

Abstract
Aim
The goal of this investigation was to determine whether epigenetic modifications in the IFNG promoter are associated with an increase of IFNG transcription in different stages of periodontal diseases.
Materials and Methods
DNA was extracted from gingival biopsy samples collected from 47 total sites from 47 different subjects: 23 periodontally healthy sites, 12 experimentally induced gingivitis sites and 12 chronic periodontitis sites. Levels of DNA methylation within the IFNG promoter containing six CpG dinucleotides were determined using pyrosequencing technology. Interferon gamma mRNA expression was analysed by quantitative polymerase chain reactions using isolated RNA from part of the biological samples mentioned above.
Results
The methylation level of all six analysed CpG sites within the IFNG promoter region in the periodontitis biopsies {52% [interquartile range, IQR (43.8%, 63%)]} was significantly lower than periodontally healthy samples {62% [IQR (51.3%, 74%)], p =0.007} and gingivitis biopsies {63% [IQR (55%, 74%)], p =0.02}. The transcriptional level of IFNG in periodontitis biopsies was 1.96-fold and significantly higher than tissues with periodontal health (p =0.04). Although the mRNA level from experimental gingivitis samples exhibited an 8.5-fold increase as compared with periodontally healthy samples, no significant methylation difference was observed in experimental gingivitis sample.
Conclusions
A hypomethylation profile within IFNG promoter region is related to an increase of IFNG transcription present in the chronic periodontitis biopsies, while such an increase of IFNG in experimentally induced gingivitis seems independent of promoter methylation alteration.
Keywords: DNA methylation, gene expression, interferon-gamma, periodontal disease, pyrosequencing
The fundamental mechanisms that lead to the development of periodontal diseases are closely related to the dynamics of the host immune and inflammatory responses to periodontal pathogens present in the dental biofilm (Gemmell & Seymour 2004). Although much is known regarding the innate immune response in periodontal disease, the specific role of T cells in modulating local Th1 and Th2 responses is not fully characterized. Cell-mediated immunity modulated by a Th1 response, which involves interferon gamma (IFN-γ) production and interleukin (IL)-2, and the humoural immune response, which is favoured by a Th2 response and driven by the secretion of IL-4, IL-6 and IL-10, are well established and these responses have been described in periodontal diseases (Yamazaki et al. 1994, Aramaki et al. 1998, Dutzan et al. 2009, Gemmell & Seymour 2004, Wright et al. 2008).
The expression of IFN-γ is noteworthy, because of not only its elevated transcriptional and translational expression in inflamed gingival tissues and gingival crevicular fluid (GCF) but also its association with advanced periodontal disease and disease progression (Gemmell & Seymour 2004, Salvi et al. 1998, Takeichi et al. 2000). In a recent molecular epidemiologic study with 6768 community-based subjects, Offenbacher et al. (2007) reported a significant increase in the GCF concentration of IFN-γ in those subjects with deep periodontal pockets and severe gingival bleeding as compared with subjects with probing depth (PD) of <3 mm. It was demonstrated previously that a high level of Th1 cytokines was found in the GCF of patients with extremely severe periodontits (terminal dentition stage), including a 10-fold increase in the concentration of IFN-γ when compared with the Th2 mediators IL-4 and IL6 (Salvi et al. 1998). The presence of high IFN-γ level is shown to enhance the phagocytic activity of monocytes and neutrophils, which helps containment of infection (Gemmell & Seymour 2004) as well as upregulates monocytic response to LPS, which results in elevated monocytic secretion of proinflammatory molecules, such as PGE2, IL-1β and TNF-α, all of which play important roles in bone loss and the disintegration of soft tissue in the periodontium (Salvi et al. 1998, Nichols & Garrison 1987). The literature demonstrates that IFN-γ can be secreted by type-1 CD4+, CD8+ T lymphocytes, NK cells, mononuclear cells and dendritic cells found in periodontal tissues (Takeichi et al. 2000, Fujihashi et al. 1996). However, the molecular signalling pathways that result in a chronically elevated level of IFN-γ expression in periodontal diseases are still the subject of investigation.
The control of inflammatory responsiveness by the host to the recurrent and omnipresent challenges imposed by the oral biofilm is complex, involving genetically determined traits, regulation by cytokine networks and changes in epigenetic patterns. For example, genetic studies suggest that specific single nucleotide polymorphism (SNP) haplotypes of IL1B in the population are associated with the level of IL-1β within the GCF (Rogus et al. 2008). IL-12 has been shown to potently enhance IFNG expression (Watford et al. 2003). Recently, alterations in epigenetic patterns have been discovered as another important mechanism for the regulation of gene expression at mucosal surfaces (Nakajima et al. 2009). In eukaryotes, DNA methylation occurs almost exclusively at the 5′ end of cytosine nucleotide within the CpG dinucleotide context (Bird & Wolffe 1999). The change of methylation status in CpG islands, which are regions of genome that contain a high percentage of CpG dinucleotides, are profoundly associated with diseases such as developmental abnormalities, cancer and chronic inflammatory states (Bird 2002, Barros & Offenbacher 2009). It has been generally accepted that an increased methylation (hypermethylation) in the gene promoter region is associated with a decrease of gene expression, while a hypomethylation pattern is closely associated with transcriptional upregulation (Jones & Laird 1999, Shuto et al. 2006). Recently, we have described an increased methylation of CpG islands within the PTGS2 promoter region in human gingival biopsies associated with a suppression of PGE2 mRNA expression (Zhang et al. 2010).
The aim of this study is to understand whether IFN-γ expression in the gingival tissues from subjects with different stages of periodontal diseases, including experimentally induced gingivitis and chronic periodontitis, is associated with an altered methylation status of the promoter region of IFNG, as evaluated in the context of IFNG SNPs known to regulate expression levels.
