together with packaging plasmids by following the protocol of the manufacturer (ViraPowerTM Lentiviral Expression Systems kit; Invitrogen). Mouse embryonic fibroblasts were passaged and plated in a six-well plate and allowed to adhere for 24 h. Cells were subjected to lentiviral infection in the presence of polybrene overnight, and media were replaced the following day. After 24 h, cells were selected by treating with media containing 1.5 μg/ml puromycin. After three to four passages, cells were considered to have stable knockdown of beclin1 as confirmed by immunoblot.
Pulse chase for long-lived proteins and long-lived protein proteolysis.
Two protocol were used to assess the breakdown of long-lived proteins. Both were adapted from Bauvy et al. (2009).
First, to confirm that methionine treatment would not cause autophagy inhibition, cells were incubated with 10 mm l-methionine (Sigma) for 10 h, and then protein was extracted for immunoblot analysis. In the first analysis, cells were incubated with media containing 2 μCi/ml of [35S]l-methionine (MP Biomedical) for 18 h. Pulse samples were then lysed using M-PER protein extraction reagent (Pierce) containing a protease inhibitor cocktail (Complete-mini; Roche). Cells for chase analysis were then washed with PBS and replaced with media containing 10 mm l-methionine for 1 h. Media were then removed and replaced again with media containing 10 mm l-methionine for 4 h. The media were then removed, and cells were lysed using M-PER protein extraction reagent (Pierce) containing a protease inhibitor cocktail (Complete-mini; Roche). Protein concentrations were then determined by the Bradford method. Equal amounts of protein (20 μg) were separated by SDS-PAGE on a 4–12% Bis/Tris gel (Invitrogen). The gels were then fixed for 1 h and then incubated in Amplify solution (GE Healthcare) for 30 min. The gels were then dried using a slab gel dryer (Savant SGD-5040; Thermo Fisher Scientific) and exposed to film at room temperature. Quantitative densiometric analyses were performed on digitized images of film using NIH ImageJ.
For the second analysis, control and presenilin-1 and -2 double knock-out (PSDKO) mice fibroblasts were pulsed with 0.2 μCi/ml [H3]l-valine (MP Biomedical) for 48 h. Cells were then washed extensively, and media were replaced with 10 mm valine for 1 h. Once more, cells were washed extensively, and media were replaced with 10 mm valine and 0.2 μm rapamycin when applicable. Subsequent time points of media and cells were collected at 4, 8, 12, and 24 h. Rapamycin-treated cells were taken at 12 h. The media was precipitated with tricholoacetic acid (TCA), and the acid-soluble radioactivity was measured. The cells were washed with cold 10% TCA with 10 mm valine and then lysed with sodium hydroxide, and the radioactivity was measured. Proteolysis is presented as the percentage of initial acid-precipitatable radioactivity transformed to acid-soluble radioactivity.
Proteasome analysis.
For immunocytochemical analysis of ubiquitin, cells were fixed on glass slides with 4% paraformaldehyde. The cells were then permeabilized and blocked using 10% NGS with Triton X-100 in PBS. The cells were then incubated overnight at 4°C in the primary antibody solution (Ubiquitin, 1:200; DAKO). After washing with PBS, the cells were incubated in the secondary antibody conjugated to Alexa Fluor-488 (Invitrogen). After washing with PBS, the slides were then coverslipped and allowed to dry for 48 h before confocal microscopy was performed on the Leica DM2500 confocal microscope (Leica Microsystems).
For fluorogenic proteasome substrate functional analysis using confocal microscopy, ZsProsensor-1 vector (Clontech) was transiently transfected using the Amaxa nucleofector system (Kit MEF 1; Lonza). Twenty-four hours after transfection, cells were fixed using 4% paraformaldehyde and coverslipped. After 48 h, confocal microscopy was performed on the Leica DM2500 confocal microscope (Leica Microsystems).
For kinetic fluorogenic proteasome activity assays, cells were homogenized in assay buffer containing 25 mm HEPES, pH 7.5, 0.5 mm EDTA, and 0.05% NP-40. Immediately before the kinetic readings, the proteasome substrates [LLVY-AMC (Leu-Leu-Val-Tyr-7-amino-4-methylcoumarin) for the chymotrypsin-like, VGR-AMC (Benzoyl-Val-Gly-Arg-amino-4-methylcoumarin) for the trypsin-like, and LLE-AMC (Benzyloxycarbonyl-Leu-Leu-Glu-amino-4-methylcoumarin) for the peptidylglutamyl hydrolytic (PGPH)/caspase-like activity] were added at 75 nm. Readings for AMC release were taken at 37°C every 1.5 min for 60–90 min (excitation, 360 nm; emission, 460 nm), on the Synergy HT using KC4 software (Bio-Tek Instruments).
γ-Secretase inhibitor treatments.
Fibroblasts and N2A neuroblastomas were treated with γ-secretase inhibitor IX [DAPT (N-[N-(3,5-difluorophenacetyl-l-alanyl)]-S-phenylglycine t-butyl ester), 500 nm, catalog #565784; Calbiochem] and γ-secretase inhibitor X [L-685,458 (1S-benzyl-4R-[1-(1S-carbamoyl-2-phenethylcarbamoyl)-1S-3-methylbutylcarbamoyl]-2R-hydroxy-5-phenylpentyl-carbamic acid tert-butyl ester), 200 nm, catalog #565771; Calbiochem] for 48 h. Protein extracts were then prepared using M-PER protein extraction reagent (Pierce) with Complete-mini protease inhibitor cocktail tablets (Roche). Successful γ-secretase inhibition was determined using Western blot analysis for C-terminal fragments of amyloid precursor protein using the antibody CT20.
Aβ ELISA.
Aβ1-40 and Aβ1-42 were measured using a sensitive sandwich ELISA system as described previously (Koike et al., 2010). Briefly, media were collected from N2A cells treated with vehicle or γ-secretase inhibitors after the full course of treatment. MaxiSorp immunoplates (Nalge Nunc International) were coated with mAB20.1 (William Van Nostrand, Stony Brook, NY) antibody at a concentration of 25 mg/ml in Coating Buffer (0.1 m NaCO3 buffer, pH 9.6) and blocked with 3% BSA. Samples were diluted at 1:5 before loading onto ELISA plates in duplicate. Standards of both Aβ40 and Aβ42 were made in antigen capture buffer (20 mm NaH2PO4, 2 mm EDTA, 0.4 m NaCl, 0.5 g of 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate, and 1% BSA, pH 7.0), loaded onto ELISA plates in duplicate, and incubated overnight at 4°C. Plates were washed and then probed with either HRP-conjugated anti-Aβ35-40 (MM32-13.1.1 for Aβ1-40) or anti-Aβ35-42 (MM40-21.3.4 for Aβ1-42) overnight at 4°C. 3,3′,5,5′-Tetramethylbenzidine was used as the chromagen, and the reaction was stopped by 30% O-phosphoric acid and read at 450 nm on a Molecular Dynamics plate reader.
Presenilin rescue into presenilin-1 knock-out cells.
pcDNA, wild-type presenilin-1 cDNA, and ΔTM1–2 presenilin-1 cDNA were transiently transfected using Lipofectamine2000 (Invitrogen) into presenilin-1 knock-out (PS1KO) mouse embryonic fibroblasts. Expression was allowed to proceed for 72 h. Protein extracts were then prepared using M-PER protein extraction reagent (Pierce) with a Complete-mini protease inhibitor cocktail tablets (Roche).
Rapamycin and lysosome inhibitor treatments.
To induce autophagy, fibroblasts were treated with rapamycin (0.2 μm; Calbiochem) for 24 h. To inhibit autophagy, fibroblasts were treated with the vacuolar ATPase inhibitor bafilomycinA1 (100 nm for 7 h; Sigma) or the microtubule destabilizer nocodazole (50 μm for 2.5 h; Sigma).
Confocal microscopy.
Images were acquired on the Leica DM2500 confocal microscope. Acquisition software used was LAS_AF (Leica Microsystems). Slides were prepared using Fluromount mounting media (Southern Biotechnology Associates), and images were acquired at room temperature. Fluorochromes used are listed under the specific experiment.
Statistics.
Data are presented as mean ± 1 SEM, with n indicating number of samples examined. An unpaired Student's t test or ANOVA was used to determine statistical significance (p < 0.05) indicated within the figure legends. Jmp8 statistical software was used.
