

Abstract
Wake neurons in the basal forebrain and brainstem provide critical inputs to optimize alertness and attention. These neurons, however, evidence heightened vulnerability to a diverse array of metabolic challenges, including aging. SIRT1 is an nicotinamide adenine dinucleotide responsive deacetylase serving diverse adaptive responses to metabolic challenges, yet this metabolic rheostat may be downregulated under conditions of significant oxidative stress. We hypothesized that SIRT1 might serve as a critical neuroprotectant for wake neurons in young animals but that this protectant would be lost upon aging, rendering the neurons more vulnerable to metabolic insults. In this collection of studies, we first established the presence of nuclear SIRT1 in wake neurons throughout the forebrain and brainstem. Supporting functional and behavioral roles for SIRT1 in wake–active neurons, transgenic whole animal, and conditional loss of brain SIRT1 in the adult mouse impart selective impairments in wakefulness, without disrupting non-rapid eye movement or rapid eye movement sleep. Populations of wake neurons, including the orexinergic, locus ceruleus, mesopontine cholinergic, and dopaminergic wake neurons, evidence loss of dendrites and neurotransmitter synthesis enzymes and develop accelerated accumulation of lipofuscin, consistent with a senescence-like phenotype in wake neurons. Normal aging results in a progressive loss of SIRT1 in wake–active neurons, temporally coinciding with lipofuscin accumulation. SIRT1 is a critical age-sensitive neuroprotectant for wake neurons, and its deficiency results in impaired wakefulness.
Introduction
Wakefulness is essential for alertness and optimal cognitive function yet is impaired in numerous metabolic conditions. Excessive daytime sleepiness and/or impaired alertness occur with increased prevalence in obesity, diabetes, depression, neurodegenerative disorders, and aging (Chasens et al., 2009; Gozal and Kheirandish-Gozal, 2009; Hawley et al., 2010). Mechanisms by which metabolic disturbances can impair wakefulness are poorly understood.
Wakefulness is modulated by multiple groups of wake–active neurons (WANs), including cholinergic neurons in the basal forebrain, midbrain and pons, the orexinergic and histaminergic neurons in the hypothalamus, and serotoninergic, dopaminergic, and noradrenergic neurons in the midbrain and pons (Fuller et al., 2006). WANs are sensitive to various metabolic perturbances, responding either by changes in activity or in measures of injury. Metabolic challenges, e.g., glucose or oxygen dyshomeostasis, influence WAN activity (Figlewicz et al., 1996; Petrisić et al., 1997). Prominent age-related changes in WANs include loss of dendrites, axonopathy, reduction in neurotransmitter synthesis enzymes, and, in select populations, lipofuscin accumulation and cell loss (Mann, 1983; Ishida et al., 2001; Zecca et al., 2004). WANs may be injured early in the course of several neurodegenerative disorders, including Alzheimer's, Parkinson's, and Huntington's diseases and amyotrophic lateral sclerosis (Chan-Palay and Asan, 1989; Zarow et al., 2003; Benarroch et al., 2009). Thus, it appears that, like wakefulness, WANs are sensitive to diverse metabolic perturbances.
Sirtuins are recognized as integral to numerous metabolic adaptations in diverse cell types. Silent information regulator 2 (SIR2) was described in yeast as extending lifespan (Gotta et al., 1997). SIRT1, the closest mammalian ortholog to yeast SIR2, functions as a nicotinamide adenine dinucleotide-dependent deacetylase for histone and non-histone targets (Imai et al., 2000). As a metabolically sensitive transcriptional regulator, SIRT1 serves to protect cells from metabolic dyshomeostasis by initiating a vast array of adaptive responses to nutrient and redox perturbations (Boily et al., 2008). SIRT1 activation of peroxisome proliferator activated receptor-γ coactivator-1α enhances mitochondrial biogenesis, optimizes mitochondrial surface/volume ratio to reduce reactive oxygen species production, and mounts an antioxidant defense (Nemoto et al., 2005; Aquilano et al., 2010). SIRT1 promotes energy homeostasis through deacetylation modifications of FOXO transcription factors (Nie et al., 2009). Autophagy, an important source of energy in neurons, is also regulated by SIRT1 (Salminen and Kaarniranta, 2009; Kume et al., 2010). Severe metabolic or redox dyshomeostasis can suppress SIRT1 activity (Zee et al., 2010).
In this series of studies, we examined populations of WANs for the presence of SIRT1, finding SIRT1 present in nuclei of WANs. We next explored the role of SIRT1 in wakefulness and in the integrity of WANs. Conditional loss of SIRT1 in the young adult mouse results in a senescence-like phenotype in WANs, including loss of key neurotransmitter enzymes or neuropeptide, loss of dendrites and axons, and accelerated accumulation of lipofuscin (aggregates of irreversibly oxidized proteins and lipids). Progressive loss of nuclear SIRT1 and lipofuscin accumulation occur across normal aging in WANs. We conclude that SIRT1 protects WANs and is essential for normal wakefulness. Age-related loss of SIRT1 likely contributes to heightened susceptibility to neurodegenerative processes.
Materials and Methods
Mice.
Studies were performed at the University of Pennsylvania with the approval of the Institutional Animal Care and Use Committee and conformed to the revised National Institutes of Health Office of Laboratory Animal Welfare Policy. Mice were housed in cages of four to five with food and water available ad libitum. Three sets of mice were used: wild-type C57BL/6J male mice (ages 2, 12, and 24 months), transgenic SIRT1 mice [wild type (WT), +/−, and −/− littermates] on an outbred background (6–8 months of age) that were generated as described previously (Boily et al., 2008), and B6;129–Sirt1tm1Ygu/J mice with a conditional deletion of exon 4 in the SIRT1 gene (SIRT1flx/flx) with WT mice of the same background strain, B6;129F1/J (6–8 months of age) (Cheng et al., 2003).
Conditional whole-brain SIRT1 knockdown.
Adeno-associated viral (AAV) vectors were designed for widespread distribution of cre-recombinase in the adult mouse brain, as described previously and validated (Cearley and Wolfe, 2007). Basically, a cytomegalovirus (CMV) promoter and the cre-recombinase transgene were incorporated into an AAV2 recombinant genome, cross-packaged into either an AAV1 or AAV9 vector. The University of Pennsylvania Vector Core performed design, packaging, purification, and measurement of vector titers. To examine the effects of SIRT1 loss in the adult mouse, SIRT1flx/flx and control mice were anesthetized with ketamine/xylazine (80 and 20 mg/kg, i.p., respectively) and injected with AAV2/1 (10 nl, >213 GC/ml) intracerebroventricularly (n = 4 SIRT1flx/flx and 4 controls) or AAV2/9 (3–5 nl, >213 GC/ml) targeting the rostral midbrain (bregma, −4.3 mm; dorsoventral, 2.7 mm; mediolateral, 0 mm) and dorsal pons bilaterally (bregma, −5.4; dorsoventral, 3.5 mm; mediolateral, 1 mm), referencing dorsoventral 0 mm to dura at injection site (n = 8 SIRT1flx/flx and 6 controls).
In situ hybridization.
To determine whether SIRT1 RNA was evident in WANs in adult mice, in situ hybridization was performed with immunolabeling of orexinergic and catecholaminergic WANs. SIRT1 cRNA probes for T3 and T7 were generated around the GenBank accession number NM_019812 using purified murine hypothalamic RNA as described recently (Ramadori et al., 2008). Appropriate amplicon size (623 kb) was confirmed with gel electrophoresis. Reverse transcriptase PCR DNA products were confirmed to share 99% sequence homology with the target SIRT1 sequence. These DNA products were used to generate RNA antisense (T7) and sense (T3) probes. C57BL/6J 8-week-old male mice (n = 6) were perfused with PBS and RNAase inhibitor; brains were immediately frozen for coronal sectioning, 10 μm. Sections from bregma −1.5 to −2.5 mm for lateral hypothalamus (LH) and −4 to −5.8 mm for ventral periaqueductal gray (VPAG) and locus ceruleus (LC) (Paxinos and Watson atlas) were placed on Superfrost slides, dried 30 min, fixed for 12 min with 4% paraformaldehyde, then washed with PBS with 0.1% Triton X-100, acetylated, and washed again before warming with prehybridization buffer. Probes were denatured at 85°C for 10 min and then added to slides in hybridization buffer in a moist chamber at 55°C overnight. The following day, washed sections were incubated with anti-digoxigenin alkaline phosphatase antibody (1:1000; catalog #1093274; BMB) and then developed the following day with 2% nitro blue tetrazolium chloride for 2 h. Sections were counterstained with either tyrosine hydroxylase antibody for dopaminergic VPAG and noradrenergic LC neurons or orexin-A antibody for orexinergic neurons (see below, Immunohistochemistry) for colocalization of SIRT1 mRNA in catecholaminergic and orexinergic wake neurons.
Implantation of electrodes for behavioral state recording.