Materials and Methods
Participants, experimental gingivitis and tissue specimens
A total of 47 participants, aged between 19 and 63 years, provided informed consent and were enrolled into this study wherein all components were approved by the Institutional Review Board (IRB) of the University of North Carolina at Chapel Hill. Exclusion criteria for recruiting participants exhibiting chronic periodontitis and periodontal health included: (1) the use of either antibiotics or non-steroidal anti-inflammatory drugs within 1 month before scheduled surgery; and (2) medical treatment for other diseases 3 months before recruitment. Measurements included PDs, clinical attachment levels (CAL), and bleeding on probing (BOP) at six sites per tooth. One interproximal gingival site was biopsied from each participant. Twelve gingival biopsies were removed during routine periodontal flap surgeries from participants clinically diagnosed with chronic periodontitis. Scaling and root planing as initial therapy were performed on those patients four to six weeks before periodontal surgeries. Those biopsied tissues were from sites exhibiting PDs of 5 mm or more, BOP and radiographic evidence of localized bone loss. Twenty-three gingival tissues were collected from different participants who were periodontally healthy or had localized mild gingivitis at non-study sites. These gingival biopsies were removed from either periodontally healthy volunteers or participants who were undergoing crown lengthening procedures at sites with PD measurements of 4 mm or less at all six interproximal probing sites, no BOP and no evidence of radiographic bone loss.
Another 12 biopsied samples were collected from different participants exhibiting experimental gingivitis. The gingivitis was induced following a 21-day stent-induced biofilm overgrowth protocol as described previously (LOE et al. 1965). In addition to the exclusion criteria mentioned above, all participants had at least four teeth in each posterior sextant. In this protocol, gingivitis participants with BOP scores of ≥10% (Loe et al. 1986) and pocket depth probing <5 mm were brought to periodontal health by initial dental prophylaxis and supragingival scaling. After 1 week following this treatment, baseline GI scores were collected and the subjects were instructed to wear two stents during routine toothbrushing and not to floss the stent-covered teeth. This stent covered the tooth surfaces and promoted biofilm overgrowth during a 3-week induction period. At day 21, biopsies were collected from one interproximal site of gingiva in one of the stent sextants.
Upon removal, all biopsied gingival tissues were divided into two comparable samples. One half, used for DNA methylation analysis, was kept in the − 80°C freezer immediately, while the other half, used for real-time polymerase chain reaction (RT-PCR), was incubated with RNA later (Applied Biosystems/Ambion, Austin, TX, USA) overnight at 4°C, and then transferred to −80°C.
DNA isolation and sodium bisulphite conversion
Genomic DNA was isolated from collected gingival tissue samples using a DNeasy Mini Kit (Qiagen, Valencia, CA, USA) according to manufacturer’s instructions. Genomic DNA was bisulphite treated using published procedures (Grunau et al. 2001). Briefly, 1–2 μg of genomic DNA in 45 μl of nuclease-free water was denatured at 42°C for 20 min. with 5 μl of freshly prepared 3 M sodium hydroxide. Denatured DNA was incubated with freshly prepared sodium bisulphite (saturated) and hydroquinone solution in the water bath at 55°C for 16 h. The bisulphite-converted DNA was purified using a Wizard DNA Clean-up Column (Promega, Madison, WI, USA) and then desulphonated by incubation with 5.5 μl of a 3 M NaOH solution at 37°C for 20 min. The bisulphate-treated DNA was finally precipitated with ethanol, and then redissolved in 25 μl of 1 mM Tris-Cl pH 8.
Bisulphite specific PCR and pyrosequencing
The detailed information of primers used in the bisulphite-specific PCR can be found in Table 1. For methylation analysis, five amplicons, which were amplified by using a HotStar Taq kit (Qiagen), include a total of six CpG sites within the proximal promoter region of IFNG. PCR conditions for each individual amplicon, in which specific CpG sites were included, can be found in Table 2. Direct quantification of the ratio of methylated to unmethylated cytosine nucleotide for each analysed CpG site present in the amplicons was determined by pyrosequencing with the PSQ HS 96 Pyrosequencing System (Qiagen) and Pyro Gold CDT Reagents (Qiagen) as described previously (Colella et al. 2003). We have also checked a polymorphism at site −179 bp in the same amplicon containing the CpG site of − 186 bp by pyrosequencer. In each pyrosequencing assay, one amplicon was used for sequencing and the corresponding sequencer can be found in Table 1. Internal controls for bisulphite conversion efficiency were included in each pyrosequencing assay. A genomic sequence that is artificially methylated on all its CpG dinucleotides (Cat. # S7821, Millipore, Billerica, MA, USA) was also used in the bisulphite conversion, PCR and pyrosequencing with the primers and sequencers mentioned above as a technical control.
Table 1.
Oligonucleotides used for bisulphite-specific polymerase chain reaction and pyrose-quencing
| CpG site (*) | Details | Sequences |
|---|---|---|
| −295 | Forward | 5′-[Biotin] TTTGTAAAGGTTTGAGAGGTTTTAGAAT-3′ |
| Reverse | 5′-CAAACCCATTATACCCACCTATACCA-3′ | |
| Sequencer | 5′-TTTTATACCTCCCCACTT-3′ | |
| −186 | Forward | 5′-TTAGAATGGTATAGGTGGGTATAATGG-3′ |
| Reverse | 5′-[Biotin] TATTATAATTAAAATTTCCTTTAAACTCCT-3′ |
|
| Sequencer | 5′-GGGTATAATGGGTTTGTT-3′ | |
| −54 | Forward | 5′-GGGTTTGTTTTATAGTTAAAGGATTTAAGG-3′ |
| Reverse | 5′-[Biotin] AATCAAAACAATATACTACACCTCCTCTAA-3′ |
|
| Sequencer | 5′-TATTTTATTTTAAAAAATTTGTG-3′ | |
| +122 ~ | Forward | 5′-[Biotin] TTTTGGATTTGATTAGTTTGATATAAGAA-3′ |
| +128 | Reverse | 5′-AAAACCCAAAACCATACAAAACTAAAA-3′ |
| Sequencer | 5′-CTAAAAAACCAAAATATAACTTAT-3′ | |
| +171 | Forward | 5′-[Biotin] TTTTGGATTTGATTAGTTTGATATAAGAA-3′ |
| Reverse | 5′-CATTTTCAACCACAAACAAATACTATTAA-3′ | |
| Sequencer | 5′-ACAACCAAAAAAACCC-3′ |
CpG sites indicate nucleotide position in relation to transcription start.