Results
Upregulation of autophagy markers in presenilin-null fibroblasts
Given previous observations that FAD-linked presenilin mutations were associated with increased lysosomal pathology (Cataldo et al., 2004) and presenilin 1-null neurons accumulated telencephalin α- and β-synuclein within degradative organelles (Esselens et al., 2004; Wilson et al., 2004), we sought to investigate the role of endogenous, wild-type presenilins in autophagic protein degradation. To this end, we used mouse embryonic fibroblasts derived from control, PS1KO, PS2KO, or PSDKO mice.
We first analyzed the steady-state levels of key autophagy-related proteins in PSDKO mouse embryonic fibroblasts. Because both presenilins are absent, neither will compensate for the loss of the other and mask the effect of presenilin loss. Microtubule-associated protein 1 LC3 is a widely used marker for autophagosomes as it is converted from a cytosolic form (LC3-I) to a lipidated form (LC3-II) and inserted into the autophagosome membrane during vesicle elongation (Kabeya et al., 2000). Using Western blot analysis, we noted a shift in molecular weight, signifying the conversion of LC3-I to LC3-II. This finding serves as an indirect measure of autophagy, because increases in LC3-II are associated with increased levels of autophagosomes (Kabeya et al., 2000). Importantly, we observed that presenilin-null fibroblasts have a twofold increase in LC3-II levels and a corresponding reduction in LC3-I levels (Fig. 1A,B), suggesting that the absence of presenilins leads to increases in the number or size of autophagosomes. Based on this observation, we proceeded to investigate the signaling pathways that regulate the induction of autophagy and hence the eventual conversion of LC3-I to LC3-II.
Figure 1.
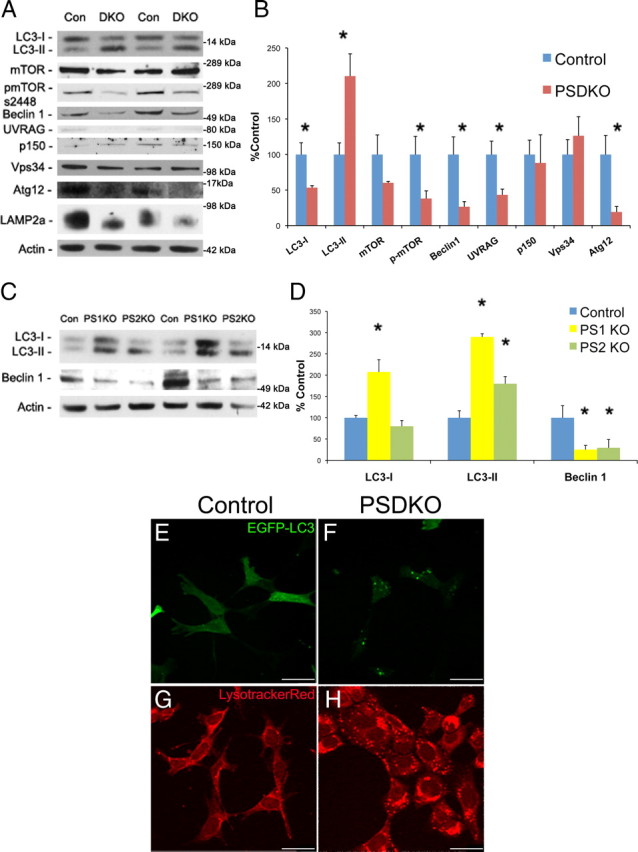
Loss of presenilin alters autophagy-related proteins and leads to a buildup of autophagosomes. A, B, Presenilin double knock-out (DKO) fibroblasts have alterations in key autophagy-related proteins by immunoblot. Quantification of Western blots in A was performed by densiometric analysis and is presented as percentage of control (Con), normalized to actin. Error bars represent ± SEM; n = 4–8; unpaired Student's t test, *p < 0.05. C, D, PS1KO and PS2KO fibroblasts have similar autophagic protein alterations as PSDKO cells using immunoblot. Quantification of Western blots in C was performed by densiometric analysis and is presented as percentage of control, normalized to actin. Error bars represent ± SEM; n = 3; one-way ANOVA analysis, post hoc comparison Tukey's HSD, *p < 0.05. E, F, PSDKO fibroblasts have increased EGFP–LC3 puncta per cell (18.24 ± 1.42 vs 37.13 ± 2.45, mean ± SEM; unpaired Student's t test, p < 0.05), representing autophagosomes, compared with controls. Magnification, 63×. Scale bars, 35 μm. Control, n = 87; PSDKO, n = 59. G, H, PSDKO fibroblasts have increased LysotrackerRed puncta per cell compared with controls (26.03 ± 3.51 vs 50.46 ± 2.35, mean ± SEM; unpaired Student's t test, p < 0.05). Magnification, 63×. Scale bars, 35 μm. Control, n = 30; PSDKO, n = 68.
One of the major regulators of autophagy induction is mTOR. Nutrient and amino acid levels control phosphorylation of mTOR to suppress or activate autophagy to regulate amino acid recycling through the lysosome system (Noda and Ohsumi, 1998; Jung et al., 2010). Phosphorylation of mTOR regulates its activity, with increased phosphorylation being associated with inhibited autophagy induction. Steady-state levels of mTOR are not significantly altered between control and presenilin-null fibroblasts, but levels of phosphorylated-mTOR (S2448) are decreased by ∼50% in the absence of presenilins (Fig. 1A,B). Decreases in phosphorylated mTOR are consistent with increased conversion of LC3-I to LC3-II.
Previous reports had shown that presenilins could regulate PI3K signaling pathways (Baki et al., 2008). Although the effects of presenilins have not been shown to specifically affect the PI3K-complex III, this complex is involved in a crucial step in the autophagic process (Burman and Ktistakis, 2010). The PI3K–complex III, which allows for autophagic vesicle nucleation to occur, comprises beclin1, UVRAG, Vps34, Atg14, and p150 (Liang et al., 1999, 2006; Zeng et al., 2006; Itakura et al., 2008; Lindmo et al., 2008; Yan et al., 2009). Presenilin-null cells displayed lowered steady-state levels of beclin1 and UVRAG but no alteration in Vps34 or p150 (Fig. 1A,B). Curiously, levels of beclin1 usually positively correlate with levels of LC3-II and autophagy (Liang et al., 1999; Spencer et al., 2009), and so diminished levels are at odds with increased LC3-II levels in the presenilin-null cells.
Given the curious reduction in PI3K complex III proteins, we next sought to confirm activity within the vesicle elongation step as we find with LC3. During phagophore vesicle elongation, Atg12 is covalently conjugated to Atg5 in an ubiquitin-like manner (Mizushima et al., 1998; Hanada et al., 2007). Although unable to detect the conjugated form by Western blot, we found decreased levels of free Atg12 in PSDKO cells, indicating that Atg12 is likely to be conjugated in the absence of presenilins (Fig. 1A,B). These data again indicate increased autophagosome elongation with presenilin deletion, confirming the result of LC3.
In addition, we find that presenilin-null fibroblasts have altered SDS-PAGE migration of LAMP2a (Fig. 1A). LAMP2a is a crucial lysosomal protein for proper autophagy and is posttranslationally modified to alter its function (Cuervo and Dice, 2000). The molecular weight shift of LAMP2a in presenilin-null cells indicates that the loss of presenilins is causing LAMP2a to function differently. Together, these results demonstrate that the absence of presenilins alters autophagy markers, indicating that endogenous presenilins play a critical role in the regulation of autophagy.
Deletion of presenilin-1 or presenilin-2 upregulates LC3-II
Having established that presenilin double knock-out leads to alterations in autophagic proteins, we wanted to determine whether ablation of either presenilin-1 or presenilin-2 could replicate these alterations. Presenilin-1 and -2 are known to have many redundant but also some unique functions, many related to their role in the γ-secretase complex (Tandon, 2002; Lai et al., 2003). To determine whether presenilin-1 or -2 or both play a role in autophagy, we used single knock-out cells and monitored the effects on key autophagy-related proteins. As was observed in the PSDKO cells, we found that ablating either PS1 or PS2 caused levels of LC3-II to increase and resulted in a decrease in beclin1 (Fig. 1C,D). Our results for a buildup of LC3-II in PS1KO cells are in accordance with the recent finding of Lee et al. (2010). These findings are significant because they provide strong evidence that both presenilins are critically involved in regulating autophagy and one cannot compensate for the loss of the other.
