

Summary
Long-term maintenance and modification of synaptic strength involve the turnover of neurotransmitter receptors. Glutamate receptors are constitutively and acutely internalized, presumptively through clathrin-mediated receptor endocytosis. Here, we show that cpg2 is a brain-specific splice variant of the syne-1 gene that encodes a protein specifically localized to a postsynaptic endocytotic zone of excitatory synapses. RNAi-mediated CPG2 knockdown increases the number of postsynaptic clathrin-coated vesicles, some of which traffic NMDA receptors, disrupts the constitutive internalization of glutamate receptors, and inhibits the activity-induced internalization of synaptic AMPA receptors. Manipulating CPG2 levels also affects dendritic spine size, further supporting a function in regulating membrane transport. Our results suggest that CPG2 is a key component of a specialized postsynaptic endocytic mechanism devoted to the internalization of synaptic proteins, including glutamate receptors. The activity dependence and distribution of cpg2 expression further suggest that it contributes to the capacity for postsynaptic plasticity inherent to excitatory synapses.
Introduction
Excitatory synaptic transmission within the brain is largely mediated by ionotropic glutamate receptors. At glutamatergic synapses, α-amino-3-hydroxy-5-methyl-isoxazole-4-proprionic acid (AMPA) and N-methyl-D-aspartate (NMDA) receptors are densely clustered apposed to presynaptic terminals (Nusser, 2000), and changes in their numbers can alter postsynaptic sensitivity to glutamate release. Therefore, the maintenance of surface glutamate receptors is a critical aspect of neuronal function, and the modification of their numbers is a potential mechanism for regulating synaptic strength during plasticity (Malinow and Malenka, 2002).
A fundamental mechanism of maintaining and modifying the number of synaptic glutamate receptors is their internalization from the synaptic membrane. AMPA receptors undergo rapid constitutive internalization that is regulated by synaptic activity (Ehlers, 2000; Lin et al., 2000). For NMDA receptors, constitutive internalization in mature neurons is slow relative to AMPA receptors and not regulated by activity but is rapid in immature neurons (Ehlers, 2000; Lin et al., 2000; Roche et al., 2001). Although NMDA receptors are thought to be relatively stable during synaptic plasticity, certain stimuli can induce their acute internalization (Nong et al., 2003; Snyder et al., 2001). Mounting evidence suggests that the internalization of glutamate receptors is a primary mechanism of long-term depression (LTD) (Beattie et al., 2000; Lin et al., 2000; Man et al., 2000; Snyder et al., 2001; Wang and Linden, 2000), an electrophysiological paradigm for synaptic plasticity wherein a specific stimulus causes a decrease in synaptic strength (Bear and Malenka, 1994).
Glutamate receptor internalization is thought to occur through clathrin-mediated endocytosis (Beattie et al., 2000; Carroll et al., 1999; Ehlers, 2000; Lin et al., 2000; Man et al., 2000; Wang and Linden, 2000), a general mechanism for the internalization of proteins from the plasma membrane (Mousavi et al., 2004). However, the specific mechanisms of glutamate receptor internalization are not well understood. In cell lines, clathrin-mediated endocytosis occurs at discrete and stable “clathrin pit zones” on the membrane (Gaidarov et al., 1999; Santini et al., 2002; Scott et al., 2002). A similar endocytic zone, segregated from the postsynaptic density (PSD), functions at the postsynaptic side of excitatory synapses and is presumed to be the site of the internalization of synaptic proteins, including glutamate receptors (Blanpied et al., 2002). Although electron microscopy has shown the presence of clathrin-coated pits and vesicles in dendritic spines (Cooney et al., 2002; Petralia et al., 2003; Spacek and Harris, 1997; Toni et al., 2001), none have been shown to traffic glutamate receptors at postsynaptic sites.
Screens for plasticity-related genes have identified multiple transcripts that encode synaptic proteins, suggesting that genes induced by activity often function in normal synaptic processes (Nedivi, 1999). candidate plasticity gene 2 (cpg2) was isolated in a screen for transcripts upregulated by kainic acid-induced seizures in the rat dentate gyrus (Nedivi et al., 1993), and its expression is regulated during development and by sensory experience (Nedivi et al., 1996). cpg2 is a splice variant of the syne-1 gene, a large gene that encodes a protein with an actin binding domain at the N terminus and a nuclear transmembrane domain at the C terminus, separated by a long helical region (Starr and Han, 2003). The cpg2 transcript is derived from a portion of the separator region (Padmakumar et al., 2004), encodes a protein with homologies to dystrophin, and contains motifs predicting a structural function, including several spectrin repeats and coiled coils (Nedivi et al., 1996). Proteins with these motifs often play a central role in organizing protein complexes (Burkhard et al., 2001; Djinovic-Carugo et al., 2002).
Here, we show that cpg2 is expressed only in the brain and encodes a protein that localizes specifically to the postsynaptic endocytic zone of excitatory synapses. We present evidence that CPG2 is a critical component of the postsynaptic endocytic pathway that mediates both constitutive and activity-regulated glutamate receptor internalization. We hypothesize that CPG2 is a key component of a specialization that is devoted to the internalization of postsynaptic proteins at synapses capable of plasticity.
Results
Analysis of cpg2 Exon/Intron Structure and Transcript Distribution
A partial cpg2 cDNA isolated in a screen for activity-regulated genes (Nedivi et al., 1993) was used as a probe to obtain a full-length cpg2 clone from a rat dentate gyrus cDNA library (Nedivi et al., 1996). The full-length cDNA contains a 2.8 kb open reading frame that predicts a protein of 941 amino acids and is flanked by a 266 nucleotide 5′ untranslated region (UTR) and a 2.3 kb 3′UTR (Nedivi et al., 1996). When using the cpg2 cDNA sequence for database searches, we identified the 18 exons of the cpg2 coding region as corresponding to exons 16 to 33 of the syne-1 gene, with the syne-1 exon 34 contained in the cpg2 3′UTR (Figure 1A). A Northern blot of total RNA from rat cerebral cortex probed with a segment of the cpg2 coding sequence showed a single 5.9 kb transcript (Figure 1B), the approximate size of the longest cpg2 cDNA clone. Inspection of the cpg2 3′UTR revealed the likely sites of syne-1 early transcription and translation termination that generate the cpg2 transcript. The cpg2 3′UTR contains an unspliced intron between exons 33 and 34 of the syne-1 gene. Exon 34 is followed by a noncanonical polyadenylation hexamer, conserved in rats and humans, which likely serves as the site of cpg2 transcription termination (Figure 1C). The unspliced intron between exons 33 and 34 contains a stop codon, seven codons downstream of the end of exon 33, which serves as the translation termination site.
Figure 1. cpg2 Is a Brain-Specific Splice Variant of syne-1.
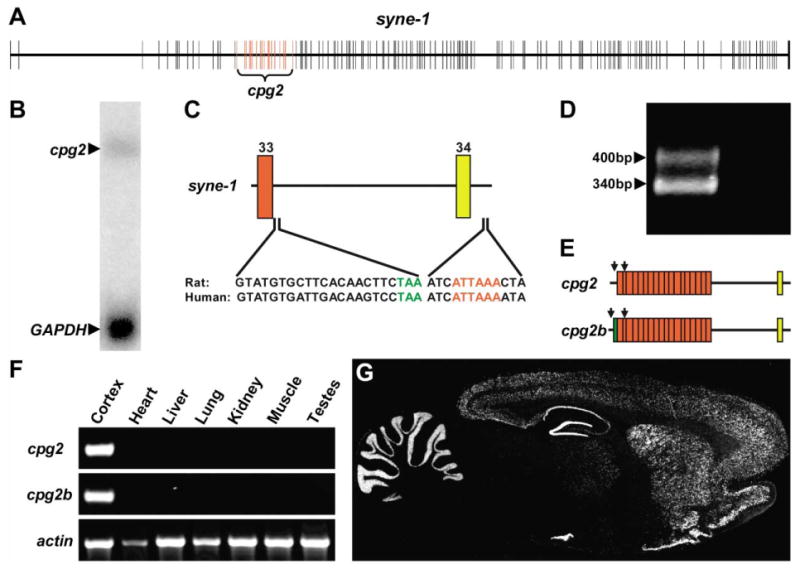
(A) Schematic of the syne-1 gene with exons marked by the vertical lines. Red lines indicate exons corresponding to the coding region of the original cpg2 transcript.
(B) Northern blot of rat brain total RNA probed with a 1 kb fragment of the cpg2 coding region reveals a 5.9 kb transcript.
(C) Schematic of the syne-1 genomic DNA sequence at the site of cpg2 transcript termination. Numbers identify the syne-1 exons. The final exon in the cpg2 coding region is marked by the red box, and a noncoding exon contained in the 3′UTR is marked in yellow. The intron between these exons is not spliced out in cpg2. Conserved rat and human sequences following the final coding exon ending with the translation stop codon (green) indicate the site of cpg2 translation termination. Sequence following the noncoding exon shows the polyadenylation hexamer (red), the site of cpg2 transcription termination.
(D) 5′RACE products of rat brain RNA using a primer in the first cpg2 coding exon, followed by PCR using nested primers in reactions with (left lane) or without (right lane) reverse transcriptase.
(E) Schematic of two cpg2 splice variants identified by sequencing of the 5′RACE fragments. The green box indicates an additional exon spliced to the 5′ end of the cpg2 transcript. Arrows indicate the locations of RT-PCR primers.
(F) Tissue RT-PCR from total RNA using primers specific to cpg2, cpg2b, and β-actin.
(G) In situ hybridization on a sagittal section through an adult rat brain using a probe from the cpg2 3′UTR.
To determine whether the original cpg2 cDNA contains the complete 5′UTR, we used 5′RACE to map the cpg2 mRNA start site and isolated two separate bands (Figure 1D). Subcloning and sequencing these bands identified two 5′ ends to the cpg2 transcript. One transcript (cpg2) corresponds to the original cpg2 cDNA isolated in the screen. A second transcript (cpg2b; accession number AY597251) contains an additional exon at the 5′ end of the cpg2 coding region (Figure 1E) encoding 24 N-terminal residues with no known structural domains. To determine the tissue distribution of the two cpg2 transcripts, we performed RT-PCR using primers that amplify the cpg2 transcripts but not other syne-1 splice variants (see Figure 1E). Both transcripts were amplified only from brain cDNA, showing that cpg2 and cpg2b are brain-specific splice variants of syne-1 (Figure 1F). Using a probe against the shared cpg2 and cpg2b 3′UTR that only recognized the cpg2 transcripts, in situ hybridization of a sagittal section from an adult rat brain showed that they are expressed in the cerebral cortex, hippocampus, striatum, and cerebellum of adult rats (Figure 1G). Thus, the two cpg2 transcripts are brain-specific splice variants of the syne-1 gene that are predominately expressed in brain regions associated with long-term potentiation (LTP) or LTD, electrophysiological paradigms of synaptic plasticity.