“−” or “+”, indicates upstream or downstream of transcription start, respectively.
Table 2.
Bisulphite-specific polymerase chain reaction conditions for CpG containing amplicons
| Amplicons [CpG site(s) included] | Initial denaturing T (°C), T (min.) | Cycles (X45) |
Final elongation T (°C), T (min.) | ||
|---|---|---|---|---|---|
| denaturing T (°C), T (s) | annealing T (°C), T (s) | elongation T (°C), T (s) | |||
| −295 | 94,15 | 94,30 | 58,30 | 72,30 | 72,10 |
| −186 | 94,15 | 94,30 | 54,30 | 72,30 | 72,10 |
| −54 | 94,15 | 94,30 | 60,30 | 72,30 | 72,10 |
| +122–+128 | 94,15 | 94,30 | 55,30 | 72,30 | 72,10 |
| +171 | 94,15 | 94,30 | 55,30 | 72,30 | 72,10 |
Quantitative real time PCR
Total RNA was isolated from gingival tissues with the use of an RNeasy Mini Kit (Qiagen). cDNA from 500 ng of total RNA was synthesized using an Omniscript Kit (Qiagen) and random decamer primers (Cat. # 5722E, Austin, TX, USA). Real-time PCR was performed with 1 μl of synthesized cDNA, 12.5 μl TaqMan Universal PCR mix, and 1.25 μl 20X assay on demand gene expression assay mix (Mm00445273_m1, Applied Biosystems, Foster City, CA, USA), in a 7000 Sequence Detection System (ABI Prism, Applied Biosystems). Each sample was performed in duplicates. The ribosomal 18S Control Reagents (part no. 4308329), which was also from Applied Biosystems, was used as an endogenous control for data normalization. The relative quantity of IFNG mRNA was calculated against 18S rRNA values according to the method recommended by Livak & Schmittgen (2001).
Immunofluorescence
After sectioning, the frozen gingival tissues from healthy gingival and chronic periodontitis tissues were fixed with 70% ethanol for 15 s and followed by acetone for another 5 min. Then, the frozen tissue slides were blocked for 1 h at room temperature, and then incubated overnight at − 4°C with monoclonal antibodies specific for CD4 (Cat. #14-0049, eBioscience, San Diego, CA, USA), CD11C (CAt.# 14-0116, eBiosciences) CD56 (Cat.# 14-0567, eBioscience). All the antibodies except for anti-CD4 were 1:50 diluted in a blocking buffer containing 8% bovine serum albumin, 1% goat serum and 1% Triton-X-100. Anti-CD4 antibody was 1:20 diluted with the same blocking buffer. After vigorous washing with 1 × PBS containing 0.1% Triton-X-100 the slides were incubated for 1 h at room temperature with a secondary antibody, Alexa Fluor 488 goat anti-mouse IgG (Molecular Probes, Invitrogen, Carlsbad, CA, USA). Sections were then washed in 0.1% Triton/PBS, mounted with ProLong Gold antifade reagent with DAPI (Invitrogen) and coverslipped. Sections were analysed using confocal microscopy (Carl Zeiss LSM 710 Confocal Microscope, Thornwood, NY, USA).
Statistical analysis
Analysis of variance was applied for the statistical analysis of clinical measurement. Fisher’s exact test was used to test gender difference among participants in different groups. Mann–Whitney/Wilcoxon two-sample test was used to compare the methylation level of each CpG site and the overall percent methylation of gingival samples among different biopsy groups. Linear regression analysis was applied for testing for significance of the slope to analyse the IFNG messenger level of different sample groups. The threshold for all statistical significance was set at a p-value <0.05.
Results
Selected characteristics between the three subsets of individuals are summarized in Table 3. There were no age differences between periodontally diseased subjects (either periodontitis or experimental gingivitis) and participants with periodontal health. As expected, in the periodontitis tissue group, both PD and CAL were greater than the periodontal health group (p<0.001 for both). Although there were more male participants in chronic periodontitis group as compared with the periodontal health group (p =0.01), there is no evidence so far that gender has a differential effect on methylation status of the IFNG promoter in periodontal diseases.
Table 3.
Demographic information of the participants and clinical parameters in the biopsied gingival sites
| Demographic/Clinical parameters | Periodontal health (n =23) | Experimental gingivitis (n =12) | Periodontitis (n =12) |
|---|---|---|---|
| Mean age (years) | 40.8 ± 11.6 | 35.8 ± 11.2 | 47.2 ± 7.4 |
| Gender | |||
| Males/Females | 6/17 | 5/7 | 9/3* |
| Probing depth (Mean±SD, mm) | 2.2 ± 0.6 | 2.24 ± 0.63 | 6.3 ± 0.8** |
| Clinic attachment level (Mean±SD, mm) | 1.2 ± 0.6 | 1.3 ± 0.45 | 4.7 ± 1.5** |
p<0.05 as compared with periodontal health.
p<0.001 as compared with periodontal health.
The positions of all six analysed CpG sites within the promoter region of the IFNG gene are depicted in Fig. 1. Because the methylation pattern of the CpG sites shown in this region has been demonstrated to be mechanistically related to the control of IFN-γ expression in various studies (White et al. 2002, White et al. 2006, Gonsky et al. 2009), we performed DNA methylation analysis on these CpG sites in our biopsied gingival tissues.
Fig. 1.

Diagrammatic representation of the IFNG promoter region (GeneBank accession no. J00219). “ “ indicates CpG dinucleotide sequences that can be potentially methylated. The position of each CpG dinucleotide site is illustrated in relation to the transcription start, which is indicated as “+1” bp.
“ indicates CpG dinucleotide sequences that can be potentially methylated. The position of each CpG dinucleotide site is illustrated in relation to the transcription start, which is indicated as “+1” bp.
Figure 2 demonstrates two representative sequencing pyrograms of the methylation level of the IFNG promoter region taken from a periodontally healthy and a chronic periodontitis sample. The pyrogram in Fig. 2a shows that, at the CpG dinucleotide at position −295 bp, 71% of the amplification products from one healthy gingival tissue sample contained a methylated cytosine nucleotide (Fig. 2a). The pyrogram in Fig. 2b demonstrates that at position −295 bp, 49% of the amplification products from one periodontitis tissue sample contained a methylated cytosine residue (Fig. 2b).