Loss of presenilin increases autophagosomes and lysosomes
As autophagosomes form, LC3 is altered to tightly associate with autophagosome membranes (Kabeya et al., 2000). Transfection of EGFP–LC3 allows for easy visualization of autophagosomes, which appear as puncta (Klionsky et al., 2007). Given the robust increases in LC3-II in the presenilin knock-out fibroblasts, we sought to determine whether this molecular change induces a cell biological change and a concomitant increase in autophagosomes. Hence, we transiently transfected control and PSDKO fibroblasts with EGFP–LC3 and observed an increase in EGFP–LC3 puncta per cell in presenilin-null cells compared with control cells (18.24 ± 1.42 vs 37.13 ± 2.45, mean ± SEM; unpaired Student's t test, p < 0.05) (Fig. 1E,F). This finding provides strong evidence that deleting presenilins causes an increase in autophagosomes and is consistent with the biochemical evidence showing that LC3-II is increased.
Alterations in autophagy cannot only be observed through changes in autophagosome number but also through changes in lysosome number (Klionsky et al., 2007). To determine whether lysosome numbers were altered in the presenilin-null cells, we used LysotrackerRed, a fluorescent dye that is taken up by acidic cellular compartments. Our studies indicate that the number of LysotrackerRed puncta is increased per cell in the presenilin-null cells compared with controls (26.03 ± 3.51 vs 50.46 ± 2.35, mean ± SEM; unpaired Student's t test, p < 0.05), which is consistent with abnormal autophagy (Fig. 1G,H). Curiously, these results differ from those of Lee et al. (2010) because they find a decrease in LysotrackerRed staining with PS1KO.
Knockdown of presenilins in neuroblastomas alters autophagy markers
Using multiple presenilin knock-out mouse embryonic fibroblast cell lines, we found alterations in autophagy and next wanted to determine whether presenilin is important for autophagy in other cell types. We used RNA interference with siRNA to transiently knockdown presenilin-1 or presenilin-2 levels in SHSY5Y neuroblastomas. Notably, we found that transiently reducing presenilin-1 or presenilin-2 with a single siRNA was sufficient to significantly alter autophagy-related proteins (data not shown). Given the previous data, we wanted to ensure that the effect on autophagy-related proteins was not attributable to off-target effects of siRNA. To combat this issue, we used esiRNA. esiRNA has the advantage of providing successful reduction in protein expression while having reduced off-target effects (Kittler et al., 2007). We find that treatment with esiRNA against presenilin-1 or presenilin-2 leads to an increase in LC3-II but do not have any alteration in LC3-I or beclin1 (Fig. 2A,B). The lack of change in beclin1 expression is in contrast to the single knock-out profile, indicating that a buildup of LC3-II is one of the first events on autophagy upon presenilin loss. Additionally, these data make it unlikely that alterations in autophagy-related proteins from RNAi knockdown are attributable to off-target effects but instead are attributable to reduction in presenilin expression. These data provide strong evidence that presenilins are important for autophagy in multiple cell types, including in a neuron-like cell type.
Figure 2.

esiRNA mediated knockdown of presenilin in neuroblastomas leads to a buildup of LC3-II. A, B, esiRNA was used to knockdown levels of PS1 and PS2 in SHSY5Y neuroblastomas. Both treatments led to an increase in LC3-II, whereas LC3-I and beclin1 were unaltered. Quantification of Western blots in A was performed by densiometric analysis and is presented as percentage of control, normalized to actin. Error bars represent ± SEM; n = 3–4; one-way ANOVA analysis, post hoc comparison Tukey's honestly significant difference (HSD) test, *p < 0.05.
Presenilin-mediated reduction of beclin1 does not cause buildup of autophagosomes
Given the consistent but unusual finding that presenilin loss reduces beclin1 while increasing levels of LC3-II, we determined whether lowering beclin1 causes LC3-II levels to increase. Hence, we used a lentiviral shRNA approach to stably knockdown beclin1 levels in wild-type fibroblasts and found that reducing beclin1 causes a decrease in LC3-I and LC3-II (Fig. 3A,B), which is consistent with previous findings (Zeng et al., 2006). Therefore, we conclude that the reduction in beclin1 that occurs with presenilin loss is not leading to their increased in LC3-II and autophagosomes. Most likely, presenilin loss is leading to dysfunction in a downstream step of autophagy and could be leading to a compensatory decrease in beclin1.
Figure 3.

Knockdown of beclin1 does not cause a buildup of LC3-II. A, B, shRNA was used to stably knockdown beclin1 in wild-type mouse embryonic fibroblasts. Beclin1 reduction leads to reduction in LC3-I and LC3-II as shown by immunoblot. Quantification of Western blots in A was performed by densiometric analysis and is presented as percentage of control, normalized to actin. Error bars represent ± SEM; n = 4; unpaired Student's t test, *p < 0.05.
Autophagy-mediated proteolyisis is decreased after loss of presenilin despite increased autophagosomes
Increases in autophagosomes can indicate either an elevation in autophagic activity or a disruption of autophagic degradation late in the pathway at the lysosome, causing a backup of autophagosomes. Consequently, we monitored the breakdown of long-lived proteins, considered a substrate of autophagy, by pulse-chase assays, as a means of gauging general autophagic activity. As a control, we first confirmed that high levels of methionine necessary during the chase period do not inhibit autophagy during the time frame of the assay (data not shown). Next, we observed radiolabeled soluble, long-lived proteins after a 4 h chase period. Longer chase time points were not used because we found evidence that they began to cause autophagy inhibition attributable to the high levels of methionine. We found a modest but significant increase in radiolabeled proteins in presenilin-null mouse embryonic fibroblasts compared with controls, whereas differences in the initial amount of labeled proteins were not found (Fig. 4A,B). To be sure of this result, we then repeated this experiment using radiolabeled [H3]l-valine and measured the percentage proteolysis over time. We found a significant reduction in proteolysis in the PSDKO cells compared with control cells at every time point measured (Fig. 4C), confirming the results of the previous assay. Additionally, we treated the cells with rapamycin, an activator of autophagy, and found that, although control cells had an increase in proteolysis because of treatments, the PSDKO cells did not (Fig. 4D). These data are consistent with a decreased ability of presenilin-null fibroblasts to breakdown long-lived proteins, even when autophagy is pharmacologically activated, most likely because of malfunction in downstream steps of the autophagic pathway.
Figure 4.

Loss of presenilins decreases proteolysis of long-lived proteins. A, B, PSDKO cells have decreased proteolysis for long-lived proteins shown by radiolabeled [S35]l-methionine. Quantification of radiographs in E was performed by densiometric analysis and is presented as percentage of control. Error bars represent ± SEM; n = 3; unpaired Student's t test, *p < 0.05. Con, Control; DKO, double knock-out. C, PSDKO fibroblasts again show a decrease in proteolysis for long-lived proteins shown by radiolabeled [H3]l-valine measured by scintillation counter. Results are presented as percentage proteolysis. Error bars represent ± SEM; n = 4; unpaired Student's t test, *p < 0.05. D, PSDKO fibroblasts do not have an increase in proteolysis with rapamycin treatment (0.2 μm, 12 h time point). Error bars represent ± SEM; n = 4; one-way ANOVA, post hoc comparison Tukey's HSD, *p < 0.05.
No changes in the ubiquitin–proteasome system in presenilin-null cells
An alternate explanation to account for the decreased proteolysis but the increased autophagosomes in presenilin-null fibroblasts would be a deficiency in the other major degradation pathway in eukaryotic cells, the ubiquitin–proteasome system (UPS). Although the pulse chase, by design, labeled long-lived proteins, it is still possible for changes in UPS activity to affect the results. Because there is much crosstalk between autophagy and the UPS, one might expect to observe a disrupted UPS in addition to disrupted autophagy (Korolchuk et al., 2010).
To determine whether presenilin knock-out cells have alterations in the proteasome, we used Western blotting and immunohistochemical analysis to monitor levels and localization of ubiquitinated proteins, whose levels change if UPS activity is altered (Sun et al., 2006). We could not detect any alterations in ubiquitinated proteins between control and PSDKO mouse embryonic fibroblasts by Western blot (100 ± 11.88 vs 76.88 ± 17.56, mean ± SEM) or with immunostaining, indicating that the UPS is unimpaired (Fig. 5A–C).