CPG2 Localizes to the Postsynaptic Site of Excitatory Synapses
Consistent with its derivation from the Syne-1 separator region, the CPG2 protein contains predicted structural or protein interaction motifs including two spectrin repeats and several coiled coils (Figure 2A). To gain insight into CPG2 function, we first determined its localization within neurons. To this end, we generated a monoclonal antibody against a peptide from CPG2. When used to probe a Western blot containing protein extracts from rat cerebral cortex, this antibody recognized a doublet band of the predicted size of CPG2 (Figure 2B, lane 1). Protein extracts from 293T cells transfected with the cpg2 cDNA showed the same-sized doublet band (Figure 2B, lane 2), suggesting that the in vivo doublet does not correspond to the translation of the cpg2 and cpg2b transcripts but rather results from posttranslational modification of CPG2 or leaky translation initiation of cpg2.
Figure 2. CPG2 Is Specifically Localized to the Postsynaptic Side of Excitatory Synapses.
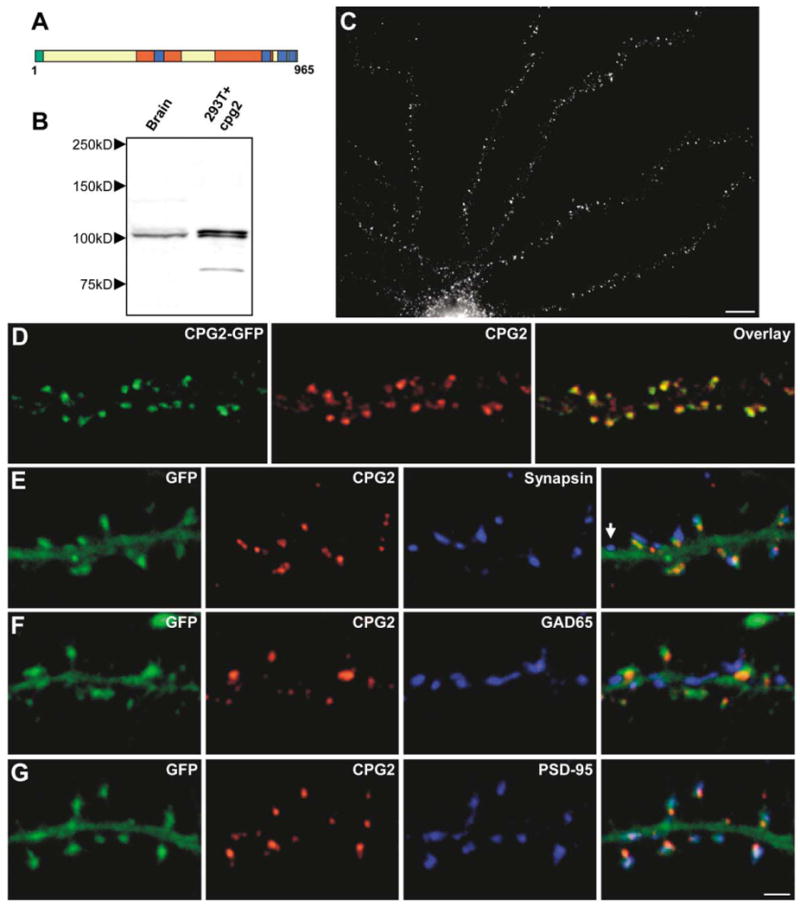
(A) Schematic of the CPG2 protein. Green, additional amino acids in CPG2B; red, spectrin repeats; blue, coiled coils.
(B) Western blot of protein extracts from rat cerebral cortex (lane 1) or 293T cells expressing cpg2 (lane 2) probed with anti-CPG2 monoclonal antibody 200A6.
(C) Cultured hippocampal neuron (24 DIV) labeled with the anti-CPG2 monoclonal antibody. Scale bar, 10 μm.
(D) Cultured hippocampal neuron infected with a CPG2-GFP-expressing lentivirus labeled for CPG2.
(E–G) Neurons infected with a GFP-expressing lentivirus were double labeled for CPG2 and synapsin I (E), GAD65 (F), or PSD-95 (G). Arrow in (E) indicates synapse without CPG2 labeling. Scale bar, 2 μm.
Immunostaining of cultured hippocampal neurons with the anti-CPG2 monoclonal antibody showed a punctate staining pattern on dendrites (Figure 2C). To further confirm the specificity of the anti-CPG2 monoclonal antibody, neurons were infected with a lentivirus expressing a CPG2-GFP fusion protein and then stained with the anti-CPG2 antibody. The CPG2-GFP signal showed an overlapping distribution with anti-CPG2 antibody labeling of CPG2 (Figure 2D), demonstrating that the CPG2 antibody recognizes endogenous CPG2 protein and the CPG2-GFP protein and results in no other detectable signal in neurons. We observed no labeling when neurons were stained with an antibody against the Syne-1 C terminus previously shown to recognize the Syne-1 protein in muscle cells (Apel et al., 2000) (data not shown). Given the Western and immunocytochemistry data, we suggest that the immunostaining patterns seen with the anti-CPG2 monoclonal antibody result from staining of the CPG2 protein and not the product of another syne-1 splice variant.
Since the punctate distribution of CPG2 in hippocampal neurons was indicative of a synaptic localization, we tested this possibility by infecting neurons with a GFP-expressing lentivirus and double staining for CPG2 and synapsin I. We found that CPG2 was localized apposed to most synapsin I-positive presynaptic terminals (89% ± 1%; n > 500 synapses, 5 neurons), often on dendritic spines (Figure 2E). These results suggest that CPG2 localizes to the postsynaptic component of a subset of synapses that are often located on dendritic spines. To determine the type of synapses containing CPG2, GFP-filled neurons were double labeled for CPG2 and the inhibitory synapse marker GAD65 or the excitatory synapse marker PSD-95. CPG2 rarely colocalized with GAD65-positive synapses (15% ± 2%; n > 500 synapses, 5 neurons) (Figure 2F) but was consistently observed at PSD-95-positive synapses (94% ± 1%; n > 500 synapses, 5 neurons) (Figure 2G). Additionally, CPG2 was not found at PSD-95-positive excitatory synapses on aspiny presumptive inhibitory neurons (data not shown). Thus, CPG2 is specifically localized to the postsynaptic side of excitatory synapses on glutamatergic neurons.
CPG2 Localizes to an Endocytic Zone of Excitatory Synapses
Close examination of the CPG2 distribution reveals that it does not colocalize with PSD-95 within spines, suggesting that CPG2 is not part of the PSD and localizes to a separate spine domain (see Figure 2G). To characterize CPG2's localization in relation to the PSD and the spine cytoskeleton, we stained neurons for actin filaments, CPG2, and PSD-95. High-magnification images of individual spines from these triple-labeled neurons show little overlap in the distribution of the three proteins, as CPG2 was often localized segregated from the PSD and actin filaments in the spine head (Figure 3A).
Figure 3. CPG2 Localizes to a Subdomain of Excitatory Synapses.
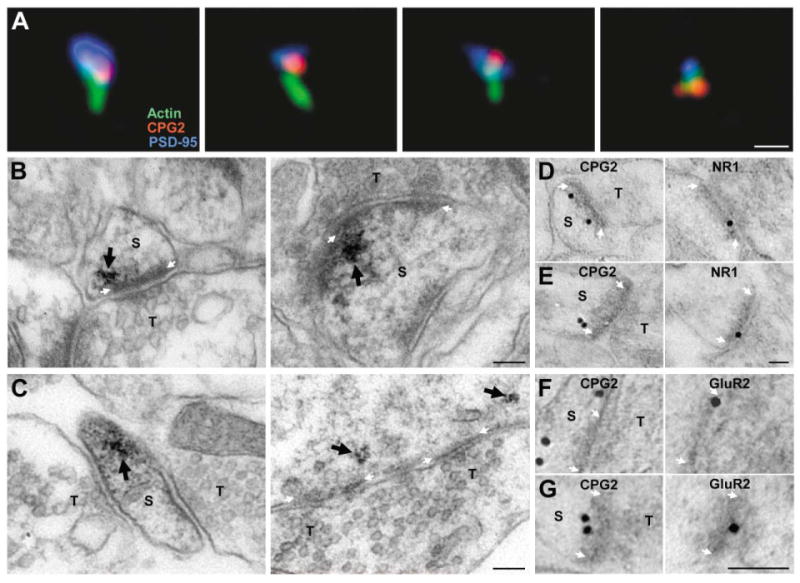
(A) Individual spines from cultured neurons stained for actin filaments (green), CPG2 (red), and PSD-95 (blue). Scale bar, 1 μm.
(B and C) Preembedding CPG2-labeled immunoEM micrographs of synapses from rat dentate gyrus or cultured hippocampal neurons. CPG2 labeling (arrow) is lateral to and underneath the PSD at synapses in vivo (B) and in vitro (C). Scale bars, 100 nm.
(D–G) Postembedding CPG2 and NR1 (D and E) or GluR2 (F and G) immunogold EM on serial sections of rat dentate gyrus. Scale bars for all images, 100 nm. S, spine; T, presynaptic terminal. White arrows mark the PSD.