Mice were implanted with skull and neck muscle silver electrodes for sleep recordings with bilateral frontal and occipital electroencephalographic and nuchal electromyographic electrodes, as detailed previously (Veasey et al., 2000). Two weeks after surgery, each mouse was connected to counterweighted recording cables with a commutator for recording digitized electroencephalographic and rectified moving average electromyographic signals (Veasey et al., 2000). Mice were given 1 week to adjust to the connecting cables, supported by the ability to stand fully upright on hindlimbs and quickly maneuver all corners of the cage. Recordings were collected across the following 2 weeks.
Behavioral state protocols and analysis.
SIRT1−/−, SIRT1+/−, and SIRT1+/+ mice underwent a 1 week baseline sleep recording of 10 s epochs scored as waking, non-rapid eye movement (NREM), or rapid eye movement (REM) sleep, as defined previously (Veasey et al., 2000), followed by a baseline multiple sleep latency test (baseline MSLT) with four 20-min nap opportunities across a 2 h period from 4:00 P.M. to 6:00 P.M., as described previously (Zhan et al., 2005). The following day mice were maintained awake for 6 h with EEG confirmation throughout deprivation, ending at 4:00 P.M., followed by a second MSLT (sleep dep MSLT). Primary endpoints were total wake time (hours) for 24 h and wake time (hours) for lights-on and lights-off periods; the baseline and sleep dep MSLT average sleep latency across the four nap opportunities. To characterize sleep/wake bout consolidation, sleep bouts were defined as >2 min consecutive NREM and/or REM sleep epochs after >1 min consecutive wake epochs that were terminated by >1 min consecutive wake epochs, and wake bouts were defined as >2 min wake after >1 min sleep epochs, terminated by >1 min sleep epochs. The arousal index was defined as the frequency (events per hours of sleep) of one or more wake epoch after >1 min sleep. To provide greater insight into wakefulness function, spectral analysis on wakefulness was performed, evaluating overall spectral power density (0–100 Hz), mean gamma power (30–100 Hz), and maximum gamma power (Brankack et al., 2010). To measure sleep homeostasis, we examined average delta power (1–4 Hz)/epoch of NREM sleep for each circadian hour. Each sleep/wake variable was averaged within a mouse, and then means across a genotype were compared using paired t tests with Bonferroni's correction.
Immunohistochemistry.
Immunohistochemistry was performed to (1) confirm SIRT1 protein in each group of WANs in SIRT1+/+ mice and establish antibody specificity with −/− mice, (2) to examine effects of SIRT1 transgenic absence or knockdown on WAN morphology, and (3) to determine whether aging alters nuclear SIRT1. After pentobarbital anesthesia, SIRT1 mutant mice and controls were transcardially perfused with 4% paraformaldehyde. Brains were postfixed in 4% paraformaldehyde and cryopreserved. Coronal sections, at 40 μm, of the entire brain were stored as 1:6 series in 0.1% sodium azide in PBS at 4°C until used. Six subtypes of wake–active neurons were studied: cholinergic neurons in the basal forebrain, midbrain, and pons, dopaminergic neurons in the ventral periaqueductal gray, histaminergic posterior hypothalamic neurons, and noradrenergic locus ceruleus, orexinergic lateral hypothalamic, and serotoninergic dorsal and median raphe neurons (Aston-Jones et al., 1991, 2001; Parmentier et al., 2002; Saper et al., 2005; Lu et al., 2006), as we have detailed previously (Zhu et al., 2007). Details of the primary antibodies to identify wake–active neurons, negative controls/antigen specificity, catalog number/source, dilutions for light microscopy, and immunofluorescence for WAN detection were published recently (Zhu et al., 2007). Primary antibodies for WANs were goat anti-choline acetyltransferase (ChAT) (AB144P; Millipore Bioscience Research Reagents), mouse anti-tyrosine hydroxylase (TH) (22941; Immunostar), mouse monoclonal anti-5-hydroxytryptamine antibody (5-HT) (20079; Immunostar), monoclonal mouse anti-Orexin A (MAB763; R & D Systems), and polyclonal rabbit anti-histidine decarboxylase (HDC) (03-16045; American Research Products), each with specificities established (Zhu et al., 2007). Secondary donkey anti-goat, anti-mouse, anti-rabbit, or anti-sheep antibodies were labeled with Alexa Fluor 488 (green; Invitrogen) for confocal microscopy, and biotinylated secondaries were labeled using diaminobenzidine (DAB) for light microscopy. SIRT1 immunoreactivity in wake neurons was detected with rabbit anti-SIRT1 antibody (2028; Cell Signaling Technology), using Alexa Fluor 594 or DAB with nickel as above. A titer of 1:100 overnight 4°C (immunofluorescence and immunohistochemistry) for SIRT1 primary provided absent signal in SIRT−/− brains yet offered robust detection of SIRT1 in wild-type mice.
To analyze colocalization of SIRT1 in wake neurons, two to four sections per mouse for each nucleus (covering rostrocaudal) were imaged with confocal microscopy (Leica SP-5, AOBS) providing a z-series of 1 μm confocal images, avoiding the top and bottom 3 μm of each section. All labeled neurons with complete nuclei were analyzed for SIRT1 with NIH ImageJ software. Using a 1:6 series of 40 μm sections ensured counting each neuron with a visualized nucleus once. The mean percentage of SIRT1-labeled WANs in 100–500 neurons per nucleus per mouse was compared using two-way ANOVA across wild-type mice for the six nuclei, as detailed previously (Zhu et al., 2007). To characterize apoptosis, we examined cleaved poly (ADP-ribose)-polymerase-1 (PARP-1) and cleaved caspase-3 in TH-labeled neurons, using recently described antibodies and protocols (Zhu et al., 2007). Both PARP-1 and cleaved caspase-3 were analyzed as two-way ANOVA for nucleus and genotype.
To determine whether transgenic absence of SIRT1 results in loss of WANs, we performed unbiased cell counts in the locus ceruleus, using the optical dissector/Cavalieri method (Nurcombe et al., 2001). Analysis was accomplished using NIH ImageJ software (available at http://rsb.info.nih.gov/ij; developed by Wayne Rasband, National Institutes of Health, Bethesda, MD). Variance in nuclear diameter (SIRT1−/− nuclei are larger) and SIRT1 deficiency-induced irregularities in somata (massive vacuoles) prevented use of the Abercrombie correction factor. All sections in a 1:3 series (six to eight nuclei per mouse) were anti-tyrosine hydroxylase labeled with Vector SG substrate and counterstained with Nissl, as described previously (Zhu et al., 2007). The x,y coordinate of the box contained the entire coronal dimensions of the LC, and the z-dimensions were defined with differential interference contrast to include from 3 to 10 μm from the coverslip (z-movement resolution, 0.1 μm). Cell count per box averaged per mouse was reported for n = 4–5 per genotype with >800 cells counted per mouse (with investigators blinded to genotype). Counts per section were analyzed as paired samples based on relative bregma position, using a paired t test.
To assess changes in WAN morphology, we analyzed dendrite density and branching area among SIRT1 transgenics and controls using NIH ImageJ software. Total area covered by LC dendrites medial to the cell bodies was measured in the two-dimensional plane using an overlaid grid with 800 μm2 gradations. Dendrite branching complexity was estimated by the mean number of LC dendrite segments per grid unit across the total counted area, in which a segment is the portion of a dendrite spanning one grid unit (800 μm2). Data were analyzed using paired t tests with pairs matched by relative bregma position.
To examine catecholaminergic neurons for evidence of lipofuscin granules, complementary light microscopy ultrastructural and confocal techniques were used (Xu et al., 2008; Jung et al., 2010). Mice at 2, 12, and 24 months were acrolein perfused, and brain sections were processed as described recently (Zhu et al., 2007). Semithin sections were imaged without labeling of tyrosine hydroxylase to interfere with cytoplasmic analysis. LC neurons were identified by size (20–25 μm diameter) and position (medial and ventral) relative to the large mesencephalic trigeminal neurons and ventricle, respectively. Light microscopy was performed on 0.3-μm-thick sections using 0.5% toluidine blue (Zhu et al., 2007) to estimate the percentage of neurons with lipofuscin aggregates, defined by size and density (Jung et al., 2010). Autofluorescence was detected using an SP-5 AOBS Leica confocal microscope. Although visible with all lasers, the 580 nm excitation showed a most intense signal paired with bandpass filtering at 605–700 nm.
Quantitative real-time PCR.