Figure 5.

No UPS dysfunction in presenilin-null cells. A, No differences in ubiquitinated proteins by immunoblot were apparent between wild-type [control (Con)] and presenilin-null [double knock-out (DKO)] fibroblasts (100 ± 11.88 vs 76.88 ± 17.56). Quantification of Western blots was performed by densiometric analysis and is presented as percentage of control, normalized to actin. n = 4; unpaired Student's t test. B, C, Confocal immunofluorescent microscopy reveals no difference in ubiquitinated proteins in wild-type (control) and PSDKO fibroblasts. Scale bar, 29 μm. D, E, Transfection with the proteasome reporter ZsProsensor shows no difference in fluorescence intensity between wild-type and presenilin-null cells by confocal microscopy. F, To assess proteasome activity, lysates of wild-type (control) and presenilin-null fibroblasts were exposed to three proteasome substrates (LLVY-AMC for the chymotrypsin-like, VGR-AMC for the trypsin-like, and LLE-AMC for the PGPH/caspase-like activity). AMC release was measured and rate of release was quantified as AFU/90 s, revealing no significant differences in proteasome activities between the two cell types. Error bars represent ± SEM; n = 3; unpaired Student's t test.
We next measured proteasome function using fluorogenic substrates of the proteasome, by transiently transfecting cells with ZsProSensor, which contains the mouse ornithine decarboxylase degradation domain (MODC d410) fused to green fluorescent reef coral protein (Zoanthus species) (Hoyt et al., 2003). PSDKO cells displayed no difference in ZsProsensor fluorescence compared with control cells (Fig. 5D,E). After that, we used a fluorogenic proteasome activity assay for the three major proteasome activities, chymotrypsin-like, trypsin-like, and caspase-like activities, and found no difference between control and presenilin-null mouse embryonic fibroblasts in any of these proteasome activities (Fig. 5F). Together, these results indicate that the buildup of long-lived proteins in presenilin-null cells is not attributable to a deficient UPS system. Therefore, presenilin loss results in a selective disruption in autophagy, whereas the proteasome remains unaffected.
Inhibition of the γ-secretase complex does not alter autophagy-related proteins
Presenilins are a critical part of the γ-secretase complex, mediating the cleavage of many intramembrane proteins (Selkoe and Wolfe, 2007). Presenilins also have functions that do not rely on their γ-secretase activity and are therefore called γ-secretase independent. Because presenilin deletion disrupts γ-secretase activity, a pharmacological approach had to be used to determine whether the role of presenilins in autophagy is γ-secretase dependent or independent. Toward this end, we treated wild-type fibroblasts with two different types of γ-secretase inhibitors for 48 h and monitored the impact of several critical autophagy-related markers by Western blot. Notably, we found no difference in LC3 or beclin1 levels between treated and untreated cells, although there was a buildup of APP C-terminal fragments as shown by CT20 immunoblot (Fig. 6A,B). To confirm these results, we performed the same treatments on N2A mouse neuroblastomas for 48 and 96 h. Once more, we found no difference between treated and untreated cells in levels of LC3 or beclin1 (Fig. 6C–F). We confirmed inhibition of γ-secretase activity attributable to inhibitor treatment through ELISAs for Aβ40 and Aβ42. Both inhibitors caused a significant decrease in both species of Aβ, demonstrating successful inhibition of the γ-secretase complex (Fig. 6G,H). These results are in conjunction with previous findings that γ-secretase inhibitors are not able to replicate the enlargement of perinuclear organelles seen with presenilin loss (Wilson et al., 2004).
Figure 6.

Inhibiting γ-secretase activity has no effect on the levels of key autophagic-related proteins. Two different γ-secretase inhibitors [inhibitor IX at 500 nm (IX) and inhibitor X at 200 nm (X)] were used to assess their effects on key autophagy-related proteins. A, B, Forty-eight hour γ-secretase inhibitor treatments in control fibroblasts had no effect on levels of LC3 and beclin1 by immunoblot despite a buildup of APP C-terminal fragments. Quantification of Western blots in A was performed by densiometric analysis and is presented as percentage of control, normalized to actin. Error bars represent ± SEM; n = 3; one-way ANOVA. Veh, Vehicle. C–F, γ-Secretase inhibitors have no effect on LC3 or beclin1 after 48 (C, D) and 96 (E, F) hours of treatment in N2A cells as shown by Western blot. Quantification of Western blots in C and E was performed by densiometric analysis and is presented as percentage of control, normalized to actin. Error bars represent ± SEM; n = 3–4; one-way ANOVA. G, H, Inhibitor concentrations used in A–F at 48 h (G) and 96 h (H) of treatment inhibit production of both Aβ40 and Aβ42 determined by ELISA. Error bars represent ± SEM; n = 4; one-way ANOVA. I, J, Both wild-type (WT) PS1 and a dominant-negative PS1, ΔTM1-2, were able to partially rescue the effect of presenilin-1 knock-out on levels of LC3-II. Quantification of Western blots in I was performed by densiometric analysis and is presented as percentage of control, normalized to actin. Error bars represent ± SEM; n = 3–4; one-way ANOVA, post hoc comparison Tukey's HSD, *p < 0.05.
Although these data point to a γ-secretase-independent function of presenilins in autophagy, we wanted to further test this hypothesis by attempting to rescue the effect of presenilin knock-out with expression of a γ-secretase dominant-negative presenilin. Therefore, we transfected pcDNA, wild-type PS1, or ΔTM1-2 PS1 into PS1KO MEFs. ΔTM1-2 PS1 expression causes a dominant-negative effect on γ-secretase activity (Murphy et al., 2000). Expression of wild-type PS1 and ΔTM1-2 PS1 led to a slight but significant decrease in LC3-II but did not have an effect on beclin1 (Fig. 6I,J). These data are in line with previous evidence that the dominant-negative D257A presenilin-1 mutant can rescue telencephalin accumulations in presenilin-1 knock-out neurons (Esselens et al., 2004). Based on these findings, we conclude that the resulting disruption in autophagy from the genetic ablation of presenilin is not attributable to the loss of γ-secretase activity but is rather attributable to a γ-secretase-independent function of presenilins.
Rapamycin increases autophagosomes in both control and PSDKO fibroblasts
Presenilin-null mouse embryonic fibroblasts have a buildup of autophagosomes as reflected by increases in LC3-II and increases in EGFP–LC3 puncta, and, because of the decreased proteolysis, this is most likely a result of dysfunction late in the autophagic pathway. We considered whether presenilin-null cells could still actively increase autophagosomes, despite being defective in autophagy, by using rapamycin. Rapamycin is a potent inhibitor of mTOR, which is a negative regulator of autophagy. To induce autophagy, we treated control and presenilin-null fibroblasts with rapamycin (0.2 μm, 24 h) and detected an increase in LC3-II in control cells, as expected, and a trend toward an increase in LC3-II in presenilin-null cells by Western blotting (p = 0.057) (Fig. 7A,B). Rapamycin treatment also caused an increase in LysotrackerRed staining (Fig. 7C–F) and EGFP–LC3 puncta (Fig. 7G–J) in both control and presenilin-null mouse embryonic fibroblasts. These results indicate that presenilin-null cells can still actively increase autophagosomes and lysosomes but that they are not functioning properly to efficiently clear proteins through the lysosome.
Figure 7.

Rapamycin further increases autophagosomes and lysosomes in presenilin-null cells. A, B, Rapamycin treatment leads to increased LC3-II levels in both control and presenilin-null cells. Quantification of Western blots in A was performed by densiometric analysis and is presented as percentage of control, normalized to actin. Error bars represent ± SEM; n = 3; one-way ANOVA, post hoc comparison Tukey's HSD, *p < 0.05. Con, Control; DKO, double knock-out; Rap, rapamycin; Veh, vehicle. C–F, Control and presenilin-null cells treated with rapamycin have increased LysotrackerRed staining compared with vehicle-treated cells. Scale bars, 14 μm. G–J, Control and presenilin-null fibroblasts treated with rapamycin have increased EGFP–LC3 puncta compared with vehicle treated. Scale bars, 35 μm.