CPG2's localization was further resolved using immunoEM. In preembedding anti-CPG2 immunoEM, approximately 47% of the labeling was associated with distal dendrites/dendritic spines, 47% was associated with cytosol/endoplasmic reticulum in the perikaryon or proximal dendrites, and 6% was associated with PSDs. No immunoreactivity was detected in presynaptic terminals. Of 100 synaptic membrane specializations that showed CPG2 labeling, 100 were asymmetrical, excitatory synapses. No symmetrical, inhibitory synapses were identified in association with CPG2 immunoreactivity. At excitatory synapses, CPG2 was localized beneath the PSD predominantly to the more lateral aspects of the PSD, both in vivo in dendritic spine (Figure 3B) and shaft (data not shown) synapses and in vitro in spine (Figure 3C, left) and shaft (Figure 3C, right) synapses from cultured hippocampal neurons. We used postembedding immunoEM against CPG2 and the NR1 NMDA receptor subunit on adjacent serial sections to define further the localization of CPG2 relative to the PSD (Figures 3D and 3E). Of 32 CPG2 immunoreactive postsynaptic zones, 21 were also immunoreactive for NR1, and the average distance between immunogold particles representing CPG2 immunoreactivity and the postsynaptic membrane thickening was 35 ± 8 nm. We also found CPG2 in dendritic spines positively labeled for the GluR2 AMPA receptor subunit using immunogold labeling of alternate sections (Figures 3F and 3G). Thus, the immunoEM confirms the immunofluorescence data and demonstrates that in vivo CPG2 is concentrated in the postsynaptic site of excitatory synapses and is localized to a distinct synaptic subdomain, lateral to and beneath the PSD.
The localization of CPG2 within spines is similar to that of a clathrin-GFP fusion protein previously shown to colocalize with endogenous clathrin in neurons and to mark an endocytic zone of excitatory synapses (Blanpied et al., 2002). To determine if CPG2 is localized to this zone, we generated a lentivirus that expresses a clathrin light chain A1-GFP fusion sequence. In neurons infected with this virus, clathrin-GFP was distributed in the dendritic shaft and within dendritic spines labeled for CPG2 and PSD-95 (Figure 4A). High-magnification views of individual spines from the clathrin-GFP-infected cells show that CPG2 and clathrin-GFP colocalize in spines to a site lateral to the PSD (Figure 4B). Within spines, CPG2 did not overlap with dynamin 3, recently shown to be localized to dendritic spines (Gray et al., 2003) (data not shown). These results are consistent with localization of dynamin 3 to the PSD and not to the endocytic zone. The colocalization studies together with the immunoEM data strongly suggest that CPG2 is localized to an endocytic zone within spines that is distinct from the postsynaptic density.
Figure 4. CPG2 Localizes to an Endocytic Zone of Excitatory Synapses.
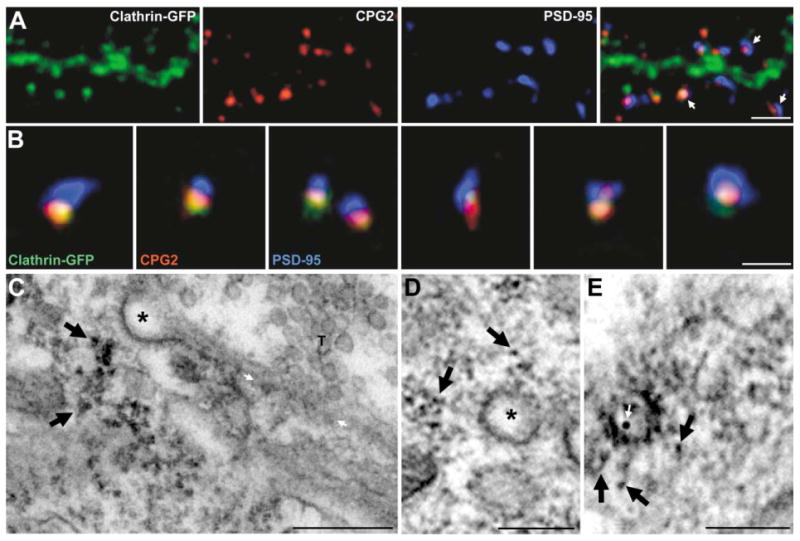
(A) Cultured hippocampal neuron infected with a lentivirus expressing a clathrin light chain A1-GFP fusion protein (green) and stained for CPG2 (red) and PSD-95 (blue). Arrows indicate dendritic spines. Scale bar, 5 μm.
(B) High-magnification views of individual spines from neurons as in (A). Scale bar, 1 μm.
(C–E) (C) Preembedding CPG2-labeled immunoEM micrograph from cultured hippocampal neurons shows CPG2 (arrows) localized lateral to the PSD and underneath a clathrin-coated pit (asterisk). White arrows denote PSD. (D) CPG2 immunoEM micrograph from cultured hippocampal neurons showing CPG2 staining (arrows) in the vicinity of a clathrin-coated vesicle (asterisk). (E) Double labeling using preembedding HRP staining for CPG2 (black arrows) and postembedding immunogold labeling for NR1 (white arrow) shows CPG2 near a clathrin-coated pit trafficking an NMDA receptor. Scale bars in (C)–(E), 100 nm.
If the region of CPG2 and clathrin colocalization is an endocytic zone, CPG2 should be associated with clathrin-coated pits and/or vesicles. Figure 4C shows an immunoEM micrograph of a synapse from a cultured neuron where CPG2 is localized near a clathrin-coated pit separated from the PSD. Figure 4D shows CPG2 in the vicinity of a clathrin-coated vesicle. Of 18 asymmetric synapses that contained a clathrin-coated pit or vesicle, all 18 contained CPG2 near (within 40 nm) these structures. Double labeling using preembedding HRP staining for CPG2 and postembedding immunogold for the NR1 subunit of the NMDA receptor shows that at least some of these CPG2-associated vesicles traffic glutamate receptors (Figure 4E). Thus, the immunoEM staining pattern is consistent with our immunofluorescence finding that CPG2 is colocalized with clathrin in the postsynaptic endocytic zone and suggests that it may be involved in the endocytosis of glutamate receptors.
CPG2 Levels Affect Dendritic Spine Size
CPG2's localization to the postsynaptic endocytic zone suggests that it may regulate the internalization of surface membrane via clathrin-mediated endocytosis. Therefore, we hypothesized that altering CPG2 levels may affect the size of dendritic spines. To test this hypothesis, we manipulated CPG2 protein levels using overexpression or RNA interference (RNAi) and examined the effects on spine morphology.
For CPG2 overexpression, we generated a lentivirus expressing a CPG2-GFP fusion protein driven by the human ubiquitin-C promoter (cpg2-gfp) that increases CPG2 levels within dendritic spines of infected neurons by 2- to 3-fold, as assessed by immunostaining (data not shown). To knock down CPG2 levels in neurons, we developed a lentivirus vector for RNAi delivery. We first generated a plasmid expressing a cpg2-specific small hairpin RNA (shRNA) driven by the mouse U6 promoter, that could effectively knock down exogenous cpg2-containing mRNA transcripts in 293T cells (data not shown; see Experimental Procedures for details). This cpg2-shRNA sequence and the U6 promoter were subcloned into the pFUGW lentivirus transfer vector upstream of the human ubiquitin-C promoter that drives expression of GFP (Figure 5A). A mutated cpg2-shRNA (Mcpg2-shRNA) lentivirus was generated as a negative control. Western blots of protein extracts from high-density cortical neurons infected with the cpg2-shRNA lentivirus showed a 90% ± 1% reduction (n = 5 cultures) in CPG2 levels relative to Mcpg2-shRNA-infected neurons (Figure 5B). In contrast, there were no changes in levels of GluR1, NR1, or β-tubulin (Figure 5B). Control and knockdown cultures showed comparable levels of GFP expression, demonstrating a similar infection rate. CPG2 knockdown was also evident at the synaptic level. Neurons infected with the control Mcpg2-shRNA virus showed a normal CPG2 subcellular distribution pattern (Figures 5C and 5E), whereas neurons infected with cpg2-shRNA showed a reduction in CPG2 levels but no apparent change in the levels or distribution of PSD-95 (Figure 5D), synapsin (Figure 5F), or actin filaments (data not shown). In addition, a lentivirus delivering a shRNA that knocks down the levels of a different candidate plasticity gene, cpg15, had no effect upon CPG2 levels (U. Putz and E.N., unpublished data). Thus, cpg2-shRNA delivered by a lentivirus vector effectively knocks down endogenous CPG2 levels in cultured neurons.
Figure 5. CPG2 Protein Levels Affect Dendritic Spine Size.
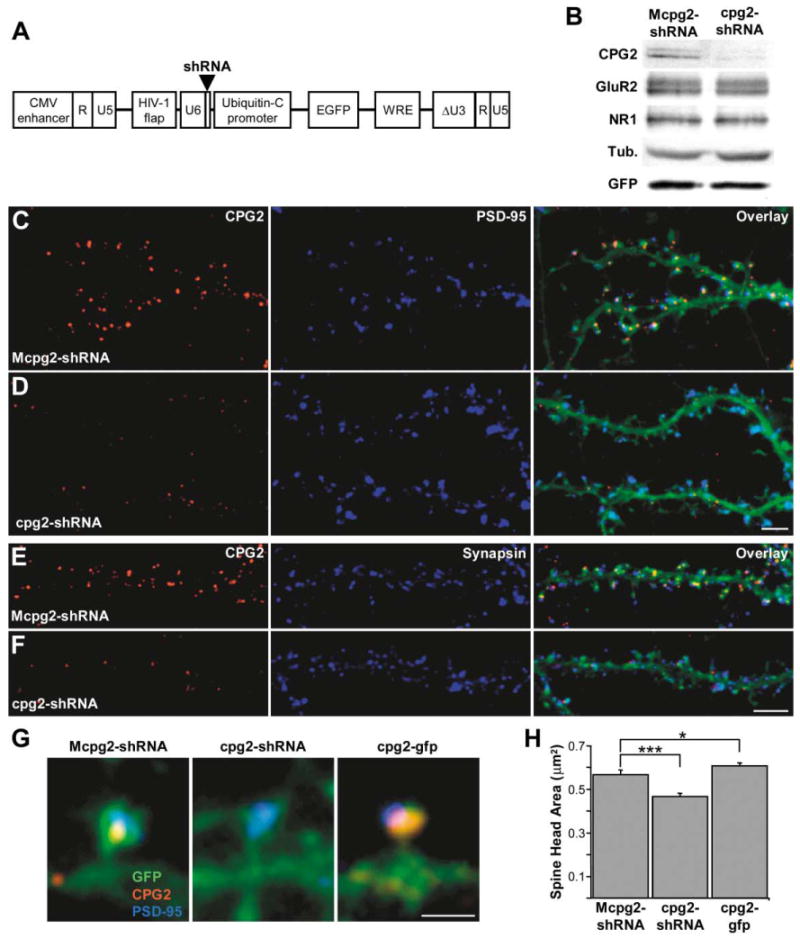
(A) Schematic of the pFUGW transfer vector used for delivery of shRNA sequences into neurons. The U6 promoter and the shRNA sequence were subcloned upstream of the ubiquitin-C promoter driving the expression of EGFP.