To examine whether loss of SIRT1 in the adult brain downregulates transcripts necessary for the production of WAN neurotransmitters, we performed tissue punch quantitative real-time PCR in WAN nuclei. During deep anesthesia with pentobarbital, brains were removed and immediately sectioned into 1 mm coronal sections on a chilled brain matrix. Scalpel dissections of the lateral hypothalamus/perifornicular region and locus ceruleus (1 mm3) in one to two sections bilaterally were placed in trizol and frozen (−70°C) before processing. RNA was purified and cDNA was created for primer/probe sets as follows: for tyrosine hydroxylase (GenBank accession number NM_009377), sense, TCT CTG TGA AGT TTG ACC CGT ACA (1372–1395); antisense, GTG GGT GGT ACC CTA TGC ATT T (1522–1501); and probe, CAG TCC TCA CAC CAT CCG GCG C (1418–1439); dopamine β hydroxylase (GenBank accession number NM_138942), sense, GAA GGA GGT GAA CAG GGA CAA C (1589–1610); antisense, TGT GGC CAG CGT CTT GTT T (1727–1709); and probe, CCA CTT CCG GGA GAT CAG AAT GC (1622–1644); orexin-A (hypocretin) (GenBank accession number NM_010410), sense, GCG CAG AGC TAG AGC ACA AT (393–412); antisense, GGG AAG TTT GGA TCA GGA CAA G (520–499); and probe, TCG CGG CTG TCC GAC CGT AAC (424–444); l-tryptophan (GenBank accession number NM_173391), sense, AAA GTC AAT TAC CCG TCC CTT CT (1437–1457); antisense, TCA AGG CAT CAC ACA CTG TGT TC (1571–1549); and probe, TCA ACC GCT ACA CGC AGA GCA TTG A (1467–1491); choline acetyltransferase (GenBank accession number D12487), sense, GGC CCT GCC AGT CAA CTC TA (557–576); antisense, CCT TGT AGC TAA GCA CAC CAG AGA (672–649); and probe, CTC GGC AGC ACT TCC AAG ACA CCA (595–618). All primer probe sets had detection of >1000 copies with linearity r2 ≥ 0.98). Taqman real-time PCR (SDS 7500 Real Time PCR System; Applied Biosystems) was used, and analysis was performed as detailed previously (Zhan et al., 2002).
Western blotting.
Immunoblots were used to provide semiquantification for SIRT1 levels in young (2 months) and old (24 months) mice and for neurotransmitter synthesis enzymes in SIRT1−/− and SIRT1+/+ littermates. Tissue dissections were obtained as above but were homogenized on ice in lysis buffer with protease inhibitors antipain, aprotinin, leupeptin, and pepstatin (Sigma-Aldrich). Twenty micrograms of total protein, measured with Pierce micro-BCA assay on centrifuged supernatants, was run individually per mouse and nucleus on SDS-PAGE gels (10% Tris-HCl; Bio-Rad). Gels were wet transferred to polyvinylidene difluoride membranes and processed for detection of TH (61 kDa), ChAT (83 kDa), and SIRT1 (120 kDa) at 1:100 using the Odyssey detection protocol with the LI-COR molecular weight marker. Mean integrated densities normalized to 20 μg of protein were analyzed with unpaired t tests.
Results
Nuclear SIRT1 is evident in all subsets of wake–active neurons
In situ hybridization was used to examine SIRT1 RNA in orexinergic and locus ceruleus wake neurons in adult B6 male mice (n = 6). SIRT1 RNA (T7, antisense) labeling was evident in all orexin-A-labeled neurons in the lateral hypothalamus and all TH-labeled neurons in the locus ceruleus (Fig. 1A). Within WANs, SIRT1 RNA was found primarily in the nucleus. In the lateral hypothalamus, immunolabeling intensity was equally strong in non-orexin-labeled neurons of similar size. Intense SIRT1 T7 labeling was present in the pyramidal neurons of the hippocampus, the piriform cortex, layers II/III of the cingulate, and motor cortex. Labeling was also evident throughout the hypothalamus, in which signal was strongest in the suprachiasmatic nuclei. In the cerebellum, T7 localized to the nuclei of Purkinje cells, and in the brainstem, T7 was most intense in regions of WANs but not limited to WANs. In the pons, intense labeling, equivalent to locus ceruleus, was evident in the majority of large neurons, including facial motor neurons. In support of probe/technique specificity, no labeling was observed for T3 (sense) polymerase in WANs or other groups of neurons. In summary, SIRT1 RNA is present in orexinergic and locus ceruleus wake neurons in addition to many other large neurons and shows a distinctive nuclear localization.
Figure 1.

SIRT1 in WANs. A, In situ hybridization with digoxigenin-labeled probes (T7, antisense; T3, sense), demonstrating SIRT1 RNA with nitro blue tetrazolium chloride (blue). Top, TH-labeled LC neurons (DAB, brown); bottom, orexin-A labeling (DAB, brown) of orexinergic neurons in the LH. Arrows highlight double-labeled orexin-A and SIRT1 RNA neurons. B, Immunolocalization of SIRT1 protein (DAB with nickel, dark blue) in top: cholinergic LDT nucleus (top, anti-ChAT); bottom, histaminergic tuberomammillary nucleus (TMN) neurons (bottom, HDC), both labeled with DAB (brown). SIRT1 null (−/−) sections are provided to support antibody specificity. SIRT1−/− mice show reduced immunoreactivity for respective neurotransmitter synthesis proteins. Scale bar, 50 μm. C, Confocal image of locus ceruleus neurons (anti-TH, red) and anti-SIRT1 (green) in a SIRT1+/+ mouse (left) and SIRT1−/− mouse (right). Scale bar, 25 μm. D, Percentage of WANs with SIRT1 immunoreactivity (mean ± SE) within populations of WANs: ChAT BF, cholinergic basal forebrain; DA PAG, dopaminergic periaqueductal gray; Hist TMN, histaminergic tuberomammillary nucleus; NA LC, noradrenergic locus ceruleus; Orex LH, orexinergic lateral hypothalamus; 5-HT DRN, serotoninergic dorsal raphe; and ChAT LDT, cholinergic laterodorsal tegmentum/pedunculopontine tegmentum, combined.
To localize SIRT1 protein within adult wake neurons and to relate findings to the other wake neuronal populations (cholinergic, histaminergic, dopaminergic, and serotoninergic), we next examined SIRT1 immunoreactivity in established WAN populations. To substantiate specificity of SIRT1 immunolabeling, mice with transgenic absence of SIRT1 (n = 5), wild-type littermates (n = 5), and SIRT1+/− mice (n = 3) were studied. In wild-type littermates, nuclear SIRT1 was evident in the vast majority of lateral basal forebrain cholinergic neurons, VPAG dopaminergic, posterior hypothalamic histaminergic, LH orexinergic, LC noradrenergic, pedunculopontine (PPT) and laterodorsal tegmentum (LDT) cholinergic, and midbrain raphe serotoninergic neurons (as shown and summarized in Fig. 1B–D). Medial forebrain cholinergic wake groups (medial septum and vertical diagonal band) had minimal SIRT1 immunoreactivity. Percentages of SIRT1-immunoreactive neurons in each WAN population are summarized in Figure 1D.
Wakefulness is impaired in SIRT1 null and heterozygous mice
To examine the role for SIRT1 in wakefulness, sleep/wake recordings were examined in mice with transgenic absence of SIRT1 (n = 8), heterozygotes (n = 3), and wild-type littermates (n = 8). A significant genotype effect was observed for behavioral state; characteristics are summarized in Figure 2. Relative to wild-type littermates, SIRT1−/− mice had markedly less spontaneous wake time (Fig. 2A). Wake time per 24 h for wild-type mice (n = 8) was (mean ± SEM) 3.4 ± 1.9 and 7.5 ± 1.3 h in SIRT1−/− mice (n = 8; p < 0.01). Wake time in SIRT1 null mice was reduced for both the lights-off (p < 0.01) and lights-on (p < 0.05) periods. In light of small sample size for the heterozygotes (n = 3), these mice were not included in statistical analyses, but data are provided in Figure 2. There were no differences in NREM sleep times across genotype for either the 24 h period or the 12 h lights-on period. Greater NREM sleep time (50% increase) was observed, however, in SIRT1−/− mice for the lights-off period (Fig. 2C). SIRT1−/− mice showed a twofold increase in REM sleep time for the 24 h period and for both lights on and lights off (p < 0.01) (Fig. 2B). The diurnal ratio of wakefulness (lights on/lights off) did not vary with genotype: controls, 0.7 ± 0.1 versus SIRT1−/−, 0.8 ± 0.2 (NS). Hourly wake percentile distributions across the 24 h period showed an expected abrupt increase in wake percentage per hour for all genotypes at lights off; however, wake predominance at lights off was of shorter duration in SIRT1−/− and SIRT1+/− mice (Fig. 2D). Using a murine sleep latency test as a correlate of sleep propensity (Veasey et al., 2004), we found a reduction in the baseline sleep latency in SIRT1−/− mice (p < 0.01). Wild-type mice showed an expected reduction in sleep latency after 6 h enforced wakefulness (p < 0.05), whereas sleep latency in SIRT1−/− mice was unaffected by sleep deprivation (Fig. 2E). Waking EEG spectral analysis was examined, revealing a maximum peak in the delta range for SIRT1−/− mice, whereas wild-type littermates showed a maximum at 6 Hz, consistent with theta frequencies (Fig. 2F). Total waking gamma power was reduced in SIRT1−/− mice relative to controls (2.9 ± 0.7 vs 5.5 ± 1.1 μV2, p < 0.05), with similar relative reductions in power across the entire gamma range.