Lysosomal inhibitors fail to increase LC3-II in PSDKO fibroblasts
The data thus far suggest that genetic ablation of presenilin leads to autophagic dysfunction late in the pathway, at the lysosome. To confirm this hypothesis, we treated the cells with the vacuolar ATPase inhibitor bafilomycinA1 and the microtubule-destabilizing drug nocodazole, both of which will inhibit fusion of autophagosome to lysosome (Yamamoto et al., 1998; Köchl et al., 2006; Klionsky et al., 2008). Cells with autophagic dysfunction at the lysosome will have a buildup in autophagosomes, and inhibition at the lysosome will not cause an additional increase in LC3-II/autophagosomes because the downstream steps of the system are already not functioning properly. Importantly, we find that presenilin-null mouse embryonic fibroblasts do not have an additional increase in LC3-II when treated with bafilomycinA1 or nocodazole (Fig. 8A–D). These data lead us to conclude a novel function of presenilins in autophagy at the level of autophagosome–lysosome interaction or lysosome function.
Figure 8.

Lysosomal inhibitors increase LC3-II in wild-type but not presenilin-null cells. A, B, Immunoblot analysis of wild-type and PSDKO fibroblasts reveals that treatment with the lysosomal inhibitor bafiloymycinA1 (100 nm) increases LC3-II only in control but not presenilin-null cells. BafA1, BafiloymycinA1; Con, control; DKO, double knock-out; Veh, vehicle. C, D, Immunoblot analysis of wild-type and PSDKO fibroblasts with another lysosomal inhibitor, nocodazole (Noc; 50 μm) also increases LC3-II only in control but not presenilin-null cells. B, D, Quantification of Western blots in A and C was performed with densiometric analysis and is presented as percentage of control, normalized to actin. Error bars represent ± SEM; n = 3; unpaired Student's t test, *p < 0.05.
Discussion
Presenilins are ubiquitous, intramembrane proteins that are known to have crucial functions in many cellular processes. Here we report the finding that endogenous, wild-type presenilins are critical mediators of cellular autophagy. Genetically ablating presenilins, together or independently, alters many key autophagic proteins and results in abnormal buildup in autophagosomes. Notably, increases in LC3-II, indicating a buildup of autophagosomes, were replicated with presenilin knockdown in neuroblastoma cells, demonstrating a role for presenilins in autophagy in multiple cell types. We found that presenilin-null cells have decreased proteolysis of long-lived proteins, even when autophagy is pharmacologically induced, suggesting that they have a buildup of autophagosomes as a result of dysfunction in autophagy after autophagosome completion. This conclusion was validated through the use of lysosomal inhibitors that did not cause additional buildup of autophagosomes in presenilin-null cells despite the ability of these cells to increase autophagosomes as shown with rapamycin treatment. Also, we found this role of presenilin to be γ-secretase independent.
Autophagy is a broad cellular process by which cytosolic contents are delivered to the lysosome for degradation. Subcategories of autophagy described in mammalian cells include microautophagy, chaperone-mediated autophagy, and macroautophagy. Macroautophagy involves the sequestration of cytosolic contents by a double-membrane structure and degradation of its contents after fusion with a lysosome and is the type of autophagy relevant to this work. Autophagy is known to play an important role in many processes, such as long-lived protein degradation, organelle degradation, aging, neurodegeneration, adaptation to cellular stress, and microbial infection (Mortimore and Pösö, 1987; Ravikumar et al., 2002; Berger et al., 2006; Hara et al., 2006; Komatsu et al., 2006; Deretic and Levine, 2009; Salminen and Kaarniranta, 2009).
Presenilins are very important proteins in maintaining proper cellular and neuronal health, with even conditional forebrain double knock-out leading to neurodegeneration (Feng et al., 2004; Chen et al., 2008). Presenilin biology is also directly related to neurodegenerative disease as mutations within presenilin can alter γ-secretase function and lead to FAD (Borchelt et al., 1996, 1997; Mann et al., 1996; Gómez-Isla et al., 1999). These same mutations also lead to exacerbated lysosomal pathology (Cataldo et al., 2004). One could postulate that their role in autophagy instigates the early age of onset found in FAD because of changes in how neurons degrade misfolded or unwanted proteins. Interestingly, presenilin mutations are also found in some cases of frontotemporal dementia, but the result of these mutations to the disease state are, as of now, unknown (Hutton, 2004; Mendez and McMurtray, 2006; Zekanowski et al., 2006; Bernardi et al., 2009). It is possible that these mutations also lead to altered autophagy and contribute to the onset and/or progression of the disease. In addition, common features of many neurodegenerative diseases include protein aggregation and dysfunction in autophagy, so it is possible that presenilins act in many neurodegenerative diseases through this role.
Presenilins have been implicated in autophagy function previously. FAD presenilin mutations lead to alterations in lysosomal pathology, indicating changes in autophagy (Cataldo et al., 2004). Also, presenilin-1 knock-out in neurons causes a buildup of telencephalin, α-synuclein, and β-synuclein within degradative organelles (Esselens et al., 2004; Wilson et al., 2004). In addition, presenilin-1 levels increase when autophagy is inhibited, demonstrating that presenilin expression or degradation is connected to autophagy function (Ohta et al., 2010). Most recently, presenilin-1 knock-out blastocysts and presenilin-1-deficient mouse brains were found to be defective in autophagy through impairment in lysosome acidification (Lee et al., 2010). They also found acidification deficits in fibroblasts of patients with FAD. In this work, we present findings that endogenous, wild-type preseniliin-1 and presenilin-2 are important for overall degradation through autophagy in multiple cell types and indicate that presenilins are key regulators of autophagolysosome formation or lysosome function.
A key alteration found in presenilin knock-out cells is an increase in autophagosomes, demonstrated through increases in LC3-II and EGFP–LC3 puncta. Traditionally, increases in LC3 are used to indicate heightened autophagy because autophagosomes increase when autophagy is activated. Given this finding, we wondered at what step in autophagy does presenilin knock-out exert its effect and what are the actual functional consequences of the increase in autophagosomes. We addressed the first question by examining proteins involved in autophagy induction and vesicle nucleation. Interestingly, although activated mTOR is reduced reflecting an increase in autophagy induction, the proteins part of the PI3K complex III, beclin1 and UVRAG, are reduced. Both single and double presenilin knock-outs have the same reduction in beclin1 whereas LC3-II is increased. Increases in beclin1 expression will cause activation of autophagy (Liang et al., 1999; Spencer et al., 2009), a result that is at odds with our findings. However, during disease states when autophagy is dysfunctional, there are increases in autophagosomes, but beclin1 levels are reduced (Pickford et al., 2008). Hence, we propose that presenilin loss leads to a disease state of autophagy in which beclin1 levels are reduced but autophagosome numbers are increased as a result of dysfunction at the lysosome.
Importantly, our data confirm a role for presenilins in autophagy in multiple cell types. We found that even transient reduction of presenilin-1 or presenilin-2 was able to cause a buildup of LC3-II. These results indicate that both presenilins function in autophagy in multiple cell types. Our data provide the first evidence of a role for presenilin-2 in autophagy.
To determine the functional consequences of autophagosome buildup in presenilin knock-out fibroblasts, we examined the degradation of long-lived proteins. These results confirmed our hypothesis that presenilin loss leads to a disease state of autophagy as a significant decrease in the breakdown of long-lived proteins was observed. Therefore, presenilin loss does not completely stop flux through the autophagic system but instead causes the system to become inefficient by disrupting autophagy after autophagosome formation.
Given the autophagic dysfunction in presenilin knock-out cells, we wanted to confirm that the dysfunction was occurring after autophagosome formation. Through rapamycin treatment, presenilin knock-out cells show ability to actively increase autophagosomes, suggesting that autophagy up to autophagosome formation is functionally able in these cells. Inhibitors of the lysosome bafilomycinA1 and nocodazole do not cause an additional increase in LC3-II to show that presenilin knock-out does, in fact, result in dysfunction in autophagy at the lysosome. With these results, presenilins could either be necessary for proper autophagolysosome formation or for proper lysosome function. In conjunction with this finding, telencephalin accumulations in presenilin-1 knock-out neurons are not acidified nor do they have lysosomal components, also demonstrating a possible dysfunction in fusion (Esselens et al., 2004). The findings of Lee et al. (2010) demonstrated a deficit in lysosomal acidification with presenilin-1 knock-out. In contrast to this, we find increased LysotrackerRed staining in PSDKO cells, indicating no deficits in lysosomal acidification, despite robust impairments in autophagic degradation. This discrepancy may arise as a result of the different cell types used or the fact that both presenilins are absent rather than just PS1. Our data raise the possibility that presenilins might function at the lysosome in another manner besides lysosome acidification. Although there may be multiple mechanisms at work, it is clear that both presenilins are required for efficient autophagic degradation and that our combined works independently highlight the important role of γ-secretase-independent presenilin regulation of protein clearance.