Fig. 2.

Representative programs of pyrosequencing. The percentage on each CpG site is the methylation percentage of mC/(mC+C) on that site. mC, methylated cytosine; C, unmethylated cytosine. The sequence shown on the top of each panel is the assayed sequence. The x-axes are the dispensation nucleotides to the sequencing reaction based on the assayed sequences. The y-axes show light emission obtained as relative light units. The methylation percentage of one specific CpG site (− 295 bp) from a healthy gingival biopsy sample (a), which shows the value of methylation percentage of 71, and a chronic periodontitis sample (b), which shows a value of methylation percentage of 49 are illustrated. This percentage is calculated from the reference peak heights, marked as dark diamonds, which are non-CpG nucleotide sequences. The red diamond marks indicate “built-in” bisulphite controls in pyrosequencing.
A lower level of methylation as determined by pyrosequencing was found at each individual CpG site within the IFNG promoter region in the DNA samples from the chronic periodontitis tissues as compared with tissues with periodontal health (Fig. 3a). Of all the analysed CpG dinucleotides, the methylation levels at site −295 bp, − 54 bp and +171 bp from the chronic periodontitis DNA samples were significantly lower than the healthy gingival samples {44.5% [inter-quartile range, IQR (37%, 50.5%)] versus 60% [IQR (51.5%, 64.5%)], p =0.002 for site −295 bp, 58.5% [IQR (55.3%, 62%)] versus 65% [IQR (58.5%, 71%)], p =0.04 for site − 54 bp, and 49% [IQR (47.5%, 50.3%)] versus 55% [IQR (52.5%, 59%)], p =0.0007 for site +171 bp}. In contrast, there is no significant difference of DNA methylation percentage in each analysed CpG site in the experimentally induced gingivitis group as compared with periodontal health samples. One of the designed amplicons encompassing one methylation site at −186 bp also contained a reported SNP at position − 179 bp. The G/T SNP (rs2069709) reported at site −179 bp is reported to be associated with an elevated level of IFNG gene expression (Smith & Humphries 2009, Bream et al. 2002). We examined this polymorphism for all the samples along with DNA methylation analysis. No minor allele (− 179T) was detected in any of the analysed samples (data not shown).
Fig. 3.

(a) The percentage of methylation in each individual CpG site from healthy gingival tissues, experimental gingivitis and chronic periodontitis biopsies. *,**,***Significantly lower methylation level in chronic periodontitis biopsies than in healthy samples at CpG site −54 (p =0.04), CpG site −295 (p =0.002) and CpG site +171 (p =0.0007), respectively. Plus error bars and minus error bars demonstrate the maximal and minimal methylation percentages, respectively, while the top line, middle line and the bottom line of boxes illustrate 75th percentile, median and 25th percentile of the methylation levels in each group analysed, respectively. (b) Percentage of overall methylation level combining all six analysed CpG sites within the IFNG promoter region in biopsied tissues exhibiting periodontal health, experimental gingivitis and chronic periodontitis. “**” and “*” indicate that the overall methylation level of IFNG promoter region in chronic periodontitis gingival tissues was statistically lower than the percentage of methylation in healthy tissues (p =0.007) and experimental gingivitits biopsies (p =0.02), respectively. The top, middle and bottom lines of the box plot indicate the 75th percentile, median and 25th percentile methylation levels, respectively. Plus error bars and minus error bars demonstrate the maximal and minimal methylation percentages, respectively.
The overall median methylation percentage of all the six CpG sites within IFNG promoter was 52% [IQR (43.8%, 63%)] in the chronic periodontitis biopsies and significantly lower than the methylation percentage in samples with periodontal health and experimental gingivitis biopsies, which were 62% [IQR (51.3%, 74%)] (p =0.007) and 63% [IQR (55%, 74%)] (p =0.02), respectively (Fig. 3b). No significant difference can be found in the IFNG promoter methylation levels comparing samples with periodontal health with samples with experimental gingivitis.
Because DNA methylation level is a critical regulatory mechanism for gene transcription, we also measured the messenger (mRNA) level of IFNG in biopsied tissues. The transcriptional level of IFNG was 1.96-fold increased and significantly higher in the periodontitis biopsy samples compared with the healthy gingival samples as determined by quantitative real-time PCR (p =0.04, Fig. 4). In contrast, there was an 8.5-fold induction of IFNG transcription in experimentally induced gingivitis samples as compared with samples with periodontal health, and this increase of messenger level was statistically significant (p =0.01).
Fig. 4.
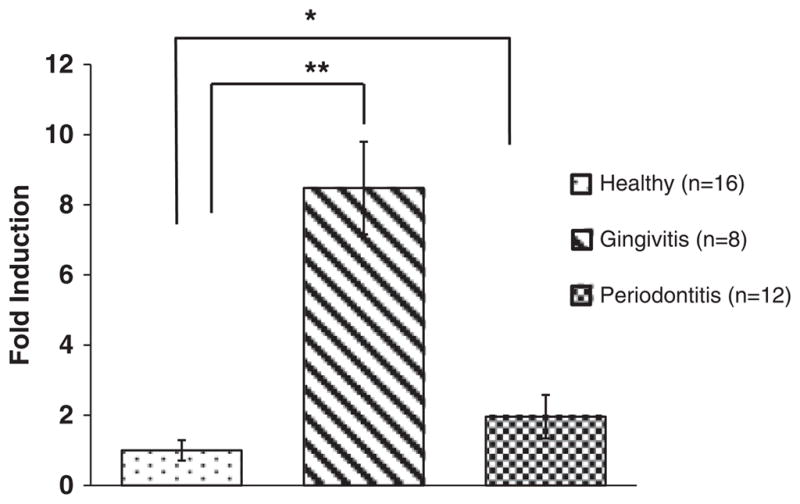
mRNA expression levels of IFNG in the healthy gingival biopsies, experimental gingivitis and chronic periodontitis samples. Fold induction is shown using 18S as an internal housekeeping gene. As compared with gingival biopsies with periodontal health, a 1.96-fold increase in the transcriptional level of IFNG is significantly higher (p =0.04) in the chronic periodontitis biopsies, which was indicated by “*”. In contrast, the transcriptional level of IFNG in experimental gingivitis showed an 8.5-fold increase and was significantly higher (p =0.01) than tissues with peridontal health, which was indicated by “**”.