Figure 2.

Effects of transgenic and conditional SIRT1 deficiency on sleep/wake activity. A, Wake time per 12 h, mean ± SEM for lights-on and lights-off periods in SIRT1+/+ (black), SIRT1+/− (blue), and SIRT1−/− (red) mice (bar, p < 0.01). B, NREM sleep time (12 h period), by genotype (bar, p < 0.05). C, REM sleep time (12 h period), by genotype (p < 0.001). D, Distribution of wake hourly percentiles across the 24 h period; lights off from 7:00 P.M. to 7:00 A.M. E, Sleep latency mean ± SEM under baseline sleep conditions (black) and after 6 h enforced wakefulness (gray). Short bar, Reduced sleep onset time in sleep-deprived wild types (*p < 0.05); long bar, genotype difference for baseline sleep latency (**p < 0.01). F, Electroencephalographic mean power density for SIRT1−/− (red) and SIRT1+/+ (blue) mice for 1 Hz frequency bands 0–30 Hz. G, Hourly wake percentile across the 24 h period for SIRT1 floxed cre-injected (SIRT1 fl-cre) mice (red) versus control cre-injected mice (black). H, Wake time, mean ± SEM in control-injected (black) and SIRT1 flox–cre mice (red) for lights-on and lights-off periods (bars, p < 0.01). I, Mean ± SEM relative NREM delta power, expressed as percentile of delta power for the last hour of baseline sleep for SIRT1 floxed (red) and control (black) mice at baseline (solid) and across 3 h of recovery sleep after 6 h sleep loss (dashed lines).
SIRT1 is necessary for WAN neurotransmitter synthesis
We next explored whether SIRT1 influences the synthesis of WAN neurotransmitters and neuromodulators. Quantitative real-time PCR on tissue micropunches of wake nuclei showed large relative reductions as summarized in Figure 3A. Copy numbers of tyrosine hydroxylase mRNA ± SEM normalized to 2 μg of RNA were significantly reduced in SIRT1−/− mice (5699 ± 205) versus wild-type littermates (170,000 ± 57,000; n = 4 per group, t = 3, p < 0.05). A similar reduction was observed in dopamine β hydroxylase mRNA in SIRT1 versus wild-type mice (2 × 107 ± 1.0 × 106 vs 7 × 105 ± 2.7 × 103, t = 2.5, p < 0.05). l-Tryptophan, necessary for serotonin synthesis, was reduced in SIRT1−/− mice (1.6 × 106 ± 2.8 × 105 vs 7.5 × 106 ± 1.5 × 106, t = 3.8, p < 0.01). A marked reduction in prepro-orexin mRNA was evident in SIRT1−/− mice (28,000 ± 3000 vs 4,000,000 ± 840,000 in wild type, t = 5, p < 0.001). ChAT, the rate-limiting enzyme for acetylcholine synthesis, trended toward reduction but was not significant (SIRT1−/−: 6000 ± 2000 vs 27,000 ± 11,000, t = 2, NS). The genotype effect on neurotransmitter enzymes persisted when normalized to β-actin mRNA. Thus, mRNA for rate-limiting enzymes for synthesis of neurotransmitters in WANs or, in the case of orexinergic neurons, the neuropeptide itself, is significantly reduced in SIRT1 null mice. To determine whether the marked reductions in mRNA result in reductions in protein, TH and ChAT were examined by Western blot for macropunches of the locus ceruleus and laterodorsal tegmentum. TH protein was significantly reduced by 50% in SIRT1−/− mice relative to controls (t = 5.8, p < 0.001) (Fig. 3B). Similarly, ChAT was reduced (∼50%) in SIRT1−/− mice (t = 3.1, p < 0.05) (Fig. 3B). For both TH and ChAT, the magnitude of mRNA reduction far exceeded the magnitude of change in protein. In summary, transgenic absence of SIRT1 downregulates mRNA and to a lesser extent protein levels of signaling key enzymes/peptides for WANs.
Figure 3.
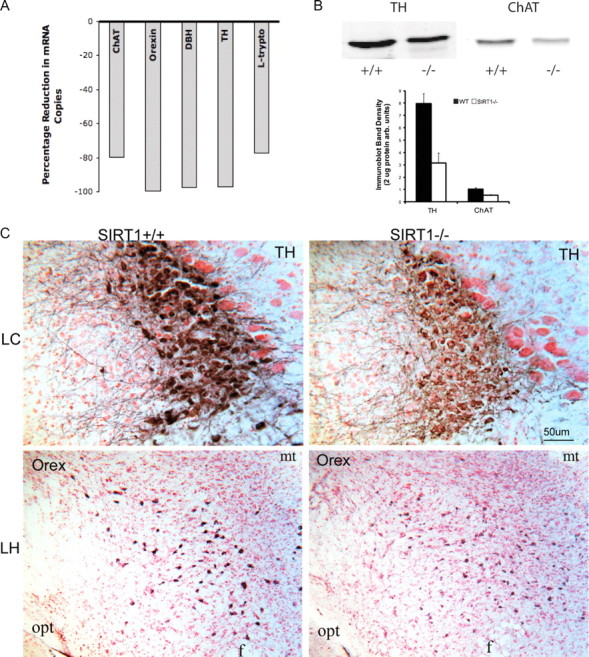
WAN neurotransmitter synthesis enzymes and orexin in SIRT1-deficient mice. A, Percentage reduction in mRNA copies for WAN neurotransmitter enzymes and prepro-orexin: ChAT, prepro-orexin (orexin), dopamine β-hydroxylase (DBH), TH, and l-tryptophan (L-trypto). B, Top, Representative TH and ChAT Western bands from the locus ceruleus and laterodorsal tegmentum from SIRT1−/− and SIRT1+/+ littermates. Bottom, Histogram integrated densities normalized to 20 μg of protein; mean ± SEM. C, Anti-TH (DAB, blue) in LC in SIRT1+/+ and SIRT1−/− mice (top) and anti-orexin A (Orex) in LH in SIRT1+/+ and SIRT1−/− mice (DAB, blue), counterstained with neutral red. Scale bar, 50 μm. Opt, Optic tract; mt, mammillothalamic tract.
AAV CMV cre-recombinase in the brain effectively reduces wake neuron SIRT1 in SIRT1flx/flx mice
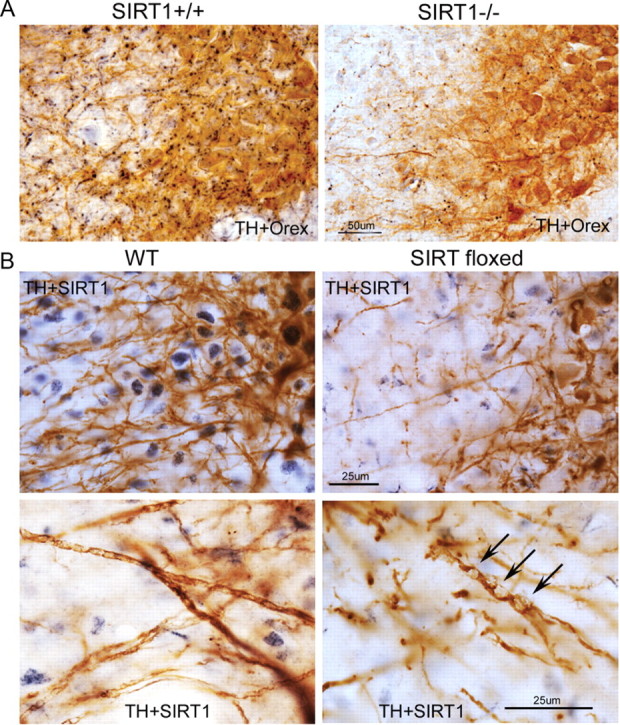
SIRT1 activity can be reduced in aging and metabolic conditions. Thus, we developed a model of acquired loss of SIRT1 to determine whether SIRT1 loss in adulthood would confer wake impairments. AAVs were designed for widespread distribution of cre-recombinase on a SIRT1–loxP mutant. Two vectors and routes were tested: both vectors included a CMV promoter and cre-recombinase transgene packaged into either an AAV1 vector for intracerebroventricular placement or an AAV9 placed in the pons via microinjection. SIRT1 immunoreactivity was noticeably reduced in all SIRT1–loxP-injected (floxed) mice (both vectors, both injection sites) throughout most of the diencephalon, mesencephalon, pons, and rostral medulla. A marked reduction in the percentage of locus ceruleus nuclei immunoreactive for SIRT1 was evident in SIRT1 floxed mice (Fig. 4A,B). Although SIRT1 loss was all or none within a given neuron, there were 5–20% of WANs (varying with region) with SIRT1 labeling. Thus, the techniques used are best characterized as a knockdown of SIRT1. Nuclear SIRT1 immunodensity was absent in ∼80% of LC neurons (t = 14, p < 0.0001), without variance across the rostrocaudal axis. In contrast, SIRT1 immunoreactivity remained present in all WANs of wild-type mice injected with the same vector. Effect of similar magnitude and direction was observed for orexinergic SIRT1, as summarized in Figure 4A. Although SIRT1 was reduced in neurons throughout the brain, SIRT1 floxed mice showed an upregulation of SIRT1 immunoreactivity throughout the cytoplasm of cells that were morphologically most consistent with microglia (Fig. 4C).