Many presenilin interacting proteins have been identified, including the other necessary members of the γ-secretase quartet. A study by the De Strooper group purified tagged presenilins from PSDKO MEFs and characterized the presenilin interactome (Wakabayashi et al., 2009). Of note, they confirmed our previous finding of presenilin interacting with the endoplasmic reticulum calcium pump SERCA (sarcoendoplasmic reticulum Ca2+-ATPase) (Green et al., 2008) but also identified several proteins involved in membrane trafficking, amino acid transport, and vacuolar ATPase subunits. It could be that these any groups of proteins are involved in the regulation of autophagic degradation by presenilin.
Presenilin knock-out will result in loss of γ-secretase activity (Herreman et al., 2000). γ-Secretase has many substrates and therefore many downstream cellular functions (Parks and Curtis, 2007; Selkoe and Wolfe, 2007), a possible mechanism by which presenilin loss causes dysfunctional autophagy. We find that γ-secretase inhibitors yield no effect on autophagic protein levels to indicate that the function of presenilins in autophagy is γ-secretase independent. This result is in conjunction with previous findings that telencephalin and synuclein accumulations found in presenilin-1 knock-out neurons is not replicated with γ-secretase inhibitors and can be rescued with expression of D257A PS1 mutant (Esselens et al., 2004; Wilson et al., 2004).
Our data combined with others show a role for presenilins in the regulation of protein turnover through the autophagosome–lysosome system by mediating autophagosome–lysosome interaction or lysosome function. Our data indicate a dysfunction in the breakdown of overall long-lived proteins with presenilin knock-out and, unlike other studies, demonstrate a role for both presenilin-1 and presenilin-2 in autophagy.
Footnotes
This work is supported by National Institutes of Health Grants 021928 and AG027544. We thank Drs. Joan Steffan, Masashi Kitasawa, Matthew Blurton-Jones, and K. C. Walls for their helpful guidance.
References
- Baki L, Neve RL, Shao Z, Shioi J, Georgakopoulos A, Robakis NK. Wild-type but not FAD mutant presenilin-1 prevents neuronal degeneration by promoting phosphatidylinositol 3-kinase neuroprotective signaling. J Neurosci. 2008;28:483–490. doi: 10.1523/JNEUROSCI.4067-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauvy C, Meijer AJ, Codogno P. Autophagy in mammalian systems, Part B. New York: Elsevier; 2009. [Google Scholar]
- Berger Z, Ravikumar B, Menzies FM, Oroz LG, Underwood BR, Pangalos MN, Schmitt I, Wullner U, Evert BO, O'Kane CJ, Rubinsztein DC. Rapamycin alleviates toxicity of different aggregate-prone proteins. Hum Mol Genet. 2006;15:433–442. doi: 10.1093/hmg/ddi458. [DOI] [PubMed] [Google Scholar]
- Bernardi L, Tomaino C, Anfossi M, Gallo M, Geracitano S, Costanzo A, Colao R, Puccio G, Frangipane F, Curcio SA, Mirabelli M, Smirne N, Iapaolo D, Maletta RG, Bruni AC. Novel PSEN1 and PGRN mutations in early-onset familial frontotemporal dementia. Neurobiol Aging. 2009;30:1825–1833. doi: 10.1016/j.neurobiolaging.2008.01.005. [DOI] [PubMed] [Google Scholar]
- Borchelt DR, Thinakaran G, Eckman CB, Lee MK, Davenport F, Ratovitsky T, Prada CM, Kim G, Seekins S, Yager D, Slunt HH, Wang R, Seeger M, Levey AI, Gandy SE, Copeland NG, Jenkins NA, Price DL, Younkin SG, Sisodia SS. Familial Alzheimer's disease-linked presenilin 1 variants elevate Abeta1-42/1-40 ratio in vitro and in vivo. Neuron. 1996;17:1005–1013. doi: 10.1016/s0896-6273(00)80230-5. [DOI] [PubMed] [Google Scholar]
- Borchelt DR, Ratovitski T, van Lare J, Lee MK, Gonzales V, Jenkins NA, Copeland NG, Price DL, Sisodia SS. Accelerated amyloid deposition in the brains of transgenic mice coexpressing mutant presenilin 1 and amyloid precursor proteins. Neuron. 1997;19:939–945. doi: 10.1016/s0896-6273(00)80974-5. [DOI] [PubMed] [Google Scholar]
- Burman C, Ktistakis NT. Regulation of autophagy by phosphatidylinositol 3-phosphate. FEBS Lett. 2010;584:1302–1312. doi: 10.1016/j.febslet.2010.01.011. [DOI] [PubMed] [Google Scholar]
- Cataldo AM, Peterhoff CM, Schmidt SD, Terio NB, Duff K, Beard M, Mathews PM, Nixon RA. Presenilin mutations in familial Alzheimer disease and transgenic mouse models accelerate neuronal lysosomal pathology. J Neuropathol Exp Neurol. 2004;63:821–830. doi: 10.1093/jnen/63.8.821. [DOI] [PubMed] [Google Scholar]
- Chen Q, Nakajima A, Choi SH, Xiong X, Tang YP. Loss of presenilin function causes Alzheimer's disease-like neurodegeneration in the mouse. J Neurosci Res. 2008;86:1615–1625. doi: 10.1002/jnr.21601. [DOI] [PubMed] [Google Scholar]
- Cuervo AM, Dice JF. Unique properties of lamp2a compared to other lamp2 isoforms. J Cell Sci. 2000;113:4441–4450. doi: 10.1242/jcs.113.24.4441. [DOI] [PubMed] [Google Scholar]
- Deretic V, Levine B. Autophagy, immunity, and microbial adaptations. Cell Host Microbe. 2009;5:527–549. doi: 10.1016/j.chom.2009.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esselens C, Oorschot V, Baert V, Raemaekers T, Spittaels K, Serneels L, Zheng H, Saftig P, De Strooper B, Klumperman J, Annaert W. Presenilin 1 mediates the turnover of telencephalin in hippocampal neurons via an autophagic degradative pathway. J Cell Biol. 2004;166:1041–1054. doi: 10.1083/jcb.200406060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng R, Wang H, Wang J, Shrom D, Zeng X, Tsien JZ. Forebrain degeneration and ventricle enlargement caused by double knockout of Alzheimer's presenilin-1 and presenilin-2. Proc Natl Acad Sci U S A. 2004;101:8162–8167. doi: 10.1073/pnas.0402733101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gómez-Isla T, Growdon WB, McNamara MJ, Nochlin D, Bird TD, Arango JC, Lopera F, Kosik KS, Lantos PL, Cairns NJ, Hyman BT. The impact of different presenilin 1 andpresenilin 2 mutations on amyloid deposition, neurofibrillary changes and neuronal loss in the familial Alzheimer's disease brain: evidence for other phenotype-modifying factors. Brain. 1999;122:1709–1719. doi: 10.1093/brain/122.9.1709. [DOI] [PubMed] [Google Scholar]
- Green KN, Demuro A, Akbari Y, Hitt BD, Smith IF, Parker I, LaFerla FM. SERCA pump activity is physiologically regulated by presenilin and regulates amyloid beta production. J Cell Biol. 2008;181:1107–1116. doi: 10.1083/jcb.200706171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanada T, Noda NN, Satomi Y, Ichimura Y, Fujioka Y, Takao T, Inagaki F, Ohsumi Y. The Atg12-Atg5 conjugate has a novel E3-like activity for protein lipidation in autophagy. J Biol Chem. 2007;282:37298–37302. doi: 10.1074/jbc.C700195200. [DOI] [PubMed] [Google Scholar]
- Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, Yokoyama M, Mishima K, Saito I, Okano H, Mizushima N. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature. 2006;441:885–889. doi: 10.1038/nature04724. [DOI] [PubMed] [Google Scholar]