(B) Western blot of protein extracts from high-density cultures infected with the control Mcpg2-shRNA or the cpg2-shRNA lentivirus, probed for CPG2, GluR2, NR1, β-tubulin, and GFP.
(C–F) Hippocampal neurons (24 DIV) infected with the Mcpg2-shRNA (C and E) or the cpg2-shRNA (D and F) lentivirus and stained for CPG2 (C–F) and PSD-95 (C and D) or synapsin I (E and F). Scale bars, 5 μm.
(G) Individual spines from neurons infected with Mcpg2-shRNA, cpg2-shRNA, and cpg2-gfp and labeled for CPG2 and PSD-95. Scale bar, 1 μm.
(H) Quantification of spine head area of Mcpg2-shRNA-, cpg2-shRNA-, and cpg2-gfp-infected neurons.
We measured spine head area from control and knockdown neurons of similar size and morphology to determine if manipulating CPG2 protein levels leads to changes in spine morphology. Figure 5G shows individual spines from neurons infected with Mcpg2-shRNA, cpg2-shRNA, and cpg2-gfp. We found that neurons infected with cpg2-shRNA had significantly smaller dendritic spine heads (−18% ± 2.4%; p < 0.0005; n = 10 cells per group, 100 spines per cell) than Mcpg2-shRNA-infected neurons, while cpg2-gfp-infected neurons had larger spine heads (+7% ± 2.1%; p < 0.05; n = 10 cells per group, 100 spines per cell) (Figure 5H). In addition, measurement of spine length from these neurons showed that dendritic spines from CPG2 knockdown neurons were 12% ± 2% (p < 0.0001; n = 10 cells per group, 100 spines per cell) shorter than those from control neurons. The localization of CPG2 to the synaptic endocytic zone and its effect on dendritic spine size raise the possibility that CPG2 may play a role in regulating membrane removal from the spine surface via vesicle-mediated endocytosis.
CPG2 Knockdown Disrupts Constitutive Glutamate Receptor Internalization
If CPG2 regulates the removal of synaptic proteins from the spine surface through vesicle-mediated endocytosis, then altering its levels may also affect the number of clathrin-coated pits and vesicles seen within spines. Using electron microscopy, we analyzed the number of synapse-associated clathrin-coated pits and vesicles in cultured hippocampal neurons infected with either the Mcpg2-shRNA or the cpg2-shRNA lentivirus. Figure 6A shows examples of spine and shaft synapses from Mcpg2-shRNA (top)- and cpg2-shRNA (bottom)-infected cultures. The number of synapse-associated clathrin-coated pits and vesicles was increased nearly 4-fold in the cpg2-shRNA-treated cultures (0.95 ± 0.18 pits or vesicles per synapse; n = 28 neurons) as compared to the Mcpg2-shRNA-treated controls (0.25 ± 0.06 pits or vesicles per synapse; n = 35 neurons; p < 0.0002) (Figure 6B). There were no obvious effects of CPG2 knockdown on presynaptic terminals. Thus, CPG2 knockdown increases the number of postsynaptic clathrin-coated pits and vesicles in the vicinity of synapses.
Figure 6. CPG2 Knockdown Disrupts Constitutive Glutamate Receptor Internalization.
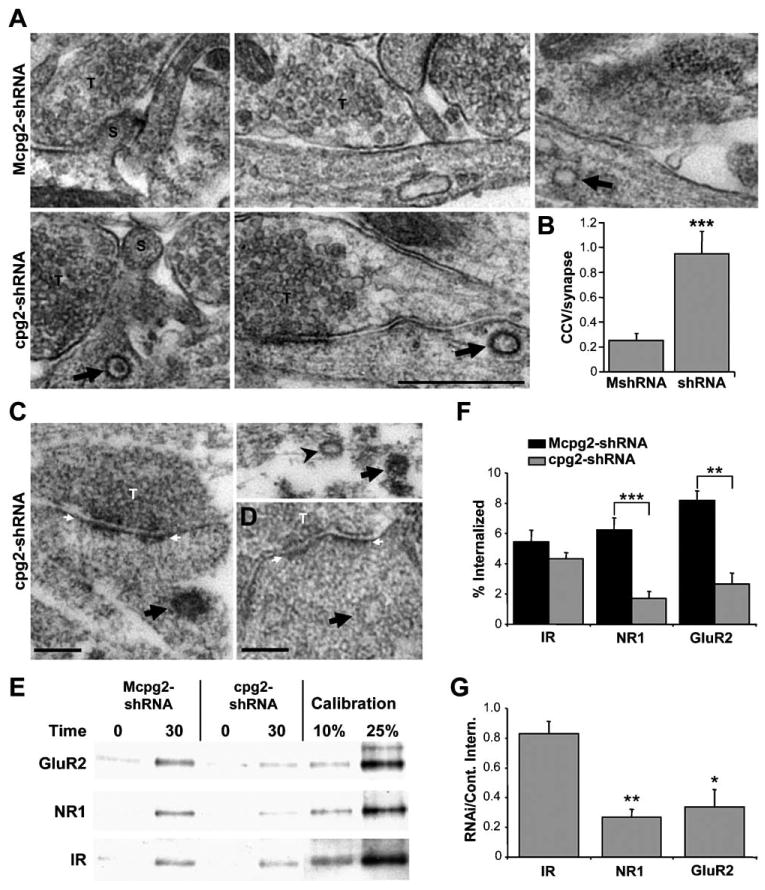
(A) EM micrographs of cultured hippocampal neurons infected with Mcpg2-shRNA or cpg2-shRNA showing clathrin-coated vesicles (arrows). S, spine; T, presynaptic terminal. Scale bar, 500 nm.
(B) Quantification of the number of synapse-associated clathrin-coated pits and vesicles in Mcpg2-shRNA-treated versus cpg2-shRNA-treated cultures.
(C) Preembedding immunostaining for NR1 on cpg2-shRNA-treated cultured hippocampal neurons. Arrows and arrowhead denote clathrin-coated vesicles positively and negatively stained from NR1, respectively.
(D) Unstained negative control showing a synapse-associated vesicle without NR1 staining. Scale bars, 100 nm. White arrows denote PSD. T, presynaptic terminal.
(E) Biotinylation internalization assay showing the internalization of GluR2, NR1, and insulin receptor β subunit (IR) after 0 and 30 min at 37°C in cortical cultures infected with Mcpg2-shRNA or cpg2-shRNA.
(F) Quantification of the percent of GluR2, NR1, and IR internalized in Mcpg2-shRNA- and cpg2-shRNA-infected neurons.
(G) Quantification of GluR2, NR1, and IR internalization in cpg2-shRNA-infected neurons as a percentage of the internalization in Mcpg2-shRNA-infected neurons. Statistical significance was determined by comparing the RNAi/control internalization values of GluR2 and NR1 with IR.
To confirm that the clathrin-coated pits and vesicles that accumulate in response to CPG2 knockdown contain synaptic proteins, we performed preembedding immunoEM for NR1 on cpg2-shRNA-treated cultures. In RNAi-treated cultures, clathrin-coated vesicles near synapses stained positively for NR1 (Figure 6C). In RNAi-treated cultures, 41% ± 11% of coated pits or vesicles stained for NR1, as opposed to only 0.5% ± 0.5% percent in control cultures (n = 12 neurons; p < 0.01). No staining was seen on similar vesicles from unstained cultures (Figure 6D). These data suggest that CPG2 regulates endocytosis of clathrin-coated pits that traffic glutamate receptors.
There are two potential mechanisms by which CPG2 knockdown can increase the number of postsynaptic clathrin-coated vesicles that traffic glutamate receptors: either it can increase the rate of their formation, or it can decrease the rate of their removal. To understand the mechanism of CPG2's regulation of glutamate receptor internalization, we performed a biochemical internalization assay. Surface proteins from cortical cultures infected with Mcpg2-shRNA or cpg2-shRNA were biotinylated with a reversible biotin and kept at 4°C to block internalization or moved to 37°C for 30 min to allow for receptor internalization (in the presence of a proteasome inhibitor). Remaining surface biotin was removed with a cell-impermeable cleavage buffer. Biotinylated proteins were then isolated, probed on a Western blot, and compared with calibration controls. In Mcpg2-shRNA-infected cultures, 6.2% ± 0.8% of surface NMDA receptors and 8.2% ± 0.6% of surface AMPA receptors were internalized after 30 min at 37°C (Figures 6E and 6F), similar to what has been previously reported for glutamate receptor internalization (Ehlers, 2000; Lin et al., 2000). In cpg2-shRNA-infected cultures, AMPA and NMDA receptor internalization was reduced to 2.7% ± 0.8% (p < 0.005; n = 3) and 1.7% ± 0.5% (p < 0.0003; n = 4), respectively (Figures 6E and 6F). In contrast, there was no significant change in the internalization of insulin receptors in the cpg2-shRNA-treated neurons (4.3% ± 0.4%) versus Mcpg2-shRNA-treated neurons (5.4% ± 0.8%; n = 4; p = 0.4) (Figures 6E and 6F). The internalization of AMPA and NMDA receptors was reduced by 66% ± 11% (p < 0.02) and 73% ± 5% (p < 0.002), respectively, in cpg2-shRNA-infected neurons (Figure 6G). These results demonstrate that the cpg2-shRNA blockade was specific to the internalization of synaptic proteins. The reduction in glutamate receptor internalization was not a result of a general reduction in GluR2 or NR1 levels, as total GluR2 and NR1 levels remained constant in Mcpg2-shRNA- and cpg2-shRNA-infected neurons (see Figure 4B). These data indicate that CPG2 is necessary for normal constitutive glutamate receptor internalization. Since CPG2 knockdown resulted in an accumulation of clathrin-coated pits and vesicles but disrupted rather than enhanced glutamate receptor internalization, CPG2 is likely involved in the clearance of internalized vesicles from the synapses rather than the formation of these vesicles (see Discussion).