Figure 4.

Conditional SIRT1 deletion in the adult mouse brain with CNS AAV cre-recombinase. A, Representative images of SIRT1 immunolabeling (DAB/nickel, black) in LC (top) and orexinergic (bottom) neurons (DAB, brown) in WT cre-injected mice and SIRT1flx/flx (SIRT1 floxed) cre-injected mice. Scale bar, 50 μm. B, Mean ± SE percentage of nuclei positive for SIRT1 immunolabeling for tyrosine hydroxylase-labeled locus ceruleus (TH+LC) neurons and orexinergic lateral hypothalamic (Orex+LH) neurons in control cre-injected (black bars) and SIRT1 floxed cre-injected (gray bars) mice. Lines represent genotype differences (p < 0.0001). C, Higher power, SIRT1 immunoreactivity (DAB/nickel, blue) in WT cre-injected and SIRT1 floxed cre-injected mice. Left, SIRT1 immunoreactivity in orexinergic neurons (DAB, brown) in WT (short arrows). Right, Glia of SIRT1 floxed mice in lateral hypothalamus (long arrows). Scale bar, 20 μm.
Wakefulness is impaired in AAV cre-floxed SIRT1 knockdown mice
To determine whether loss of SIRT1 in the adult brain impairs wakefulness, wake activity was examined in SIRT1 floxed and wild-type controls, as above for transgenics. Total wake time per 24 h was significantly and equally reduced in both sets of SIRT1 floxed mice and thus were combined (n = 8–9 per group; mean ± SEM, 9.3 ± 1.5 vs 12.1 ± 0.9 h, t = 5.2, p < 0.001) (Fig. 2G,H). SIRT1 floxed mice did not show differences in total (24 h) REM sleep (1.3 ± 0.1 vs 1.2 ± 0.1 h, t = 0.8, NS). NREM sleep was, however, increased in SIRT1 floxed mice (13.4 ± 0.4 vs 10.6 ± 0.6 h in control mice, t = 4.2, p < 0.01). Hourly distributions of wake across the 24 h cycle (Fig. 2G) showed similar reductions (∼20%) for most time points of the 24 h period with the exception of the onset of lights off when wakefulness increased comparably in the two strains. Sleep was less fragmented in the SIRT1 floxed mice: SIRT1 floxed arousal index, 9.6/hr ± 2 sleep vs control arousal index, 23/hr ± 5 sleep, t = 5, p < 0.01. Durations of wake bouts did not differ with SIRT1 flox condition, whereas sleep bouts (NREM + REM sleep) were longer in SIRT1 floxed mice (SIRT1 floxed, 41 ± 3 min vs control, 16 ± 3 min, t = 7, p < 0.001). In summary, brain SIRT1 knockdown in the adult mouse results in significant reductions in wake without disrupting sleep or wake consolidation. We next examined the homeostatic response to sleep in SIRT1flx/flx versus control-injected mice. Mice typically show a decline in delta power (electroencephalographic mean power density between 1 and 4 Hz/epoch NREM sleep) across the rest–predominant lights-on period (Franken et al., 2001). Across baseline sleep/wake activity, there was a strain difference in the delta power decline across the lights-on period (Fig. 2I). In SIRT1 floxed, the slope was −3 ± 1 and in controls was −27 ± 3 (p < 0.01), consistent with less homeostatic drive at baseline in SIRT1 floxed mice. In response to 6 h enforced wakefulness, control mice showed a large increase in the relative delta power at the beginning of recovery and at least for 6 h into recovery. SIRT1 floxed mice showed minimal increase in delta power in response to sleep deprivation, further supporting a blunted homeostatic drive in SIRT1-deficient mice (Fig. 2I). After sleep deprivation, a greater pressure for sleep is realized in the ensuing lights-off period. Wake time across the first 5 h of the lights-off period after sleep deprivation was reduced in control mice after sleep deprivation (1.5 h less wake time) with no change in SIRT1 conditional knock-out mice wake time in the first 5 h after sleep deprivation. In summary, in mice with conditional knockdown of SIRT1 in the brain, baseline wake time is markedly reduced without disruptions in sleep, and yet, by three measures of sleep homeostasis, homeostatic response to prolonged wakefulness was severely diminished in SIRT1-deficient mice.
SIRT1 regulates wake neuron morphology
We next determined whether SIRT1 deficiency results in WAN loss, focusing on the locus ceruleus as a population of WANs with pronounced morphologic changes in SIRT1-deficient mice. Using optical dissector/Cavalieri counting (Williams and Rakic, 1988; Nurcombe et al., 2001) across previously defined coordinates for the locus ceruleus (Zhu et al., 2007), there was loss of locus ceruleus neurons evident in SIRT1−/− mice. SIRT1−/− mice averaged 126 ± 7 (SEM) neurons per section, and SIRT1+/+ mice averaged 136.5 ± 9 neurons per section (t = 0.8, NS). There was no evidence for significant apoptosis in SIRT1−/− mice. Cleaved (activated) caspase-3 immunoreactivity, particularly in the dopaminergic and serotoninergic neurons, was weakly evident but present exclusively in the cytoplasm of WANs. Scant nuclear cleaved PARP-1 was evident in LC WANs of SIRT1−/− mice. SIRT1 floxed mice showed minimal upregulation in cleaved PARP-1 without nuclear cleaved caspase-3. Thus, neither transgenic nor conditional SIRT1 deficiency influences WAN cell number or apoptosis.
Significant effects, however, were observed in WAN morphology in both SIRT1−/− and SIRT1 floxed mice, in which effects on bouton and dendritic densities and nuclear size were prominent. Within the region of LC dendrites, SIRT1 null mice showed a marked reduction in orexinergic boutons in the LC nucleus: SIRT1 boutons/1000 μm2, 34 ± 4 versus controls, 91 ± 5 (t = 9, p < 0.0001). In SIRT1−/− mice, orexinergic boutons appeared close to LC cell bodies with very few boutons in the dendritic region of the locus ceruleus (Fig. 5A). Intriguingly, orexinergic projections in the hypothalamus and anterior cingulate cortex were unchanged in SIRT1−/− and floxed mice relative to controls. Dendrites in SIRT1−/− mice showed numerous changes in morphology. Most prominently, the area encompassed by LC TH dendrite segments was reduced in SIRT1−/− mice (0.074 ± 0.008 mm2 vs controls, 0.124 ± 0.007 mm2, t = 4.3, p < 0.001), with fewer LC TH-positive dendritic segments in the SIRT1−/− mice (150 ± 22 dendritic segments/mm2 vs control, 299 ± 24 dendritic segments/mm2, t = 4.7, p < 0.001) (Fig. 5B,C). Similarly, the density of LC TH dendrite segments varied with genotype both in the region most proximal to the LC cell bodies and distally, suggesting loss of both length and complexity of dendrites in SIRT1−/− mice. Control animals evidenced small vacuoles present in rare dendrites. In contrast, SIRT1 floxed mice and SIRT1−/− mice showed numerous large vacuoles distorting dendrites and cytoplasm of the pontine WANs identified with large nuclei (Fig. 5B). In both transgenic SIRT1−/− mice and SIRT1 floxed mice, the nuclei were enlarged in catecholaminergic and cholinergic neurons relative to control mice. In SIRT1 floxed mice, the nuclei/cytoplasm area of LC neurons was 1.1 ± 0.11 and in control-injected mice was 0.54 ± 0.04 (t = 5.7, p < 0.0001). Nuclear area in SIRT1 floxed mice was larger than in control mice by 160% (t = 6.3, p < 0.001) (Fig. 5A).
Figure 5.
Effects on SIRT1 deficiency on wake neuron morphology. A, Orexinergic projections (nickel, black) into the region of LC dendrites (DAB, brown) in SIRT1+/+ and SIRT1−/− mice. Scale bar, 50 μm. B, LC dendrites in WT cre-injected and SIRT1 floxed cre-injected mice. Top, Locus ceruleus TH-labeled dendrites in WT cre-injected mouse (left) and SIRT1 floxed cre-injected mouse (right). Scale bar, 25 μm. Neuronal SIRT1 immunoreactivity (DAB, dark blue) in WT and scant glial labeling in SIRT floxed mouse. Bottom two panels show LC–TH dendrites in WT (left) and SIRT1 flox–cre mouse (right) with large vacuoles (arrows). Scale bar, 25 μm.