We also performed an immunofluorescence experiment to identify the IFN-γ-secreting cell populations and compare them between chronic periodontitis samples and tissues with periodontal health. In chronic periodontitis tissues more CD4+ T cells were present in the epithelial and lamina propia as compared with the tissues with periodontal health (Fig. 5a and b). In addition, we also found more CD56+ (Fig. 5c and d) and CD11C (data not shown) cells, which are markers for NK and dendritic cells, respectively, infiltrated in the chronically diseased biopsies than samples in healthy gingival tissues.
Fig 5.
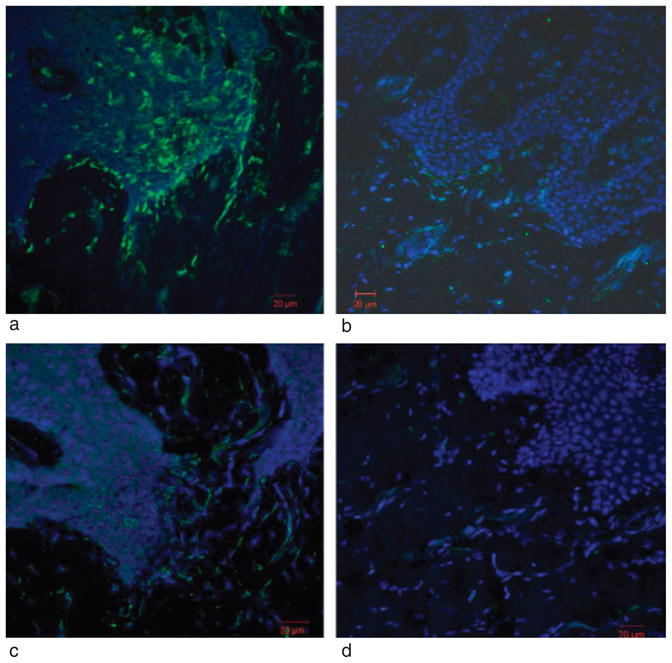
Immunofuorescence staining of CD4+ and CD56+ cells in one representative chronic periodontitis biopsy and one biopsy with periodontal health. (a) and (b) Biopsy section from either a site with chronic periodontits (a) or a site with periodontal health (b) showing CD4+ (green) T cells within the gingival epithelium and lamina propria. (c) and (d) A chronic periodontitis (c) and a healthy gingival tissue (d) were stained for CD56+ (green) NK cells. Nuclei were stained with DAPI (blue).
Discussion
Although B-cell mediated adaptive immune responses dominate in chronic periodontitis, differential distribution of Th1 or Th2 cytokine secretion profiles have been reported to be related to the severity and/or progression of periodontal diseases (Salvi et al. 1998, Garlet et al. 2003). Among the Th1 cytokines, IFN-γ plays a central role in the containment of infection and represents one of the most efficient cytokines for triggering antimicrobial activity in both macrophages and neutrophils (Huang et al. 1993, Meda et al. 1994, Farrar & Schreiber 1993). In addition to priming the antimicrobial activities of phagocytes, overexpression of IFN-γ levels may also lead to direct and indirect host tissue destruction through the activation of these phagocytes (Boehm et al. 1997).
In our study, we noted a significant 1.96-fold increase in IFN-γ messenger levels in chronically inflamed periodontal biopsies as compared with periodontally healthy samples, consistent with several earlier reports (Offenbacher et al. 2007, Dutzan et al. 2009). Studies have shown an increased IFN-γ level within the GCF and higher IFN-γ expression in gingival tissues from progressive periodontal lesions (Dutzan et al. 2009). By contrast, other studies have suggested that elevated IFN-γ is a characteristic of the stable periodontal lesion and that active lesions favour a Th2 cell response (Lappin et al. 2001, Gemmell et al. 2002, Reinhardt et al. 1989). We also found that the transcriptional level of IFNG was 8.5-fold higher in the experimentally induced gingivitis samples as compared with tissues with periodontal health. Such a more pronounced transcriptional expression seems independent of IFNG methylation in the analysed promoter region. Although the inflammatory lesion from experimental gingivitis is histologically similar to the chronic periodontitis in that inflammatory infiltrates dominated by lymphocytes and antigen-presenting cells such as macrophage and dendritic cells are noticeably present (Zachrisson 1968, Seymour et al. 1988), the mechanisms for IFN-γ production may be possibly different in the different stages of periodontal diseases. For example, a cytokine-dominating mechanism such as high IL-12 secretion from antigen-presenting cells and IL-18 may be possibly responsible for the observed high-level production of IFN-γ in Th1-committed cells in experimental gingivitis lesions (Yang et al. 1999, Berenson et al. 2004). In addition, p38 and JNK signalling pathways have also been shown to be critically involved in IFN-γ production (Lu et al. 2001). However, this study supports that a decreased methylation profile within the IFNG promoter region may contribute to a higher gene transcriptional level, as found in chronically diseased gingival tissues. We hypothesized here that the promoter region of IFNG in infiltrating Th1 cells may be already epigenetically modified before migration into the lesion after prolonged and systemic exposure to periodontal pathogens present in the oral biofilm in chronic periodontitis patients.
It has been extensively reported that genetic polymorphisms can regulate cytokine expression levels. Bream et al. (2002) reported a guanidine to thymidine transition at site −179bp (G/T) within the IFNG proximal promoter; these authors reported that, as compared with −179G allele, the − 179T allele exhibited a 6–13-fold increase in the expression of IFNG in a promoter assay. The absence of SNP at position −179 bp within the IFNG promoter in all the gingival biopsies analysed by pyrosequencing supports the argument that the differences in IFNG expression in the chronic periodontitis biopsies were less likely attributable to IFNG polymorphisms, but more likely due to epigenetic influences conferred by prolonged environmental exposures.