- Herreman A, Serneels L, Annaert W, Collen D, Schoonjans L, De Strooper B. Total inactivation of gamma-secretase activity in presenilin-deficient embryonic stem cells. Nat Cell Biol. 2000;2:461–462. doi: 10.1038/35017105. [DOI] [PubMed] [Google Scholar]
- Hoyt MA, Zhang M, Coffino P. Ubiquitin-independent mechanisms of mouse ornithine decarboxylase degradation are conserved between mammalian and fungal cells. J Biol Chem. 2003;278:12135–12143. doi: 10.1074/jbc.M211802200. [DOI] [PubMed] [Google Scholar]
- Hutton M. Presenilin mutations associated with fronto-temporal dementia. Ann Neurol. 2004;55:604–606. doi: 10.1002/ana.20103. [DOI] [PubMed] [Google Scholar]
- Itakura E, Kishi C, Inoue K, Mizushima N. Beclin 1 forms two distinct phosphatidylinositol 3-kinase complexes with mammalian Atg14 and UVRAG. Mol Biol Cell. 2008;19:5360–5372. doi: 10.1091/mbc.E08-01-0080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung CH, Ro SH, Cao J, Otto NM, Kim DH. mTOR regulation of autophagy. FEBS Lett. 2010;584:1287–1295. doi: 10.1016/j.febslet.2010.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, Kominami E, Ohsumi Y, Yoshimori T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000;19:5720–5728. doi: 10.1093/emboj/19.21.5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kittler R, Surendranath V, Heninger AK, Slabicki M, Theis M, Putz G, Franke K, Caldarelli A, Grabner H, Kozak K, Wagner J, Rees E, Korn B, Frenzel C, Sachse C, Sönnichsen B, Guo J, Schelter J, Burchard J, Linsley PS, et al. Genome-wide resources of endoribonuclease-prepared short interfering RNAs for specific loss-of-function studies. Nat Methods. 2007;4:337–344. doi: 10.1038/nmeth1025. [DOI] [PubMed] [Google Scholar]
- Klionsky DJ, Cuervo AM, Seglen PO. Methods for monitoring autophagy from yeast to human. Autophagy. 2007;3:181–206. doi: 10.4161/auto.3678. [DOI] [PubMed] [Google Scholar]
- Klionsky DJ, Elazar Z, Seglen PO, Rubinsztein DC. Does bafilomycin A1 block the fusion of autophagosomes with lysosomes? Autophagy. 2008;4:849–950. doi: 10.4161/auto.6845. [DOI] [PubMed] [Google Scholar]
- Köchl R, Hu XW, Chan EYW, Tooze SA. Microtubules facilitate autophagosome formation and fusion of autophagosomes with endosomes. Traffic. 2006;7:129–145. doi: 10.1111/j.1600-0854.2005.00368.x. [DOI] [PubMed] [Google Scholar]
- Koike MA, Green KN, Blurton-Jones M, Laferla FM. Oligemic hypoperfusion differentially affects tau and amyloid-β. Am J Pathol. 2010;177:300–310. doi: 10.2353/ajpath.2010.090750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komatsu M, Waguri S, Chiba T, Murata S, Iwata J, Tanida I, Ueno T, Koike M, Uchiyama Y, Kominami E, Tanaka K. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature. 2006;441:880–884. doi: 10.1038/nature04723. [DOI] [PubMed] [Google Scholar]
- Korolchuk VI, Menzies FM, Rubinsztein DC. Mechanisms of cross-talk between the ubiquitin-proteasome and autophagy-lysosome systems. FEBS Lett. 2010;584:1393–1398. doi: 10.1016/j.febslet.2009.12.047. [DOI] [PubMed] [Google Scholar]
- Kovacs DM, Fausett HJ, Page KJ, Kim TW, Moir RD, Merriam DE, Hollister RD, Hallmark OG, Mancini R, Felsenstein KM, Hyman BT, Tanzi RE, Wasco W. Alzheimer-associated presenilins 1 and 2: neuronal expression in brain and localization to intracellular membranes in mammalian cells. Nat Med. 1996;2:224–229. doi: 10.1038/nm0296-224. [DOI] [PubMed] [Google Scholar]
- LaFerla FM. Calcium dyshomeostasis and intracellular signalling in Alzheimer's disease. Nat Rev Neurosci. 2002;3:862–872. doi: 10.1038/nrn960. [DOI] [PubMed] [Google Scholar]
- Lai MT, Chen E, Crouthamel MC, DiMuzio-Mower J, Xu M, Huang Q, Price E, Register RB, Shi XP, Donoviel DB, Bernstein A, Hazuda D, Gardell SJ, Li YM. Presenilin-1 and presenilin-2 exhibit distinct yet overlapping gamma-secretase activities. J Biol Chem. 2003;278:22475–22481. doi: 10.1074/jbc.M300974200. [DOI] [PubMed] [Google Scholar]
- Lanz TA, Himes CS, Pallante G, Adams L, Yamazaki S, Amore B, Merchant KM. The gamma-secretase inhibitor N-[N-(3,5-difluorophenacetyl)-L-alanyl]-S-phenylglycine t-butyl ester reduces A beta levels in vivo in plasma and cerebrospinal fluid in young (plaque-free) and aged (plaque-bearing) Tg2576 mice. J Pharmacol Exp Ther. 2003;305:864–871. doi: 10.1124/jpet.102.048280. [DOI] [PubMed] [Google Scholar]
- Lee JH, Yu WH, Kumar A, Lee S, Mohan PS, Peterhoff CM, Wolfe DM, Martinez-Vicente M, Massey AC, Sovak G, Uchiyama Y, Westaway D, Cuervo AM, Nixon RA. Lysosomal proteolysis and autophagy require presenilin 1 and are disrupted by Alzheimer-related PS1 mutations. Cell. 2010;141:1146–1158. doi: 10.1016/j.cell.2010.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang C, Feng P, Ku B, Dotan I, Canaani D, Oh BH, Jung JU. Autophagic and tumour suppressor activity of a novel Beclin1-binding protein UVRAG. Nat Cell Biol. 2006;8:688–699. doi: 10.1038/ncb1426. [DOI] [PubMed] [Google Scholar]
- Liang XH, Jackson S, Seaman M, Brown K, Kempkes B, Hibshoosh H, Levine B. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature. 1999;402:672–676. doi: 10.1038/45257. [DOI] [PubMed] [Google Scholar]
- Lindmo K, Brech A, Finley KD, Gaumer S, Contamine D, Rusten TE, Stenmark H. The PI 3-kinase regulator Vps15 is required for autophagic clearance of protein aggregates. Autophagy. 2008;4:500–506. doi: 10.4161/auto.5829. [DOI] [PubMed] [Google Scholar]
- Mann DM, Iwatsubo T, Cairns NJ, Lantos PL, Nochlin D, Sumi SM, Bird TD, Poorkaj P, Hardy J, Hutton M, Prihar G, Crook R, Rossor MN, Haltia M. Amyloid beta protein (Abeta) deposition in chromosome 14-linked Alzheimer's disease: predominance of Abeta42(43) Ann Neurol. 1996;40:149–156. doi: 10.1002/ana.410400205. [DOI] [PubMed] [Google Scholar]
- Mendez MF, McMurtray A. Frontotemporal dementia-like phenotypes associated with presenilin-1 mutations. Am J Alzheimer's Dis Other Demen. 2006;21:281–286. doi: 10.1177/1533317506290448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima N, Noda T, Yoshimori T, Tanaka Y, Ishii T, George MD, Klionsky DJ, Ohsumi M, Ohsumi Y. A protein conjugation system essential for autophagy. Nature. 1998;395:395–398. doi: 10.1038/26506. [DOI] [PubMed] [Google Scholar]
- Mortimore GE, Pösö AR. Intracellular protein catabolism and its control during nutrient deprivation and supply. Annu Rev Nutr. 1987;7:539–564. doi: 10.1146/annurev.nu.07.070187.002543. [DOI] [PubMed] [Google Scholar]
- Murphy MP, Uljon SN, Fraser PE, Fauq A, Lookingbill HA, Findlay KA, Smith TE, Lewis PA, McLendon DC, Wang R, Golde TE. Presenilin 1 regulates pharmacologically distinct gamma-secretase activities. Implications for the role of presenilin in gamma-secretase cleavage. J Biol Chem. 2000;275:26277–26284. doi: 10.1074/jbc.M002812200. [DOI] [PubMed] [Google Scholar]
- Noda T, Ohsumi Y. Tor, a phosphatidylinositol kinase homologue, controls autophagy in yeast. J Biol Chem. 1998;273:3963–3966. doi: 10.1074/jbc.273.7.3963. [DOI] [PubMed] [Google Scholar]