The ultrastructure of LC neurons was examined in SIRT1 floxed and control AAV cre-injected mice. In semithin sections, LC neurons were identified by size and position relative to TH+-labeled neurons and medial to the large mesencephalic nucleus of the trigeminal nerve neurons. Striking differences were observed with marked increases in both lysosomes and lipofuscin aggregates (Fig. 6A,B). Rare lipofuscin aggregates were evident in control-treated mice, whereas numerous aggregates were present in the same WAN populations identified with nuclear enlargement and dendritic and cytoplasmic vacuolization in SIRT1 floxed mice (LC, VPAG, PPT, and LDT). Aggregates showed maximal emission between 605 and 700 nm, ranging in size up to 6 μm (Fig. 6B). LC cytoplasm in SIRT1 floxed mice showed a homogeneic cytoplasmic density not observed in control-injected mice.
Figure 6.
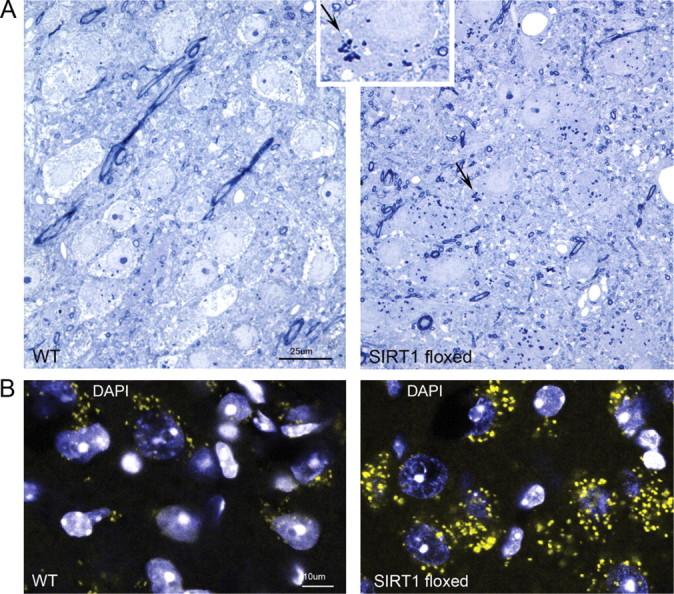
Lipofuscin aggregates in wake–active neurons in SIRT1-deficient mice. A, Toluidine blue-stained 0.3 μm sections of the LC highlight differences in cytoplasm densities in SIRT1 floxed cre-injected mice. Large dense irregular bodies are consistent with lipofuscin bodies (as highlighted with arrow and inset at higher magnification). B, Confocal image using laser excitation at 580 nm and emission spectrum at 605–700 nm shows accumulation of autofluorescence aggregates in LC neurons with nuclei counterstained with 4′,6-diamidino-2-phenylindole (DAPI) (blue).
Nuclear SIRT1 declines across aging in most wake–active neurons, coinciding with accumulation of lipofuscin aggregates
B6 adult male mice were examined at 2, 12, 17, and 24 months (n = 4 per age group) for changes in nuclear SIRT1 protein in WANs. In LC neurons, a decline in nuclear SIRT1 was observed from 2 to 12 months (t = 9, p < 0.001) and from 2 to 24 months (t = 13, p < 0.001) and between 12 and 24 months (t = 5, p < 0.001) (Fig. 7A,B). At 2 months, there was scant cytoplasmic SIRT1 in VPAG or tyrosine hydroxylase LC neurons. At 12 and 24 months, autofluorescence within the cytoplasm prevented analysis of cytoplasmic SIRT1. The autofluorescence was consistent with lipofuscin aggregates in size (0.5–6 μm), distribution (throughout cytoplasm), and maximum emission (600–705 nm, varying by spectral analysis from the Alexa Fluor 488 or 594 emission spectra), similar to aggregates observed in SIRT1-deficient mice. Age-dependent reduction in nuclear SIRT1 was also evident in the VPAG neurons (Fig. 7C) and in the orexinergic and mesopontine cholinergic neurons. In contrast, serotoninergic dorsal raphe neurons exhibited increased SIRT1 immunodensity in the nucleus with aging. Western blots were performed to substantiate the LC and VPAG nuclear SIRT1 findings, examining SIRT1 in young (2 months, n = 4) and old (24 months, n = 4) mice and showed reduced overall SIRT1 (by 25% in micropunches, t = 4, p < 0.05) at 24 months in the LC and VPAG, supporting an age-associated SIRT1 loss in WANs.
Figure 7.

Age-related changes in nuclear SIRT1 in wake–active neurons. A, LC neurons across ages 2 (2M), 12 (12M), and 24 (24M) months. Left, Anti-tyrosine hydroxylase (red) and anti-SIRT1 (green) labeling for the three age groups in LC sections. Right, Autofluorescence (yellow) in non-immunolabeled sections in LC, counterstained with 4′,6-diamidino-2-phenylindole (DAPI). B, Mean ± SE age effect on nuclear SIRT1 immunodensity in LC neurons. Lines signify statistical significances (p < 0.01). C, Age-related SIRT1 immunoreactivity in dopaminergic ventral periaqueductal gray wake neurons for 2 and 12 months. Arrow highlights dendritic SIRT1 at 12 months.
Discussion
In this series of studies, we identified SIRT1 in most populations of WANs and demonstrated essential roles for SIRT1 in wakefulness behavior and in the function and health of WANs. SIRT1 was found in the lateral basal forebrain cholinergic, histaminergic, and orexinergic neurons and brainstem dopaminergic periaqueductal gray, noradrenergic locus ceruleus, serotoninergic dorsal raphe, and cholinergic pedunculopontine and laterodorsal tegmentum neurons, in which SIRT1 localized predominantly to the nucleus in young adult mice. Loss of SIRT1, transgenically or acquired, imparted profound wake impairments, without disturbing sleep consolidation. Delta power in recovery sleep was markedly blunted in SIRT1-deficient mice, supporting an influential role for SIRT1 (either directly or indirectly) in sleep homeostasis. SIRT1 deficiency resulted in significant morphological changes to WANs, including loss of both orexinergic axons projecting to the locus ceruleus and locus ceruleus dendrites. WAN neurotransmitter synthesis enzymes and wake neuropeptide were suppressed in all WAN populations. Conditional SIRT1 loss resulted in accelerated accumulation of lipofuscin aggregates throughout the cytoplasm, consistent with a senescence-like phenotype in these WANs. In wild-type mice, aging was associated with a progressive loss of nuclear SIRT1 temporally coinciding with lipofuscin accumulation. Here, we present the significance of the behavioral, molecular, and morphological findings relevant to SIRT1 loss.
In this collection of studies, we demonstrate that brain SIRT1 is essential for wakefulness. Impaired wakefulness, however, does not necessarily signify WAN dysfunction. Wake impairments may also develop as a consequence of heightened sleep drive, cortical neuron dysfunction, or sleep/wake instability (Fuller et al., 2006). The blunted delta power across the lights-on period and in response to sleep loss in SIRT1 floxed mice argues against heightened sleep drive in SIRT1 deficiency (Franken et al., 2001). SIRT1-deficient mice are easy to arouse with gentle handling, and the cortical electroencephalogram on awakening shows increased frequencies. These findings are inconsistent with a fixed cortical injury. Having focused on WANs, we cannot exclude important effects of SIRT1 loss on sleep–active neurons. Although not quantified, we observed qualitatively comparable effects of SIRT1 loss on melanin-concentrating hormone neurons, a population of REM sleep-on neurons in the lateral hypothalamus. The overall phenotype of SIRT1 deficiency, however, is one of reduced wakefulness. Sleep state instability was not observed in SIRT1-deficient mice. In fact, these mice showed longer sleep bouts with less fragmentation than controls. Collectively, the findings substantiate WAN functional dependency on SIRT1 and suggest an imbalance between SIRT1 reliance for wake–active and sleep–active neurons.
The composite of SIRT1 deficiency effects on sleep and wake architecture indicate involvement of specific populations of WANs. Previous studies involving lesions of various WAN populations have examined the roles specific WAN groups play in arousal, attention, and wakefulness. In general, substantial lesions in any one WAN group have minimal effects on total wakefulness, with the exception of the dopaminergic and orexinergic WANs. Neuron-specific loss of dopaminergic neurons results in reduced total wakefulness (Lu et al., 2006), and lesioning orexinergic neurons in the adult rat reduces wakefulness in the lights-off period (Blanco-Centurion et al., 2007). Composite lesioning of locus ceruleus, histaminergic, and cholinergic basal forebrain does not significantly influence total wake time, although this is expected to affect attention and alertness (Blanco-Centurion et al., 2007). Our findings of reduced total wakefulness time in SIRT1-deficient mice support dysfunction of dopaminergic and possibly orexinergic neurons. The vacuolization and dendrite loss in dopaminergic neurons and the prominent loss of orexinergic boutons projecting to the locus ceruleus support dysfunction of these groups. The increased REM sleep supports monoaminergic neuron dysfunction (Fuller et al., 2006). The impaired sleep homeostasis in SIRT1-deficient mice was quite striking and is consistent with locus ceruleus dysfunction (Cirelli et al., 2005). Together, we believe that dysfunction to multiple groups of WANs contributes to the sleep/wake phenotype for SIRT1 deficiency.