In this study, the general demethylation pattern across all six CpG sites within the IFNG promoter region in the periodontal lesions could be due to chronic inflammation or the direct action and/or invasion of periodontal pathogens. Hypomethylation in chronically diseased gingival tissues could reflect a dilution of the tissue DNA pool by an influx of non-methylated DNA-bearing cells or the loss of methylation from the resident cells. We have found that more CD4+ T cells, CD11C+ cells and CD56+ NK cells infiltrated in the periodontitis gingival samples than tissues in healthy gingival samples. Therefore, the observed lower methylation pattern in the IFNG promoter region in periodontitis may be associated with altered methylation patterns on those cells capable of IFN-γ production. Nares et al. (2009) showed a marked infiltration by various inflammatory cell types in periodontally inflamed tissues in comparison with healthy gingival tissues, and also demonstrated that IFN-γ immunostaining was related to the presence of monocytes, macrophages and lymphocytes in periodontally diseased tissues. This evidence is consistent with our hypothesis that in periodontally diseased gingival tissues, hypomethylation status of IFNG promoter in these inflammatory cell types contributes to the observed higher IFN-γ expression in comparison with healthy gingival tissues. This association has also been reported and discussed by others; Gonsky et al. (2009), examining patients with inflammatory bowel disease, collected intestinal specimens from patients undergoing surgical resection of the colon, and showed that the infiltrated T cells isolated from the lamina propria in the mucosal tissues exhibited lower level of IFNG methylation in comparison with T cells isolated from peripheral blood from the same patients, suggesting that the epigenetic methylation status of IFNG plays a mechanistic role in the modulation of IFNG secretion in the mucosa. In another recent study on the methylation levels of IFNG promoter in human dental pulp tissues, the authors found an elevated level of hypomethylation in symptomatic pulpitis as compared with control pulp samples from impacted teeth, and also associated the IFNG hypomethylation levels with an increased number of infiltrating mononuclear cells in the inflamed tissues (Cardoso et al. 2010).
A hypomethylation pattern of the IFNG promoter region has also been reported by several groups as the hallmark of T cell commitment to a Th1 phenotype (White et al. 2002, White et al. 2006, Kwon et al. 2008). In this study, although the methylation level of IFNG from periodontal lesions is significantly lower than control sites [52% (IQR, 43.8%, 63%) versus 62% (IQR, 51.3%, 74%) p =0.007], the magnitude of the difference (~ 10%) is not large, probably due to the fact that the modulation of methylation patterns of IFN-γ-competent cell populations, which are infiltrated inflammatory cells, constitute a relatively small percentage of the total cell numbers within the periodontal lesion. Therefore, the magnitude of methylation difference between chronic periodontitis biopsies and gingival tissue with periodontal health would be expected to be relatively small, diluted by the overwhelming presence of non-IFN-γ competent cell populations in the tissue. However, this small decrease in methylation can account for a greater induction of messenger level of IFNG because it may be targeted to the IFN-γ-producing cells. Gonsky et al. (2009) reported that a 5% lower methylation level observed in the IFNG promoter region of lamina propria T cells from patients with inflammatory bowel disease as compared with controls was associated with an almost 3-fold induction of IFNG transcription. Thus, the difference of the IFNG methylation level that we observed in the oral mucosa is in close agreement with the levels observed in inflammatory bowel disease. However, the causal relationship between a small change of methylation in the promoter region of IFNG and an increase of its transcription level requires further mechanistic evidence supported by experiments such as chromatin immunoprecipitation, electrophoretic mobility shift assay, etc.
The findings from this study indicate the potential role of local epigenetic effects that result in regional modifications of the tissue DNA methylation status in the pathogenesis of chronic inflammatory periodontal disease. The observed hypomethylation of the IFNG promoter is a characteristic of a chronic inflammatory state. The study of methylation in inflamed tissues under chronic infection is still at an early stage and altered methylation patterns likely affect many genes in disease. Additional research is needed to elucidate the potential diagnostic utility of epigenetic markers as a determinant of disease progression, as well as response to treatment. Because the DNA methylation status can be modified by certain drugs, the possibility of reversing epigenetic modifications may have profound effects on periodontal treatment responses.
Acknowledgments
source of funding statement
This work was supported by grants: GCRC: RR00046 and UL1RR025747 (National Institutes of Health/National Center for Research Resources, Bethesda, MD).
Footnotes
Conflict of interest
The authors declare no conflicts of interests.