If CPG2 knockdown inhibits the internalization of glutamate receptors, one might expect an accumulation of surface receptors. To examine the amount of surface AMPA and NMDA receptors, we performed a surface protein biotinylation assay. Surface proteins from control or CPG2 knockdown neurons were biotinylated, separated from total protein using Neutravidin, then run on SDS-PAGE gels side by side with total protein from the same cells and probed on Western blots with antibodies against GluR2 or NR1. We then compared the ratios of surface receptors to total receptors in cpg2-shRNA-treated and control cultures. Knockdown of CPG2 resulted in a significant increase in the surface to total protein ratios for both GluR2 (+25% ± 10%; n = 5; p < 0.05) and NR1 (+20% ± 7%; n = 4; p < 0.05) (Figures 7A and 7B), demonstrating that CPG2 knockdown does result in an increase in surface receptors. Since biotinylation assays measure the amount of both synaptic and extrasynaptic receptors, we further examined the levels of surface AMPA receptors by staining fixed, unperme-abilized control and knockdown neurons with an antibody against an extracellular epitope of the GluR1 AMPA receptor subunit. When we quantified the intensity of GluR1 punctae, we found that there was a similar increase in synaptic GluR1 levels in CPG2 knockdown neurons (+18% ± 5%; p < 0.05; n = 8 to 10 cells per group) (Figures 7C and 7D). Thus, CPG2 knockdown increases the levels of synaptic glutamate receptors, supporting the conclusion that CPG2 plays a role in their internalization.
Figure 7. CPG2 Knockdown Disrupts Activity-Induced Internalization of AMPA Receptors.
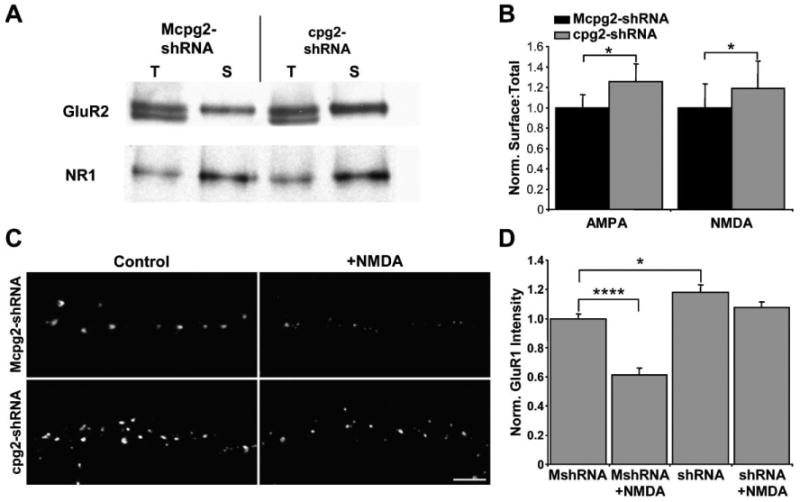
(A) Biotinylation assay showing surface (S) and total protein (T) levels of GluR2 and NR1 in Mcpg2-shRNA- and cpg2-shRNA-infected neurons.
(B) Quantification of surface to total ratios of GluR2 and NR1 in Mcpg2-shRNA- and cpg2-shRNA-infected neurons.
(C) Immunostaining of surface GluR1 in Mcpg2-shRNA- and cpg2-shRNA-infected neurons with or without NMDA treatment. Scale bar, 5 μm.
(D) Quantification of the intensity of surface synaptic GluR1 punctae as shown in (C).
CPG2 Knockdown Disrupts Activity-Induced AMPA Receptor Internalization
LTD is thought to occur in part by the rapid clathrin-mediated internalization of AMPA receptors following the activation of a specific set of signaling pathways (Beattie et al., 2000; Lin et al., 2000; Man et al., 2000; Snyder et al., 2001; Wang and Linden, 2000). To see if CPG2 plays a potential role in LTD, we examined the effect of CPG2 knockdown on the activity-induced internalization of AMPA receptors. Control and CPG2 knockdown neurons were incubated with the anti-GluR1 antibody and then in control media or media plus NMDA, followed by fixation and secondary staining for surface GluR1. The intensity of GluR1 punctae was then quantified. In control neurons, there was a 40% ± 5% (p < 0.000001; n = 8 to 10 cells per group) reduction in surface, synaptic AMPA receptors following NMDA treatment. In contrast, CPG2 knockdown inhibited the NMDA-induced internalization of surface AMPA receptors (−8% ± 3%; p > 0.1; n = 8 cells per group) (Figures 7C and 7D). Thus, CPG2 is critical for the activity-dependent internalization of AMPA receptors and may be a crucial component of the LTD pathway.
Discussion
Clathrin-mediated endocytosis is a general mechanism that regulates the internalization of proteins from the cell surface in eukaryotic cells (Kirchhausen, 2000; Mousavi et al., 2004), and its basic machinery is conserved across species and cell types (Schmid, 1997). In cases where specialized endocytic functions are required, specific mechanisms have evolved for regulating endocytosis. In the presynaptic terminal, for example, there are novel mechanisms and proteins for meeting the demands of synaptic transmission, often consisting of terminal-specific isoforms or splice variants of general endocytic proteins (Jarousse and Kelly, 2001; Slepnev and De Camilli, 2000). Given the importance of glutamate receptor internalization for maintaining and modifying synaptic strength (Carroll et al., 2001; Malinow and Malenka, 2002), neurons may have developed novel specific mechanisms for regulating the endocytosis of glutamate receptors. A postsynaptic endocytic zone has been shown to be present and stable on dendritic spine and shaft synapses (Blanpied et al., 2002). Although segregated from the PSD, it is likely an integral synaptic component (Blanpied et al., 2002), and our data strongly suggest that it is the site of glutamate receptor internalization. Since CPG2 is primarily localized to the postsynaptic endocytic zone of excitatory synapses, it is not a constitutive part of the ubiquitous endocytic machinery. While the disruption of components of the general endocytic machinery has been shown to disrupt glutamate receptor internalization (Carroll et al., 1999; Luscher et al., 1999; Metzler et al., 2003; Zhou et al., 2001), CPG2 is the first known protein that is specifically involved in the internalization of postsynaptic proteins. We hypothesize that CPG2 is a component of a specialized postsynaptic endocytic mechanism dedicated to the internalization of synaptic proteins, including glutamate receptors.
Knockdown of CPG2 led to an increase in postsynaptic clathrin-coated vesicles, some trafficking NMDA receptors, and a disruption of glutamate receptor internalization. This phenotype is similar to that seen in presynaptic terminals of organisms with mutations of endophilin or synaptojanin, where there is an accumulation of clathrin-coated vesicles and a disruption of the synaptic vesicle cycle, due to a defect in clathrin-coated vesicle uncoating (Cremona et al., 1999; Harris et al., 2000; Kim et al., 2002; Verstreken et al., 2002, 2003). Since CPG2 contains multiple protein interaction motifs, including two spectrin-like repeats and several coiled coils, we propose a model in which CPG2 interacts with other proteins at the postsynaptic endocytic zone that are necessary for the downstream processing, such as the uncoating, of glutamate receptor-trafficking vesicles. According to this model, following CPG2 knockdown, the CPG2-associated protein network is disrupted, inhibiting vesicle clearance and causing their accumulation. Due to the slowed kinetics of this late step, the entire endocytic pathway is slowed, disrupting further glutamate receptor internalization. Future experiments should reveal the proteins with which CPG2 interacts at synapses and at what point they function to regulate the vesicle cycle. This would clarify whether CPG2 is an active regulator of the endocytic pathway or is a component of the overall endocytic machinery unique to the postsynaptic density.
In CPG2 knockdown neurons, there is an increase in synaptic glutamate receptors, which is likely due to the disruption of glutamate receptor endocytosis. Although some reports have indicated that endocytosis inhibition does not affect the number of surface AMPA receptors (Lledo et al., 1998; Man et al., 2000), our data are consistent with results showing that acute blockade of endocytosis causes an increase in synaptic transmission (Luscher et al., 1999). However, the increase in surface receptors is moderate (20%) despite a greater than 70% disruption in glutamate receptor internalization. This discrepancy may be due to the tight link, previously proposed, between the rates of receptor insertion into and removal from the synaptic membrane (Ehlers, 2000; Liang and Huganir, 2001). Considering there is a decrease in glutamate receptor internalization in these neurons, there must be a compensatory decrease in glutamate receptor insertion for steady-state amount of surface receptors to remain relatively constant. Thus, CPG2 knockdown appears to slow not only the internalization, but also the insertion, of glutamate receptors, suggesting that CPG2 may be necessary for the rapid cycling of synaptic glutamate receptors.
Given a disruption of membrane internalization and an increase in synaptic glutamate receptors in CPG2 knockdown neurons, one might expect an increase in dendritic spine size as membrane continues to be inserted into the spines during receptor exocytosis. However, we observed the opposite phenotype, with dendritic spines on CPG2 knockdown neurons nearly twenty percent smaller than in control neurons. According to our model of CPG2 function, the reduction in spine size may result from an increase in membrane retention within internalized clathrin-coated vesicles, as the clathrin coat cannot be removed and the membrane cannot be recovered from these vesicles. However, since the cytoskeleton is critical for both spine morphology and endocytosis (Engqvist-Goldstein and Drubin, 2003; Rao and Craig, 2000), it is possible that CPG2 regulates the spine cytoskeleton and that its knockdown affects both spine size and endocytosis in parallel. A large proportion of AMPA receptors are inserted into the plasma membrane at extrasynaptic sites, possibly between spines (Passafaro et al., 2001). While normally there is a tight correlation between spine size and synaptic AMPA receptor numbers (Kasai et al., 2003), in CPG2 knockdown neurons, the total amount of synaptic glutamate receptors increases despite smaller spines, suggesting that CPG2 knockdown may disrupt the spine size/receptor number relationship.
cpg2 was isolated in a screen for seizure-induced genes and is regulated by physiological activity (Nedivi et al., 1993, 1996). In general, screens for activity-regulated genes have isolated a number of synaptic proteins that are the components of the basic transmission machinery, indicating that the synaptic rearrangements that occur during plasticity likely involve an augmentation of normal synaptic processes (Nedivi, 1999). Since CPG2 is important for normal synaptic function and for the activity-induced internalization of glutamate receptors that may underlie LTD, an increase in cpg2 expression may belie an increase in synapse formation during synaptic plasticity. However, following LTP, there is an increase in the number of clathrin-coated pits and vesicles in dendritic spines, which may reflect an enhancement of postsynaptic protein cycling during plasticity (Toni et al., 2001). The critical role of CPG2 in activity-dependent glutamate receptor endocytosis may explain the increased need for cpg2 expression during periods of synaptic plasticity. As the product of an activity-regulated transcript that is expressed solely in brain regions with significant synaptic plasticity mechanisms, CPG2 may underlie an adaptation of the clathrin-mediated endocytosis pathway that enables the capacity for postsynaptic plasticity in excitatory synapses.