WANs in SIRT1-deficient mice evidenced pronounced loss of mRNA and protein for neurotransmitter synthesis enzymes specific to each group of WANs or, in the case of orexin, for the neuropeptide itself. Interestingly, mRNA declined to a much greater extent than protein. This may signify either alterations in miRNA or a decline in protein clearance through proteosomal degradation or macroautophagy pathways. Both modes of protein clearance may be affected by loss of SIRT1 (Lee et al., 2008; Min et al., 2010). Although poorly understood, previous studies support the concept that SIRT1 regulates neurotransmitter responsiveness, synthesis, and neurotransmission. Increasing brain SIRT1 upregulates orexin B receptors in the hypothalamus in response to feeding restriction, and low SIRT1 diminishes responses (Satoh et al., 2010). A relationship between SIRT1, tyrosine hydroxylase, and its neurotransmitter dopamine has been demonstrated. Nicotinamide, an inhibitor of SIRT1 activity, reduces tyrosine hydroxylase in parallel with reductions in dopamine in the forebrain (Lee et al., 2009). In neuroblastoma cells, SIRT1 influences differentiation, including tyrosine hydroxylase upregulation (Kim et al., 2009). In addition, SIRT1 influences the differentiation of neural progenitor cells (Prozorovski et al., 2008), in which nuclear translocation is necessary for differentiation effects, suggesting a role as a histone deacetylase in modulating transcription (Hisahara et al., 2008). The collective findings suggest that SIRT1 plays an integral role in regulating WAN neurotransmitter synthesis, in which SIRT1 deficiency manifests in a loss of neurotransmitters and a de-differentiated state in WANs. This, we believe, contributes to SIRT1 deficiency wake impairments.
Conditional knock-out of brain SIRT1 results in a marked increase in lipofuscin aggregates, presumably over the course of several weeks. Lipofuscin normally accumulates slowly across aging in mice across 2 years. The accelerated lipofuscin accumulation in the present study indicates that endogenous SIRT1 normally protects against lipofuscin accumulation. Lipofuscin is an amalgam of highly oxidized and heavily cross-linked proteins and lipids, and, because the proteins cannot be degraded, lipofuscin accumulates over time (Jung et al., 2007). Accelerated accumulation of lipofuscin in SIRT1-deficient mice implies mitochondrial injury and increased oxidization of proteins and lipids, severe enough to induce cross-linking (Jung et al., 2007). SIRT1 reduces mitochondrial superoxide production, increases numbers of mitochondria, and initiates an antioxidant transcriptional response (Nemoto et al., 2005; Aquilano et al., 2010), whereas lipofuscin impairs proteosomal degradation and autophagy, increasing oxidative stress (Jung et al., 2007). Thus, loss of SIRT1 is expected to play a feedforward role in oxidative injury in all WANs susceptible to lipofuscin accumulation, thereby rapidly accelerating lipofuscin accumulation and oxidative stress.
We have identified age-related loss of nuclear SIRT1 in catecholaminergic, cholinergic, and orexinergic wake neurons but not in serotoninergic WANs. The mechanisms by which SIRT1 declines with age are not known, but impressive reductions are evident by midlife for the mouse, 12 months of age. HIC1 is a SIRT1 transcriptional repressor that is regulated by redox state (Zhang et al., 2007). Thus, age-related redox imbalance may repress SIRT1 transcription. In aged mice, we found increased cytoplasmic SIRT1 in neurons with reduced or absent nuclear SIRT1, suggesting an age-related translocation of SIRT1 to the cytoplasm. Age-related accumulation of lipofuscin has been demonstrated for the locus ceruleus (Sulzer et al., 2008), and we observed temporally related reductions in nuclear SIRT1 at 12 months of age when age-related reductions in wakefulness are expected to occur (Colas et al., 2005; Hasan et al., 2010). The differential loss of SIRT1 across aging in cholinergic, orexinergic, and catecholaminergic to a greater extent than serotoninergic neurons should provide a key to discovering mechanisms of SIRT1 loss with aging. Whether augmenting SIRT1 activity in WANs can abrogate the age-related wake impairments and lipofuscin accumulation will be important to determine.
Insomnia is the most common subjective complaint regarding age-related effects on sleep/wake behaviors. We know very little, however, about the effect of aging on human electroencephalographic activity across the entire 24 h period. Electrographic 24 h data were recently analyzed in several strains of mice across 2–24 months of age (Hasan et al., 2010). Age-related effects on wakefulness and the waking EEG included progressive declines in wakefulness time per 24 h in all strains through 12 months of age, with wake reductions most pronounced in the active (lights-off) period. In addition, there was an age-related reduction in sleep homeostasis, less sleep fragmentation in older mice, and age-related power density changes in the B6 mice that are remarkably similar to what we observe in SIRT1-deficient mice. Thus, many of the sleep/wake findings observed in aged mice are similar to what we have observed in SIRT1-deficient mice. These findings support the concept that wake impairments in aging may occur, at least in part, as a consequence of age-related SIRT1 loss. It is likely, however, that fragmented sleep with advanced age occurs through a SIRT1-independent mechanism.
We have established that SIRT1, present in WANs, is essential for normal wakefulness and the integrity of WANs. The neuroprotectant role for SIRT1 in WANs is supported by the observation that loss of SIRT1 in the adult brain results in a senescence-like phenotype in WANs with a behavioral correlate. It is anticipated that age-related loss of nuclear SIRT1 contributes to a relatively heightened susceptibility of WANs in aging, metabolic disturbances, and numerous neurodegenerative disorders.
Footnotes
This work was supported from the National Institutes of Health/National Heart, Lung, and Blood Institute Grants HL079555, HL096037, and HL07713. We are grateful to Jennifer Montoya for help in preparing this manuscript and Dr. Jini Naidoo for brain sections in aged mice.
The authors declare no competing financial interests.
References
- Aquilano K, Vigilanza P, Baldelli S, Pagliei B, Rotilio G, Ciriolo MR. Peroxisome proliferator-activated receptor gamma co-activator 1alpha (PGC-1alpha) and sirtuin 1 (SIRT1) reside in mitochondria: possible direct function in mitochondrial biogenesis. J Biol Chem. 2010;285:21590–21599. doi: 10.1074/jbc.M109.070169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benarroch EE, Schmeichel AM, Dugger BN, Sandroni P, Parisi JE, Low PA. Dopamine cell loss in the periaqueductal gray in multiple system atrophy and Lewy body dementia. Neurology. 2009;73:106–112. doi: 10.1212/WNL.0b013e3181ad53e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanco-Centurion C, Gerashchenko D, Shiromani PJ. Effects of saporin-induced lesions of three arousal populations on daily levels of sleep and wake. J Neurosci. 2007;27:14041–14048. doi: 10.1523/JNEUROSCI.3217-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boily G, Seifert EL, Bevilacqua L, He XH, Sabourin G, Estey C, Moffat C, Crawford S, Saliba S, Jardine K, Xuan J, Evans M, Harper ME, McBurney MW. SirT1 regulates energy metabolism and response to caloric restriction in mice. PLoS One. 2008;3:e1759. doi: 10.1371/journal.pone.0001759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brankack J, Kukushka VI, Vyssotski AL, Draguhn A. EEG gamma frequency and sleep-wake scoring in mice: comparing two types of supervised classifiers. Brain Res. 2010;1322:59–71. doi: 10.1016/j.brainres.2010.01.069. [DOI] [PubMed] [Google Scholar]
- Cearley CN, Wolfe JH. A single injection of an adeno-associated virus vector into nuclei with divergent connections results in widespread vector distribution in the brain and global correction of a neurogenetic disease. J Neurosci. 2007;27:9928–9940. doi: 10.1523/JNEUROSCI.2185-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan-Palay V, Asan E. Alterations in catecholamine neurons of the locus coeruleus in senile dementia of the Alzheimer type and in Parkinson's disease with and without dementia and depression. J Comp Neurol. 1989;287:373–392. doi: 10.1002/cne.902870308. [DOI] [PubMed] [Google Scholar]