References
- Aramaki M, Nagasawa T, Koseki T, Ishikawa I. Presence of activated B-1 cells in chronic inflamed gingival tissue. Journal of Clinical Immunology. 1998;18:421–429. doi: 10.1023/a:1023234823783. [DOI] [PubMed] [Google Scholar]
- Barros SP, Offenbacher S. Epigenetics: connecting environment and genotype to phenotype and disease. Journal of Dental Research. 2009;88:400–408. doi: 10.1177/0022034509335868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berenson LS, Ota N, Murphy KM. Issues in T-helper 1 development–resolved and unresolved. Immunological Reviews. 2004;202:157–174. doi: 10.1111/j.0105-2896.2004.00208.x. [DOI] [PubMed] [Google Scholar]
- Bird AP, Wolffe AP. Methylation-induced repression–belts, braces, and chromatin. Cell. 1999;99:451–454. doi: 10.1016/s0092-8674(00)81532-9. [DOI] [PubMed] [Google Scholar]
- Bird A. DNA methylation patterns and epigenetic memory. Genes and Development. 2002;16:6–21. doi: 10.1101/gad.947102. [DOI] [PubMed] [Google Scholar]
- Boehm U, Klamp T, Groot M, Howard JC. Cellular responses to interferon-gamma. Annual Review of Immunology. 1997;15:749–795. doi: 10.1146/annurev.immunol.15.1.749. [DOI] [PubMed] [Google Scholar]
- Bream JH, Ping A, Zhang X, Winkler C, Young HA. A single nucleotide polymorphism in the proximal IFN-gamma promoter alters control of gene transcription. Genes and Immunity. 2002;3:165–169. doi: 10.1038/sj.gene.6363870. [DOI] [PubMed] [Google Scholar]
- Cardoso FP, Viana MB, Sobrinho AP, Diniz MG, Brito JA, Gomes CC, Moreira PR, Gomez RS. Methylation pattern of the IFN-gamma gene in human dental pulp. Journal of Endodontics. 2010;36:642–646. doi: 10.1016/j.joen.2009.12.017. [DOI] [PubMed] [Google Scholar]
- Colella S, Shen L, Baggerly KA, Issa JP, Krahe R. Sensitive and quantitative universal pyrosequencing methylation analysis of CpG sites. BioTechniques. 2003;35:146–150. doi: 10.2144/03351md01. [DOI] [PubMed] [Google Scholar]
- Dutzan N, Vernal R, Hernandez M, Dezerega A, Rivera O, Silva N, Aguillon JC, Puente J, Pozo P, Gamonal J. Levels of interferon-gamma and transcription factor T-bet in progressive periodontal lesions in patients with chronic periodontitis. Journal of Periodontology. 2009;80:290–296. doi: 10.1902/jop.2009.080287. [DOI] [PubMed] [Google Scholar]
- Farrar MA, Schreiber RD. The molecular cell biology of interferon-gamma and its receptor. Annual Review of Immunology. 1993;11:571–611. doi: 10.1146/annurev.iy.11.040193.003035. [DOI] [PubMed] [Google Scholar]
- Fujihashi K, Yamamoto M, Hiroi T, Bamberg TV, McGhee JR, Kiyono H. Selected Th1 and Th2 cytokine mRNA expression by CD4(+) T cells isolated from inflamed human gingival tissues. Clinical and Experimental Immunology. 1996;103:422–428. doi: 10.1111/j.1365-2249.1996.tb08297.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garlet GP, Martins W, Jr, Ferreira BR, Milanezi CM, Silva JS. Patterns of chemokines and chemokine receptors expression in different forms of human periodontal disease. Journal of Periodontal Research. 2003;38:210–217. doi: 10.1034/j.1600-0765.2003.02012.x. [DOI] [PubMed] [Google Scholar]
- Gemmell E, Seymour GJ. Immunoregulatory control of Th1/Th2 cytokine profiles in periodontal disease. Periodontology 2000. 2004;35:21–41. doi: 10.1111/j.0906-6713.2004.003557.x. [DOI] [PubMed] [Google Scholar]
- Gemmell E, Yamazaki K, Seymour GJ. Destructive periodontitis lesions are determined by the nature of the lymphocytic response. Critical reviews in oral biology and medicine: an official publication of the American Association of Oral Biologists. 2002;13:17–34. doi: 10.1177/154411130201300104. [DOI] [PubMed] [Google Scholar]
- Gonsky R, Deem RL, Targan SR. Distinct methylation of IFNG in the gut. Journal of Interferon and Cytokine Research: The Official Journal of the International Society for Interferon and Cytokine Research. 2009;29:407–414. doi: 10.1089/jir.2008.0109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grunau C, Clark SJ, Rosenthal A. Bisulphite genomic sequencing: systematic investigation of critical experimental parameters. Nucleic Acids Research. 2001;29:E65–5. doi: 10.1093/nar/29.13.e65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang S, Hendriks W, Althage A, Hemmi S, Bluethmann H, Kamijo R, Vilcek J, Zinkernagel RM, Aguet M. Immune response in mice that lack the interferon-gamma receptor. Science. 1993;259:1742–1745. doi: 10.1126/science.8456301. [DOI] [PubMed] [Google Scholar]
- Jones PA, Laird PW. Cancer epigenetics comes of age. Nature Genetics. 1999;21:163–167. doi: 10.1038/5947. [DOI] [PubMed] [Google Scholar]
- Kwon NH, Kim JS, Lee JY, Oh MJ, Choi DC. DNA methylation and the expression of IL-4 and IFN-gamma promoter genes in patients with bronchial asthma. Journal of Clinical Immunology. 2008;28:139–146. doi: 10.1007/s10875-007-9148-1. [DOI] [PubMed] [Google Scholar]
- Lappin DF, MacLeod CP, Kerr A, Mitchell T, Kinane DF. Anti-inflammatory cytokine IL-10 and T cell cytokine profile in periodontitis granulation tissue. Clinical and Experimental Immunology. 2001;123:294–300. doi: 10.1046/j.1365-2249.2001.01448.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods (San Diego, Calif) 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Loe H, Anerud A, Boysen H, Morrison E. Natural history of periodontal disease in man. Rapid, moderate and no loss of attachment in Sri Lankan laborers 14 to 46 years of age. Journal of Clinical Periodontology. 1986;13:431–445. doi: 10.1111/j.1600-051x.1986.tb01487.x. [DOI] [PubMed] [Google Scholar]
- Loe H, Theilade E, Jensen SB. Experimental gingivitis in man. Journal of Periodontology. 1965;36:177–187. doi: 10.1902/jop.1965.36.3.177. [DOI] [PubMed] [Google Scholar]
- Lu B, Yu H, Chow C, Li B, Zheng W, Davis RJ, Flavell RA. GADD45gamma mediates the activation of the p38 and JNK MAP kinase pathways and cytokine production in effector TH1 cells. Immunity. 2001;14:583–590. doi: 10.1016/s1074-7613(01)00141-8. [DOI] [PubMed] [Google Scholar]