Experimental Procedures
RNA Analysis
Northern blot hybridization was done as described (Sambrook et al., 1989), with 32P-labeled probes synthesized with the High Prime labeling kit (Roche) using a 1 kb fragment from the coding region of rat cpg2 and a 300 bp mouse GAPDH cDNA fragment excised from pTRI-GAPDH-mouse (Ambion).
5′RACE was performed using GeneRacer Kit (Invitrogen) with a cpg2-specific primer (GCTCCACGGACTCTCGCCGCCCATA). These 5′ fragments were PCR amplified using nested primers specific to the ligated linker and to cpg2 (GACTCAGCGAGGACCAGCAGGGACAGTA). In the negative control, the reverse transcriptase was omitted. The two PCR products were gel purified, subcloned using the TOPO TA Cloning Kit (Invitrogen), and sequenced (Retrogen).
For RT-PCR, tissues dissected from Sprague-Dawley rats were frozen on dry ice and ground into a fine powder. Total RNA extraction using Trizol (Invitrogen) was followed by cDNA synthesis with cDNA Synthesis Kit (Invitrogen), and fragments of the cDNA transcripts were amplified by PCR (30 cycles). For cpg2 and cpg2b, the same antisense primer (CCATCGGCTTTGACGTACAT) was used in combination with unique sense primers (cpg2, AAAGGCTTCCATCGGTCGTT; cpg2b, TCAGCCTCTTGTTTCCTCGT). The control reaction used β-actin primers (sense, AAATGCTGCACTGTGCGGC; antisense, GTTTTATAGGACGCCACAGC). Sense and antisense primers were from separate exons to avoid amplification of genomic DNA.
In situ hybridization was performed as described (Lee and Nedivi, 2002) on sagittal sections of adult rat brain, using a 2 kb probe against the 3′UTR of the cpg2 transcript.
Western Blot
Hippocampus protein extracts were generated by homogenizing rat hippocampi in 10 mM Tris (pH 7.4), 1 mM EGTA, 0.5 mM DTT plus a protease inhibitor cocktail (1:200; Sigma). HEK293T-cpg2 protein extracts were generated from HEK293T cells transfected with cpg2, subcloned into pcDNA3 (Promega) using Lipofectamine 2000 (Invitrogen). After 24 hr, cells were rinsed and lysed in 150 mM NaCl, 50 mM Tris-HCl (pH 7.6), and 0.5% NP-40 (Sigma). Cell lysates were centrifuged, and supernatants were diluted in SDS-PAGE sample buffer.
Mouse monoclonal anti-CPG2 antibody 200A6.1 (IgG1) was generated against the peptide RKLESTLTGLEQSRERQERR (ResGen). Antibody was purified from hybridoma supernatants with Protein G columns (Invitrogen). Protein extracts from rat cerebral cortex (40 μg) and HEK293T cells were separated on an 8% SDS-PAGE gel, transferred to nitrocellulose, blocked with 10% nonfat milk, and probed with the anti-CPG2 antibody (3 μg/ml). The blot was incubated in horseradish peroxidase-conjugated goat anti-mouse secondary antibodies (Sigma; 1:5000) and visualized with ECL (Amer-sham). For culture Western blotting, high-density cortical cultures (1.5 × 106 cells/6 cm dish) were infected at 6 DIV with ∼1.5 × 107 pfu of Mcpg2-shRNA or cpg2-shRNA lentivirus. At 21 DIV, cells were lysed with 500 μl of 0.5% SDS-RIPA buffer (1% Triton X-100, 0.5% SDS, 0.5% deoxycholic acid, 50 mM NaPO4, 150 mM NaCl, 2 mM EDTA, 50 mM NaF). Protein was run on an 8% SDS-PAGE gel and probed with the anti-CPG2 monoclonal antibody (3 μg/ml), a mouse anti-GluR2 antibody (1:1500; Chemicon), a mouse anti-NR1 antibody (1:500; Chemicon), a rabbit anti-β-tubulin antibody (1:200; Santa Cruz), and a rabbit anti-GFP antibody (1:100; Clontech).
Neuronal Cultures
Embryonic medium-density hippocampal cultures were made as described (Brewer et al., 1993). Glass coverslips (Carolina Biological Supply) were incubated in poly-D-lysine (40 μg/ml) and laminin (2.5 μg/ml) (BD Biosciences) overnight, rinsed with water, and incubated in plating medium: Neurobasal (Gibco) plus 0.5 mM L-glutamine, 12.5 μM L-glutamate, and B27 (Gibco). Hippocampi from E18 embryos were isolated, digested in 0.25% trypsin (Gibco) plus 0.1% DNase (Sigma), and triturated with fire-polished pipettes in 0.1% DNase. Cells were then centrifuged, resuspended in plating medium, and plated at 1.4 × 104 cells/cm2. After 4 days, one half of the medium was replaced with plating medium without additional L-glutamate. For postnatal high-density cortical cultures, cortices from P1 rats were plated onto poly-D-lysine and laminin-coated 6 cm dishes at 1.5 × 106 cells per dish in Neurobasal A medium (Invitrogen), supplemented with 0.5 mM L-glutamine and B27.
Immunocytochemistry
Cells were fixed in 4% formaldehyde for 20 min, permeabilized in 0.3% Triton X-100 for 5 min, and blocked in 10% goat serum for 1 hr. Cultures were then incubated overnight with primary antibodies, rinsed in PBS, incubated with secondary antibodies, and mounted with Fluoromount G (Southern Biotechnology). Primary antibodies were as follows: mouse anti-CPG2 monoclonal (1.5 μg/ml), rabbit anti-synapsin (1:2000; Sigma), rabbit anti-PSD-95 (1:300; Zymed), rabbit anti-GAD65 (1:2000; Chemicon), and rabbit anti-Syne-1 (Apel et al., 2000). Secondary antibodies were as follows: goat anti-mouse Alexa Fluor 555 (1:1000; Molecular Probes) and goat anti-rabbit Alexa Fluor 647 (1:500; Molecular Probes). For actin filament staining, Alexa Fluor 488-conjugated phalloidin (1:500; Molecular Probes) was added to the secondary antibody mixture. Fluorescent images of neurons were imported into Spot 3.5 (Diagnostic Instruments) with a Diagnostic Instruments Spot2 digital camera mounted on a Nikon Eclipse E600 using a 60×/1.40 Plan Apo oil immersion objective (Nikon). For quantifications, spine head area from GFP-filled neurons was measured blind to treatment using Scion Image Beta 4.0.2. Student's t test was used to compare groups.
Electron Microscopic Immunocytochemistry
Cultures fixed with 4% paraformaldehyde and 0.1% glutaraldehyde or vibratome sections of perfusion-fixed rat hippocampi were immunostained for CPG2 (1.5 μg/ml) or NR1 (1:1000; Chemicon) overnight at room temperature. They were then incubated with biotinylated horse anti-mouse secondary antibodies (1:250; Vector) for 2 hr followed by incubation in Avidin biotin-peroxidase (1:200; Vector) for 2 hr at room temperature. Tissue bound peroxidase was then visualized with a diaminobenzidine (DAB; 5 mg/ml, H202 0.03%) reaction. Coverslips were osmicated, dehydrated, and embedded in durcupan (FLUKA, ACM). Ultrathin sections (60 nm thick) were prepared on an Ultracut UCT, Leica ultramicrotome and analyzed in a Tecnai 10 electron microscope. For clathrin-coated pit and vesicle quantifications, neurons randomly selected from unstained Mcpg2-shRNA (n = 35 cells)- and cpg2-shRNA (n = 28 cells)-infected cultures were photographed at magnification of 55,400. Part of the perykarion and adjacent neuropil of cells were analyzed in a 49 μm2 region per cell. The number of endocytic pits and vesicles in association with asymmetrical synaptic membrane specializations (within 500 nm) were quantified blind to the treatment group.
For CPG2, NR1, and GluR2 postembedding immunoEM, we used lowicryl-embedded hippocampal sections (Nyiri et al., 2003). Sections were washed in phosphate buffer overnight and then placed into increasing concentration sucrose solutions (0.5, 1, and 2 M sucrose for 0.5, 1, and 2 hr, respectively) for cryoprotection. Sections were then slammed onto copper blocks cooled in liquid nitrogen and, following low-temperature dehydration and freeze substitution, embedded in Lowicryl HM 20 resin (Chemische Werke Lowi). Postembedding immunocytochemistry was carried out on 60 nm thick serial sections placed on pioloform-coated nickel grids. Serial sections on two consecutive grids were incubated on drops of blocking solution for 1 hr (20% bovine serum albumin), followed by incubation on drops of either anti-CPG2, anti-NR1, or anti-GluR2 primary antibodies overnight at room temperature. The primary and secondary antibodies (15 nm gold-conjugated anti-mouse IgG; 1:20; Amer-sham) were diluted in TBS plus 0.03% Triton X-100 and 2% human serum albumin (Sigma). Following several washes in TBS, the sections were washed in water and then contrasted with saturated aqueous uranyl acetate followed by lead citrate.
RNA Interference
Five separate DNA oligonucleotides containing a 19 nucleotide sequence specific to cpg2, a 9 nucleotide loop region (TTCAAGAGA), and the 19 nucleotide antisense cpg2 sequence were annealed to their antisense counterparts and ligated into the pSilencer1.0-U6 plasmid (Ambion) downstream of the U6 promoter. These shRNA constructs were cotransfected with a cpg2-IRES-GFP construct into 293T cells at a 30:1 ratio using Lipofectamine 2000 (Invitrogen). After 48 hr, GFP fluorescence was examined. One shRNA (cpg2-shRNA) resulted in >80% reduction in GFP levels (data not shown). The effective RNAi target sequence was GCAGACTGCTGAGATGTAC. For a negative control, a mutated shRNA (Mcpg2-shRNA) was generated with the orientation of six interior nucleotides of the target sequence reversed: GCAGACAGTCGTGATGTAC.