- Ohta K, Mizuno A, Ueda M, Li S, Suzuki Y, Hida Y, Hayakawa-Yano Y, Itoh M, Ohta E, Kobori M, Nakagawa T. Autophagy impairment stimulates PS1 expression and γ-secretase activity. Autophagy. 2010;6:345–352. doi: 10.4161/auto.6.3.11228. [DOI] [PubMed] [Google Scholar]
- Onodera J, Ohsumi Y. Autophagy is required for maintenance of amino acid levels and protein synthesis under nitrogen starvation. J Biol Chem. 2005;280:31582–31586. doi: 10.1074/jbc.M506736200. [DOI] [PubMed] [Google Scholar]
- Page RM, Baumann K, Tomioka M, Pérez-Revuelta BI, Fukumori A, Jacobsen H, Flohr A, Luebbers T, Ozmen L, Steiner H, Haass C. Generation of Abeta38 and Abeta42 is independently and differentially affected by familial Alzheimer disease-associated presenilin mutations and gamma-secretase modulation. J Biol Chem. 2008;283:677–683. doi: 10.1074/jbc.M708754200. [DOI] [PubMed] [Google Scholar]
- Palacino JJ, Berechid BE, Alexander P, Eckman C, Younkin S, Nye JS, Wolozin B. Regulation of amyloid precursor protein processing by presenilin 1 (PS1) and PS2 in PS1 knockout cells. J Biol Chem. 2000;275:215–222. doi: 10.1074/jbc.275.1.215. [DOI] [PubMed] [Google Scholar]
- Parks AL, Curtis D. Presenilin diversifies its portfolio. Trends Genet. 2007;23:140–150. doi: 10.1016/j.tig.2007.01.008. [DOI] [PubMed] [Google Scholar]
- Pasternak SH, Bagshaw RD, Guiral M, Zhang S, Ackerley CA, Pak BJ, Callahan JW, Mahuran DJ. Presenilin-1, nicastrin, amyloid precursor protein, and gamma-secretase activity are co-localized in the lysosomal membrane. J Biol Chem. 2003;278:26687–26694. doi: 10.1074/jbc.m304009200. [DOI] [PubMed] [Google Scholar]
- Pickford F, Masliah E, Britschgi M, Lucin K, Narasimhan R, Jaeger PA, Small S, Spencer B, Rockenstein E, Levine B, Wyss-Coray T. The autophagy-related protein beclin 1 shows reduced expression in early Alzheimer disease and regulates amyloid β accumulation in mice. J Clin Invest. 2008;118:2190–2199. doi: 10.1172/JCI33585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravikumar B, Duden R, Rubinsztein DC. Aggregate-prone proteins with polyglutamine and polyalanine expansions are degraded by autophagy. Hum Mol Genet. 2002;11:1107–1117. doi: 10.1093/hmg/11.9.1107. [DOI] [PubMed] [Google Scholar]
- Salminen A, Kaarniranta K. Regulation of the aging process by autophagy. Trends Mol Med. 2009;15:217–224. doi: 10.1016/j.molmed.2009.03.004. [DOI] [PubMed] [Google Scholar]
- Sarkar S, Ravikumar B, Floto RA, Rubinsztein DC. Rapamycin and mTOR-independent autophagy inducers ameliorate toxicity of polyglutamine-expanded huntingtin and related proteinopathies. Cell Death Differ. 2009;16:46–56. doi: 10.1038/cdd.2008.110. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ, Wolfe MS. Presenilin: running with scissors in the membrane. Cell. 2007;131:215–221. doi: 10.1016/j.cell.2007.10.012. [DOI] [PubMed] [Google Scholar]
- Spasic D, Tolia A, Dillen K, Baert V, De Strooper B, Vrijens S, Annaert W. Presenilin-1 maintains a nine-transmembrane topology throughout the secretory pathway. J Biol Chem. 2006;281:26569–26577. doi: 10.1074/jbc.M600592200. [DOI] [PubMed] [Google Scholar]
- Spencer B, Potkar R, Trejo M, Rockenstein E, Patrick C, Gindi R, Adame A, Wyss-Coray T, Masliah E. Beclin 1 gene transfer activates autophagy and ameliorates the neurodegenerative pathology in α-synuclein models of Parkinson's and Lewy body diseases. J Neurosci. 2009;29:13578–13588. doi: 10.1523/JNEUROSCI.4390-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun F, Anantharam V, Zhang D, Latchoumycandane C, Kanthasamy A, Kanthasamy AG. Proteasome inhibitor MG-132 induces dopaminergic degeneration in cell culture and animal models. Neurotoxicology. 2006;27:807–815. doi: 10.1016/j.neuro.2006.06.006. [DOI] [PubMed] [Google Scholar]
- Tandon A, Fraser P. The presenilins. Genome Biol. 2002;3:1–9. doi: 10.1186/gb-2002-3-11-reviews3014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakabayashi T, Craessaerts K, Bammens L, Bentahir M, Borgions F, Herdewijn P, Staes A, Timmerman E, Vandekerckhove J, Rubinstein E, Boucheix C, Gevaert K, De Strooper B. Analysis of the gamma-secretase interactome and validation of its association with tetraspanin-enriched microdomains. Nat Cell Biol. 2009;11:1340–1346. doi: 10.1038/ncb1978. [DOI] [PubMed] [Google Scholar]
- Wilson CA, Murphy DD, Giasson BI, Zhang B, Trojanowski JQ, Lee VM. Degradative organelles containing mislocalized alpha-and beta-synuclein proliferate in presenilin-1 null neurons. J Cell Biol. 2004;165:335–346. doi: 10.1083/jcb.200403061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia W, Zhang J, Ostaszewski BL, Kimberly WT, Seubert P, Koo EH, Shen J, Selkoe DJ. Presenilin 1 regulates the processing of beta-amyloid precursor protein C-terminal fragments and the generation of amyloid beta-protein in endoplasmic reticulum and Golgi. Biochemistry. 1998;37:16465–16471. doi: 10.1021/bi9816195. [DOI] [PubMed] [Google Scholar]
- Yamamoto A, Tagawa Y, Yoshimori T, Moriyama Y, Masaki R, Tashiro Y. Bafilomycin A1 prevents maturation of autophagic vacuoles by inhibiting fusion between autophagosomes and lysosomes in rat hepatoma cell line, H-4-II-E cells. Cell Struct Funct. 1998;23:33–42. doi: 10.1247/csf.23.33. [DOI] [PubMed] [Google Scholar]
- Yan Y, Flinn RJ, Wu H, Schnur RS, Backer JM. hVps15, but not Ca2+/CaM, is required for the activity and regulation of hVps34 in mammalian cells. Biochem J. 2009;417:747–755. doi: 10.1042/BJ20081865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, Huang J, Geng J, Nair U, Klionsky DJ. Atg22 recycles amino acids to link the degradative and recycling functions of autophagy. Mol Biol Cell. 2006;17:5094–5104. doi: 10.1091/mbc.E06-06-0479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu WH, Kumar A, Peterhoff C, Shapiro Kulnane L, Uchiyama Y, Lamb BT, Cuervo AM, Nixon RA. Autophagic vacuoles are enriched in amyloid precursor protein-secretase activities: implications for β-amyloid peptide over-production and localization in Alzheimer's disease. Int J Biochem Cell Biol. 2004;36:2531–2540. doi: 10.1016/j.biocel.2004.05.010. [DOI] [PubMed] [Google Scholar]
- Zekanowski C, Golan MP, Krzyśko KA, Lipczyńska-Łojkowska W, Filipek S, Kowalska A, Rossa G, Pepłońska B, Styczyńska M, Maruszak A, Religa D, Wender M, Kulczycki J, Barcikowska M, Kuźnicki J. Two novel presenilin 1 gene mutations connected with frontotemporal dementia-like clinical phenotype: genetic and bioinformatic assessment. Exp Neurol. 2006;200:82–88. doi: 10.1016/j.expneurol.2006.01.022. [DOI] [PubMed] [Google Scholar]
- Zeng X, Overmeyer JH, Maltese WA. Functional specificity of the mammalian Beclin-Vps34 PI 3-kinase complex in macroautophagy versus endocytosis and lysosomal enzyme trafficking. J Cell Sci. 2006;119:259–270. doi: 10.1242/jcs.02735. [DOI] [PubMed] [Google Scholar]