- Meda L, Gasperini S, Ceska M, Cassatella MA. Modulation of proinflammatory cytokine release from human polymorphonuclear leukocytes by gamma interferon. Cellular Immunology. 1994;157:448–461. doi: 10.1006/cimm.1994.1241. [DOI] [PubMed] [Google Scholar]
- Nakajima T, Yamashita S, Maekita T, Niwa T, Nakazawa K, Ushijima T. The presence of a methylation fingerprint of Helicobacter pylori infection in human gastric mucosae. International Journal of Cancer. Journal International du Cancer. 2009;124:905–910. doi: 10.1002/ijc.24018. [DOI] [PubMed] [Google Scholar]
- Nares S, Moutsopoulos NM, Angelov N, Rangel ZG, Munson PJ, Sinha N, Wahl SM. Rapid myeloid cell transcriptional and proteomic responses to periodontopathogenic Porphyromonas gingivalis. The American Journal of Pathology. 2009;174:1400–1414. doi: 10.2353/ajpath.2009.080677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols FC, Garrison SW. Interferon-gamma potentiation of lipopolysaccharide-induced eicosanoid release from human monocytes. Journal of Interferon Research. 1987;7:121–129. doi: 10.1089/jir.1987.7.121. [DOI] [PubMed] [Google Scholar]
- Offenbacher S, Barros SP, Singer RE, Moss K, Williams RC, Beck JD. Periodontal disease at the biofilm-gingival interface. Journal of Periodontology. 2007;78:1911–1925. doi: 10.1902/jop.2007.060465. [DOI] [PubMed] [Google Scholar]
- Reinhardt RA, McDonald TL, Bolton RW, DuBois LM, Kaldahl WB. IgG subclasses in gingival crevicular fluid from active versus stable periodontal sites. Journal of Periodontology. 1989;60:44–50. doi: 10.1902/jop.1989.60.1.44. [DOI] [PubMed] [Google Scholar]
- Rogus J, Beck JD, Offenbacher S, Huttner K, Iacoviello L, Latella MC, de Gaetano M, Wang HY, Kornman KS, Duff GW. IL1B gene promoter haplotype pairs predict clinical levels of interleukin-1beta and C-reactive protein. Human Genetics. 2008;123:387–398. doi: 10.1007/s00439-008-0488-6. [DOI] [PubMed] [Google Scholar]
- Salvi GE, Brown CE, Fujihashi K, Kiyono H, Smith FW, Beck JD, Offenbacher S. Inflammatory mediators of the terminal dentition in adult and early onset periodontitis. Journal of Periodontal Research. 1998;33:212–225. doi: 10.1111/j.1600-0765.1998.tb02193.x. [DOI] [PubMed] [Google Scholar]
- Seymour GJ, Gemmell E, Walsh LJ, Powell RN. Immunohistological analysis of experimental gingivitis in humans. Clinical and Experimental Immunology. 1988;71:132–137. [PMC free article] [PubMed] [Google Scholar]
- Shuto T, Furuta T, Oba M, Xu H, Li JD, Cheung J, Gruenert DC, Uehara A, Suico MA, Okiyoneda T, Kai H. Promoter hypomethylation of Toll-like receptor-2 gene is associated with increased proinflammatory response toward bacterial peptidoglycan in cystic fibrosis bronchial epithelial cells. Federation of American Societies for Experimental Biology Journal. 2006;20:782–784. doi: 10.1096/fj.05-4934fje. [DOI] [PubMed] [Google Scholar]
- Smith AJ, Humphries SE. Cytokine and cytokine receptor gene polymorphisms and their functionality. Cytokine and Growth Factor Reviews. 2009;20:43–59. doi: 10.1016/j.cytogfr.2008.11.006. [DOI] [PubMed] [Google Scholar]
- Takeichi O, Haber J, Kawai T, Smith DJ, Moro I, Taubman MA. Cytokine profiles of T-lymphocytes from gingival tissues with pathological pocketing. Journal of Dental Research. 2000;79:1548–1555. doi: 10.1177/00220345000790080401. [DOI] [PubMed] [Google Scholar]
- Watford WT, Moriguchi M, Morinobu A, O’Shea JJ. The biology of IL-12: coordinating innate and adaptive immune responses. Cytokine and Growth Factor Reviews. 2003;14:361–368. doi: 10.1016/s1359-6101(03)00043-1. [DOI] [PubMed] [Google Scholar]
- White GP, Hollams EM, Yerkovich ST, Bosco A, Holt BJ, Bassami MR, Kusel M, Sly PD, Holt PG. CpG methylation patterns in the IFNgamma promoter in naive T cells: variations during Th1 and Th2 differentiation and between atopics and non-atopics. Pediatric Allergy and Immunology : Official Publication of the European Society of Pediatric Allergy and Immunology. 2006;17:557–564. doi: 10.1111/j.1399-3038.2006.00465.x. [DOI] [PubMed] [Google Scholar]
- White GP, Watt PM, Holt BJ, Holt PG. Differential patterns of methylation of the IFN-gamma promoter at CpG and non-CpG sites underlie differences in IFN-gamma gene expression between human neonatal and adult CD45RO- T cells. Journal of Immunology (Baltimore, Md.:1950) 2002;168:2820–2827. doi: 10.4049/jimmunol.168.6.2820. [DOI] [PubMed] [Google Scholar]
- Wright HJ, Matthews JB, Chapple IL, Ling-Mountford N, Cooper PR. Periodontitis associates with a type 1 IFN signature in peripheral blood neutrophils. Journal of Immunology (Baltimore, Md.:1950) 2008;181:5775–5784. doi: 10.4049/jimmunol.181.8.5775. [DOI] [PubMed] [Google Scholar]
- Yamazaki K, Nakajima T, Gemmell E, Polak B, Seymour GJ, Hara K. IL-4- and IL-6-producing cells in human periodontal disease tissue Journal Of Oral Pathology & Medicine: official publication of the International Association of Oral Pathologists and the. American Academy of Oral Pathology. 1994;23:347–353. doi: 10.1111/j.1600-0714.1994.tb00074.x. [DOI] [PubMed] [Google Scholar]
- Yang J, Murphy TL, Ouyang W, Murphy KM. Induction of interferon-gamma production in Th1 CD4+ T cells: evidence for two distinct pathways for promoter activation. European Journal of Immunology. 1999;29:548–555. doi: 10.1002/(SICI)1521-4141(199902)29:02<548::AID-IMMU548>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- Zachrisson BU. A histological study of experimental gingivitis in man. Journal of Periodontal Research. 1968;3:293–302. doi: 10.1111/j.1600-0765.1968.tb01935.x. [DOI] [PubMed] [Google Scholar]
- Zhang S, Barros SP, Niculescu MD, Moretti AJ, Preisser JS, Offenbacher S. Alteration of PTGS2 promoter methylation in chronic periodontitis. Journal of Dental Research. 2010;89:133–137. doi: 10.1177/0022034509356512. [DOI] [PMC free article] [PubMed] [Google Scholar]