Lentivirus Generation
For GFP cell labeling, lentivirus particles were generated from the pFUGW transfer vector, in which GFP is downstream of the ubiquitin-C promoter (Lois et al., 2002). For the CPG2-GFP fusion protein, the cpg2 coding sequence was subcloned into pFUGW downstream of and in frame with the GFP coding sequence. The shRNA sequences with the U6 promoter were amplified from the pSilencer1.0 plasmid by PCR and subcloned into pFUGW immediately upstream of the ubiquitin-C promoter. The clathrin A1 light chain-GFP fusion sequence from EGFP-C1 (Blanpied et al., 2002) was subcloned into pFUGW downstream of the human ubiquitin-C promoter. Lentivirus particles were produced by cotransfecting the transfer vector, the HIV-1 packaging vector Δ8.9, and the VSVG envelope glycoprotein vector into 293T cells with Lipofectamine 2000 (Invitrogen). Culture medium was collected 48 to 60 hr after transfection, filtered, and centrifuged at 25,000 RPM for 90 min at 4°C. Supernatants were discarded, and the virus pellets were rehydrated overnight in PBS. Viral particles were resuspended, aliquoted, and frozen at −80°C. Viral titers were determined by application of serial dilutions to 293T cells and quantifications of GFP expression 72 hr later. Typical titers were ∼1 × 106 pfu/μl.
The overexpression of CPG2 with the cpg2-gfp lentivirus lead to significant toxicity within neurons, likely due to its misexpression. Therefore, analysis of CPG2 overexpression was limited to experiments where it was possible to use a low titer. For example, measuring CPG2's effects upon dendritic spine size could be done on isolated healthy neurons overexpressing CPG2 at lower levels. In contrast, the effects of CPG2 overexpression could not be tested in the biochemical assays or in the immunoEM experiments, as these required near 100% infection efficiency, which would result in multiple infections and in significant attrition in cell density.
Biotinylation Internalization Assay
High-density cultures were infected at 6 DIV with the Mcpg2-shRNA or cpg2-shRNA lentivirus vectors, resulting in >95% GFP-expressing cells. At 21 DIV, cultures were incubated in the protease inhibitor leupeptin (100 μg/ml; Sigma) for 30 min and then incubated in 1.5 mg/ml sulfo-NHS-SS-biotin in DPBS for 20 min at 4°C. Unbound biotin was quenched with DPBS plus 50 mM glycine. Cultures were then either left at 4°C or returned to the original media (plus leupeptin) and incubated at 37°C for 30 min. Extracellular biotin was then cleaved by three washes for 15 min each in stripping buffer: 50 mM glutathione, 90 mM NaCl, 10 mM EDTA, 75 mM NaOH, 1% BSA (pH 8.75). Stripping buffer was quenched with 5 mg/ml iodacetamide. Calibration cultures were kept at 4°C in DPBS, without stripping. Cultures were then lysed in 0.5% SDS-RIPA buffer and scraped into eppendorf tubes, and protein extracts were centrifuged. Protein (300 μg) was added to 1 ml 0.1% SDS-RIPA buffer, and biotinylated proteins were isolated with neutravidin-agarose slurry (Pierce). Biotinylated proteins were separated on an 8% SDS-PAGE gel, transferred to nitrocellulose, and probed with monoclonal antibodies against NR1 (1:500; Chemicon), GluR2 (1:1500; Chemicon), and the insulin receptor β-subunit (1:200; Oncogene). For internalization quantifications, pixel density for each band was determined with Scion Image Beta 4.0.2. Internalized proteins were quantified by comparing their pixel densities with those of the total surface protein calibration. Unstripped protein levels (<5%) were subtracted from internalized protein levels. Groups were compared with Student's t tests. For total surface protein biotinylation assay, 75 μg of unstripped surface biotinylated proteins from Mcpg2-shRNA- and cpg2-shRNA-infected cultures were pelleted with neutravidin, run on an SDS-PAGE gel next to 30 μg of total protein, and probed for NR1 and GluR2. Surface to total protein ratios were quantified as shown above.
Chemical LTD Assay
At 19–21 DIV, cultures were incubated in TTX (2 μM; Tocris) for 1 hr and then in a rabbit anti-GluR1 antibody (10 μg/ml; Calbiochem) for 20 min. Cultures were incubated in TTX (2 μM) alone or TTX plus NMDA (50 μM) for 4 min and then returned to normal conditioned media and incubated for 15 additional minutes. Cultures were fixed in cold 4% paraformaldehyde and 4% sucrose for 20 min and stained with goat anti-rabbit-Alexa Fluor 555 antibodies (Molecular Probes) for 1 hr at room temperature. For analysis, the average pixel intensity of the GluR1 clusters from neurons from each treatment group was measured using Scion Image Beta 4.0.2.
Acknowledgments
We thank Dr. Carlos Lois for the lentivirus vectors, advice on lentivirus design, and use of his equipment; Dr. Michael Ehlers for the clathrin-GFP construct; Dr. Joshua Sanes for the anti-Syne-1 antibody; Dr. Gabor Nyiri for lowicryl-embedded hippocampal sections; Dr. Ulrich Putz for the cpg15-shRNA lentivirus; Wei-Chung Lee and Elizabeth Lester for assistance with the in situ hybridizations; and Shifali Arora for spine size quantifications and subcloning assistance. We would also like to thank Dr. Troy Littleton, Dr. Michele Jacobs, Dr. Tadahiro Fujino, and Wei-Chung Lee for critical comments on the manuscript. This work was supported by NEI (E.N.), NCRR (T.L.H.), and NIDDK (T.L.H.).
Footnotes
Accession Numbers: The GenBank accession number for the cpg2b sequence reported in this paper is AY597251.
References
- Apel ED, Lewis RM, Grady RM, Sanes JR. Syne-1, a dystrophin- and Klarsicht-related protein associated with synaptic nuclei at the neuromuscular junction. J Biol Chem. 2000;275:31986–31995. doi: 10.1074/jbc.M004775200. [DOI] [PubMed] [Google Scholar]
- Bear MF, Malenka RC. Synaptic plasticity: LTP and LTD. Curr Opin Neurobiol. 1994;4:389–399. doi: 10.1016/0959-4388(94)90101-5. [DOI] [PubMed] [Google Scholar]
- Beattie EC, Carroll RC, Yu X, Morishita W, Yasuda H, von Zastrow M, Malenka RC. Regulation of AMPA receptor endocytosis by a signaling mechanism shared with LTD. Nat Neurosci. 2000;3:1291–1300. doi: 10.1038/81823. [DOI] [PubMed] [Google Scholar]
- Blanpied TA, Scott DB, Ehlers MD. Dynamics and regulation of clathrin coats at specialized endocytic zones of dendrites and spines. Neuron. 2002;36:435–449. doi: 10.1016/s0896-6273(02)00979-0. [DOI] [PubMed] [Google Scholar]
- Brewer GJ, Torricelli JR, Evege EK, Price PJ. Optimized survival of hippocampal neurons in B27-supplemented Neurobasal, a new serum-free medium combination. J Neurosci Res. 1993;35:567–576. doi: 10.1002/jnr.490350513. [DOI] [PubMed] [Google Scholar]
- Burkhard P, Stetefeld J, Strelkov SV. Coiled coils: a highly versatile protein folding motif. Trends Cell Biol. 2001;11:82–88. doi: 10.1016/s0962-8924(00)01898-5. [DOI] [PubMed] [Google Scholar]
- Carroll RC, Beattie EC, Xia H, Luscher C, Altschuler Y, Nicoll RA, Malenka RC, von Zastrow M. Dynamin-dependent endocytosis of ionotropic glutamate receptors. Proc Natl Acad Sci USA. 1999;96:14112–14117. doi: 10.1073/pnas.96.24.14112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll RC, Beattie EC, von Zastrow M, Malenka RC. Role of AMPA receptor endocytosis in synaptic plasticity. Nat Rev Neurosci. 2001;2:315–324. doi: 10.1038/35072500. [DOI] [PubMed] [Google Scholar]
- Cooney JR, Hurlburt JL, Selig DK, Harris KM, Fiala JC. Endosomal compartments serve multiple hippocampal dendritic spines from a widespread rather than a local store of recycling membrane. J Neurosci. 2002;22:2215–2224. doi: 10.1523/JNEUROSCI.22-06-02215.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cremona O, Di Paolo G, Wenk MR, Luthi A, Kim WT, Takei K, Daniell L, Nemoto Y, Shears SB, Flavell RA, et al. Essential role of phosphoinositide metabolism in synaptic vesicle recycling. Cell. 1999;99:179–188. doi: 10.1016/s0092-8674(00)81649-9. [DOI] [PubMed] [Google Scholar]
- Djinovic-Carugo K, Gautel M, Ylanne J, Young P. The spectrin repeat: a structural platform for cytoskeletal protein assemblies. FEBS Lett. 2002;513:119–123. doi: 10.1016/s0014-5793(01)03304-x. [DOI] [PubMed] [Google Scholar]
- Ehlers MD. Reinsertion or degradation of AMPA receptors determined by activity-dependent endocytic sorting. Neuron. 2000;28:511–525. doi: 10.1016/s0896-6273(00)00129-x. [DOI] [PubMed] [Google Scholar]
- Engqvist-Goldstein AE, Drubin DG. Actin assembly and endocytosis: from yeast to mammals. Annu Rev Cell Dev Biol. 2003;19:287–332. doi: 10.1146/annurev.cellbio.19.111401.093127. [DOI] [PubMed] [Google Scholar]
- Gaidarov I, Santini F, Warren RA, Keen JH. Spatial control of coated-pit dynamics in living cells. Nat Cell Biol. 1999;1:1–7. doi: 10.1038/8971. [DOI] [PubMed] [Google Scholar]
- Gray NW, Fourgeaud L, Huang B, Chen J, Cao H, Oswald BJ, Hemar A, McNiven MA. Dynamin 3 is a component of the postsynapse, where it interacts with mGluR5 and Homer. Curr Biol. 2003;13:510–515. doi: 10.1016/s0960-9822(03)00136-2. [DOI] [PubMed] [Google Scholar]
- Harris TW, Hartwieg E, Horvitz HR, Jorgensen EM. Mutations in synaptojanin disrupt synaptic vesicle recycling. J Cell Biol. 2000;150:589–600. doi: 10.1083/jcb.150.3.589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarousse N, Kelly RB. Endocytotic mechanisms in synapses. Curr Opin Cell Biol. 2001;13:461–469. doi: 10.1016/s0955-0674(00)00237-4. [DOI] [PubMed] [Google Scholar]
- Kasai H, Matsuzaki M, Noguchi J, Yasumatsu N, Nakahara H. Structure-stability-function relationships of dendritic spines. Trends Neurosci. 2003;26:360–368. doi: 10.1016/S0166-2236(03)00162-0. [DOI] [PubMed] [Google Scholar]
- Kim WT, Chang S, Daniell L, Cremona O, Di Paolo G, De Camilli P. Delayed reentry of recycling vesicles into the fusion-competent synaptic vesicle pool in synaptojanin 1 knockout mice. Proc Natl Acad Sci USA. 2002;99:17143–17148. doi: 10.1073/pnas.222657399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirchhausen T. Clathrin. Annu Rev Biochem. 2000;69:699–727. doi: 10.1146/annurev.biochem.69.1.699. [DOI] [PubMed] [Google Scholar]
- Lee WC, Nedivi E. Extended plasticity of visual cortex in dark-reared animals may result from prolonged expression of cpg15-like genes. J Neurosci. 2002;22:1807–1815. doi: 10.1523/JNEUROSCI.22-05-01807.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang F, Huganir RL. Coupling of agonist-induced AMPA receptor internalization with receptor recycling. J Neuro-chem. 2001;77:1626–1631. doi: 10.1046/j.1471-4159.2001.00377.x. [DOI] [PubMed] [Google Scholar]
- Lin JW, Ju W, Foster K, Lee SH, Ahmadian G, Wyszynski M, Wang YT, Sheng M. Distinct molecular mechanisms and divergent endocytotic pathways of AMPA receptor internalization. Nat Neurosci. 2000;3:1282–1290. doi: 10.1038/81814. [DOI] [PubMed] [Google Scholar]
- Lledo PM, Zhang X, Sudhof TC, Malenka RC, Nicoll RA. Postsynaptic membrane fusion and long-term potentiation. Science. 1998;279:399–403. doi: 10.1126/science.279.5349.399. [DOI] [PubMed] [Google Scholar]
- Lois C, Hong EJ, Pease S, Brown EJ, Baltimore D. Germline transmission and tissue-specific expression of transgenes delivered by lentiviral vectors. Science. 2002;295:868–872. doi: 10.1126/science.1067081. [DOI] [PubMed] [Google Scholar]
- Luscher C, Xia H, Beattie EC, Carroll RC, von Zastrow M, Malenka RC, Nicoll RA. Role of AMPA receptor cycling in synaptic transmission and plasticity. Neuron. 1999;24:649–658. doi: 10.1016/s0896-6273(00)81119-8. [DOI] [PubMed] [Google Scholar]
- Malinow R, Malenka RC. AMPA receptor trafficking and synaptic plasticity. Annu Rev Neurosci. 2002;25:103–126. doi: 10.1146/annurev.neuro.25.112701.142758. [DOI] [PubMed] [Google Scholar]
- Man HY, Lin JW, Ju WH, Ahmadian G, Liu L, Becker LE, Sheng M, Wang YT. Regulation of AMPA receptor-mediated synaptic transmission by clathrin-dependent receptor internalization. Neuron. 2000;25:649–662. doi: 10.1016/s0896-6273(00)81067-3. [DOI] [PubMed] [Google Scholar]
- Metzler M, Li B, Gan L, Georgiou J, Gutekunst CA, Wang Y, Torre E, Devon RS, Oh R, Legendre-Guillemin V, et al. Disruption of the endocytic protein HIP1 results in neurological deficits and decreased AMPA receptor trafficking. EMBO J. 2003;22:3254–3266. doi: 10.1093/emboj/cdg334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mousavi SA, Malerod L, Berg T, Kjeken R. Clathrin-dependent endocytosis. Biochem J. 2004;377:1–16. doi: 10.1042/BJ20031000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nedivi E. Molecular analysis of developmental plasticity in neocortex. J Neurobiol. 1999;41:135–147. [PMC free article] [PubMed] [Google Scholar]
- Nedivi E, Hevroni D, Naot D, Israeli D, Citri Y. Numerous candidate plasticity-related genes revealed by differential cDNA cloning. Nature. 1993;363:718–722. doi: 10.1038/363718a0. [DOI] [PubMed] [Google Scholar]
- Nedivi E, Fieldust S, Theill LE, Hevron D. A set of genes expressed in response to light in the adult cerebral cortex and regulated during development. Proc Natl Acad Sci USA. 1996;93:2048–2053. doi: 10.1073/pnas.93.5.2048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nong Y, Huang YQ, Ju W, Kalia LV, Ahmadian G, Wang YT, Salter MW. Glycine binding primes NMDA receptor internalization. Nature. 2003;422:302–307. doi: 10.1038/nature01497. [DOI] [PubMed] [Google Scholar]
- Nusser Z. AMPA and NMDA receptors: similarities and differences in their synaptic distribution. Curr Opin Neurobiol. 2000;10:337–341. doi: 10.1016/s0959-4388(00)00086-6. [DOI] [PubMed] [Google Scholar]
- Nyiri G, Stephenson FA, Freund TF, Somogyi P. Large variability in synaptic N-methyl-D-aspartate receptor density on interneurons and a comparison with pyramidal-cell spines in the rat hippocampus. Neuroscience. 2003;119:347–363. doi: 10.1016/s0306-4522(03)00157-x. [DOI] [PubMed] [Google Scholar]
- Padmakumar VC, Abraham S, Braune S, Noegel AA, Tunggal B, Karakesisoglou I, Korenbaum E. Enaptin, a giant actin-binding protein, is an element of the nuclear membrane and the actin cytoskeleton. Exp Cell Res. 2004;295:330–339. doi: 10.1016/j.yexcr.2004.01.014. [DOI] [PubMed] [Google Scholar]
- Passafaro M, Piech V, Sheng M. Subunit-specific temporal and spatial patterns of AMPA receptor exocytosis in hippocampal neurons. Nat Neurosci. 2001;4:917–926. doi: 10.1038/nn0901-917. [DOI] [PubMed] [Google Scholar]
- Petralia RS, Wang YX, Wenthold RJ. Internalization at glutamatergic synapses during development. Eur J Neurosci. 2003;18:3207–3217. doi: 10.1111/j.1460-9568.2003.03074.x. [DOI] [PubMed] [Google Scholar]
- Rao A, Craig AM. Signaling between the actin cytoskeleton and the postsynaptic density of dendritic spines. Hippocampus. 2000;10:527–541. doi: 10.1002/1098-1063(2000)10:5<527::AID-HIPO3>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- Roche KW, Standley S, McCallum J, Dune Ly C, Ehlers MD, Wenthold RJ. Molecular determinants of NMDA receptor internalization. Nat Neurosci. 2001;4:794–802. doi: 10.1038/90498. [DOI] [PubMed] [Google Scholar]
- Sambrook J, Fritsch EF, Maniatis T. Molecular Cloning: A Laboratory Manual. Second. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- Santini F, Gaidarov I, Keen JH. G protein-coupled receptor/arrestin3 modulation of the endocytic machinery. J Cell Biol. 2002;156:665–676. doi: 10.1083/jcb.200110132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid SL. Clathrin-coated vesicle formation and protein sorting: an integrated process. Annu Rev Biochem. 1997;66:511–548. doi: 10.1146/annurev.biochem.66.1.511. [DOI] [PubMed] [Google Scholar]
- Scott MG, Benmerah A, Muntaner O, Marullo S. Recruitment of activated G protein-coupled receptors to pre-existing clathrin-coated pits in living cells. J Biol Chem. 2002;277:3552–3559. doi: 10.1074/jbc.M106586200. [DOI] [PubMed] [Google Scholar]
- Slepnev VI, De Camilli P. Accessory factors in clathrin-dependent synaptic vesicle endocytosis. Nat Rev Neurosci. 2000;1:161–172. doi: 10.1038/35044540. [DOI] [PubMed] [Google Scholar]
- Snyder EM, Philpot BD, Huber KM, Dong X, Fallon JR, Bear MF. Internalization of ionotropic glutamate receptors in response to mGluR activation. Nat Neurosci. 2001;4:1079–1085. doi: 10.1038/nn746. [DOI] [PubMed] [Google Scholar]
- Spacek J, Harris KM. Three-dimensional organization of smooth endoplasmic reticulum in hippocampal CA1 dendrites and dendritic spines of the immature and mature rat. J Neurosci. 1997;17:190–203. doi: 10.1523/JNEUROSCI.17-01-00190.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starr DA, Han M. ANChors away: an actin based mechanism of nuclear positioning. J Cell Sci. 2003;116:211–216. doi: 10.1242/jcs.00248. [DOI] [PubMed] [Google Scholar]
- Toni N, Buchs PA, Nikonenko I, Povilaitite P, Parisi L, Muller D. Remodeling of synaptic membranes after induction of long-term potentiation. J Neurosci. 2001;21:6245–6251. doi: 10.1523/JNEUROSCI.21-16-06245.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verstreken P, Kjaerulff O, Lloyd TE, Atkinson R, Zhou Y, Mein-ertzhagen IA, Bellen HJ. Endophilin mutations block clathrin-mediated endocytosis but not neurotransmitter release. Cell. 2002;109:101–112. doi: 10.1016/s0092-8674(02)00688-8. [DOI] [PubMed] [Google Scholar]
- Verstreken P, Koh TW, Schulze KL, Zhai RG, Hiesinger PR, Zhou Y, Mehta SQ, Cao Y, Roos J, Bellen HJ. Synaptojanin is recruited by endophilin to promote synaptic vesicle uncoating. Neuron. 2003;40:733–748. doi: 10.1016/s0896-6273(03)00644-5. [DOI] [PubMed] [Google Scholar]
- Wang YT, Linden DJ. Expression of cerebellar long-term depression requires postsynaptic clathrin-mediated endocytosis. Neuron. 2000;25:635–647. doi: 10.1016/s0896-6273(00)81066-1. [DOI] [PubMed] [Google Scholar]
- Zhou Q, Xiao M, Nicoll RA. Contribution of cytoskeleton to the internalization of AMPA receptors. Proc Natl Acad Sci USA. 2001;98:1261–1266. doi: 10.1073/pnas.031573798. [DOI] [PMC free article] [PubMed] [Google Scholar]