- Chasens ER, Sereika SM, Burke LE. Daytime sleepiness and functional outcomes in older adults with diabetes. Diabetes Educ. 2009;35:455–464. doi: 10.1177/0145721709333857. [DOI] [PubMed] [Google Scholar]
- Cheng HL, Mostoslavsky R, Saito S, Manis JP, Gu Y, Patel P, Bronson R, Appella E, Alt FW, Chua KF. Developmental defects and p53 hyperacetylation in Sir2 homolog (SIRT1)-deficient mice. Proc Natl Acad Sci U S A. 2003;100:10794–10799. doi: 10.1073/pnas.1934713100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cirelli C, Huber R, Gopalakrishnan A, Southard TL, Tononi G. Locus ceruleus control of slow-wave homeostasis. J Neurosci. 2005;25:4503–4511. doi: 10.1523/JNEUROSCI.4845-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colas D, Cespuglio R, Sarda N. Sleep wake profile and EEG spectral power in young or old senescence accelerated mice. Neurobiol Aging. 2005;26:265–273. doi: 10.1016/j.neurobiolaging.2004.03.004. [DOI] [PubMed] [Google Scholar]
- Figlewicz DP, Brot MD, McCall AL, Szot P. Diabetes causes differential changes in CNS noradrenergic and dopaminergic neurons in the rat: a molecular study. Brain Res. 1996;736:54–60. doi: 10.1016/0006-8993(96)00727-5. [DOI] [PubMed] [Google Scholar]
- Franken P, Chollet D, Tafti M. The homeostatic regulation of sleep need is under genetic control. J Neurosci. 2001;21:2610–2621. doi: 10.1523/JNEUROSCI.21-08-02610.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuller PM, Gooley JJ, Saper CB. Neurobiology of the sleep-wake cycle: sleep architecture, circadian regulation, and regulatory feedback. J Biol Rhythms. 2006;21:482–493. doi: 10.1177/0748730406294627. [DOI] [PubMed] [Google Scholar]
- Gotta M, Strahl-Bolsinger S, Renauld H, Laroche T, Kennedy BK, Grunstein M, Gasser SM. Localization of Sir2p: the nucleolus as a compartment for silent information regulators. EMBO J. 1997;16:3243–3255. doi: 10.1093/emboj/16.11.3243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gozal D, Kheirandish-Gozal L. Obesity and excessive daytime sleepiness in prepubertal children with obstructive sleep apnea. Pediatrics. 2009;123:13–18. doi: 10.1542/peds.2008-0228. [DOI] [PubMed] [Google Scholar]
- Hasan S, Dauvilliers Y, Mongrain V, Franken P, Tafti M. Age-related changes in sleep in inbred mice are genotype dependent. Neurobiol Aging. 2010 doi: 10.1016/j.neurobiolaging.2010.05.010. [DOI] [PubMed] [Google Scholar]
- Hawley CJ, Gale TM, Sivakumaran T, Paul S, Kondan VR, Farag A, Shahzad J. Excessive daytime sleepiness in psychiatric disorders: prevalence, correlates and clinical significance. Psychiatry Res. 2010;175:138–141. doi: 10.1016/j.psychres.2008.10.037. [DOI] [PubMed] [Google Scholar]
- Hisahara S, Chiba S, Matsumoto H, Tanno M, Yagi H, Shimohama S, Sato M, Horio Y. Histone deacetylase SIRT1 modulates neuronal differentiation by its nuclear translocation. Proc Natl Acad Sci U S A. 2008;105:15599–15604. doi: 10.1073/pnas.0800612105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai S, Armstrong CM, Kaeberlein M, Guarente L. Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature. 2000;403:795–800. doi: 10.1038/35001622. [DOI] [PubMed] [Google Scholar]
- Ishida Y, Shirokawa T, Miyaishi O, Komatsu Y, Isobe K. Age-dependent changes in noradrenergic innervations of the frontal cortex in F344 rats. Neurobiol Aging. 2001;22:283–286. doi: 10.1016/s0197-4580(00)00203-7. [DOI] [PubMed] [Google Scholar]
- Jung T, Bader N, Grune T. Lipofuscin: formation, distribution, and metabolic consequences. Ann N Y Acad Sci. 2007;1119:97–111. doi: 10.1196/annals.1404.008. [DOI] [PubMed] [Google Scholar]
- Jung T, Höhn A, Grune T. Lipofuscin: detection and quantification by microscopic techniques. Methods Mol Biol. 2010;594:173–193. doi: 10.1007/978-1-60761-411-1_13. [DOI] [PubMed] [Google Scholar]
- Kim MJ, Ahn K, Park SH, Kang HJ, Jang BG, Oh SJ, Oh SM, Jeong YJ, Heo JI, Suh JG, Lim SS, Ko YJ, Huh SO, Kim SC, Park JB, Kim J, Kim JI, Jo SA, Lee JY. SIRT1 regulates tyrosine hydroxylase expression and differentiation of neuroblastoma cells via FOXO3a. FEBS Lett. 2009;583:1183–1188. doi: 10.1016/j.febslet.2009.03.007. [DOI] [PubMed] [Google Scholar]
- Kume S, Uzu T, Horiike K, Chin-Kanasaki M, Isshiki K, Araki S, Sugimoto T, Haneda M, Kashiwagi A, Koya D. Calorie restriction enhances cell adaptation to hypoxia through Sirt1-dependent mitochondrial autophagy in mouse aged kidney. J Clin Invest. 2010;120:1043–1055. doi: 10.1172/JCI41376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee IH, Cao L, Mostoslavsky R, Lombard DB, Liu J, Bruns NE, Tsokos M, Alt FW, Finkel T. A role for the NAD-dependent deacetylase Sirt1 in the regulation of autophagy. Proc Natl Acad Sci U S A. 2008;105:3374–3379. doi: 10.1073/pnas.0712145105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JY, Ahn K, Jang BG, Park SH, Kang HJ, Heo JI, Ko YJ, Won MH, Tae-Cheon Kang, Jo SA, Kim MJ. Nicotinamide reduces dopamine in postnatal hypothalamus and causes dopamine-deficient phenotype. Neurosci Lett. 2009;461:163–166. doi: 10.1016/j.neulet.2009.06.005. [DOI] [PubMed] [Google Scholar]
- Lu J, Jhou TC, Saper CB. Identification of wake–active dopaminergic neurons in the ventral periaqueductal gray matter. J Neurosci. 2006;26:193–202. doi: 10.1523/JNEUROSCI.2244-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann DM. The locus coeruleus and its possible role in ageing and degenerative disease of the human central nervous system. Mech Ageing Dev. 1983;23:73–94. doi: 10.1016/0047-6374(83)90100-8. [DOI] [PubMed] [Google Scholar]
- Min SW, Cho SH, Zhou Y, Schroeder S, Haroutunian V, Seeley WW, Huang EJ, Shen Y, Masliah E, Mukherjee C, Meyers D, Cole PA, Ott M, Gan L. Acetylation of tau inhibits its degradation and contributes to tauopathy. Neuron. 2010;67:953–966. doi: 10.1016/j.neuron.2010.08.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemoto S, Fergusson MM, Finkel T. SIRT1 functionally interacts with the metabolic regulator and transcriptional coactivator PGC-1α. J Biol Chem. 2005;280:16456–16460. doi: 10.1074/jbc.M501485200. [DOI] [PubMed] [Google Scholar]
- Nie Y, Erion DM, Yuan Z, Dietrich M, Shulman GI, Horvath TL, Gao Q. STAT3 inhibition of gluconeogenesis is downregulated by SirT1. Nat Cell Biol. 2009;11:492–500. doi: 10.1038/ncb1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nurcombe V, Wreford NG, Bertram JF. Stereological estimation of neuronal number. The optical disector/cavalieri combination. Methods Mol Biol. 2001;169:251–268. doi: 10.1385/1-59259-060-8:251. [DOI] [PubMed] [Google Scholar]
- Petrisić MS, Augood SJ, Bicknell RJ. Monoamine transporter gene expression in the central nervous system in diabetes mellitus. J Neurochem. 1997;68:2435–2441. doi: 10.1046/j.1471-4159.1997.68062435.x. [DOI] [PubMed] [Google Scholar]
- Prozorovski T, Schulze-Topphoff U, Glumm R, Baumgart J, Schröter F, Ninnemann O, Siegert E, Bendix I, Brüstle O, Nitsch R, Zipp F, Aktas O. Sirt1 contributes critically to the redox-dependent fate of neural progenitors. Nat Cell Biol. 2008;10:385–394. doi: 10.1038/ncb1700. [DOI] [PubMed] [Google Scholar]
- Ramadori G, Lee CE, Bookout AL, Lee S, Williams KW, Anderson J, Elmquist JK, Coppari R. Brain SIRT1: anatomical distribution and regulation by energy availability. J Neurosci. 2008;28:9989–9996. doi: 10.1523/JNEUROSCI.3257-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salminen A, Kaarniranta K. SIRT1: regulation of longevity via autophagy. Cell Signal. 2009;21:1356–1360. doi: 10.1016/j.cellsig.2009.02.014. [DOI] [PubMed] [Google Scholar]
- Satoh A, Brace CS, Ben-Josef G, West T, Wozniak DF, Holtzman DM, Herzog ED, Imai S. SIRT1 promotes the central adaptive response to diet restriction through activation of the dorsomedial and lateral nuclei of the hypothalamus. J Neurosci. 2010;30:10220–10232. doi: 10.1523/JNEUROSCI.1385-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulzer D, Mosharov E, Talloczy Z, Zucca FA, Simon JD, Zecca L. Neuronal pigmented autophagic vacuoles: lipofuscin, neuromelanin, and ceroid as macroautophagic responses during aging and disease. J Neurochem. 2008;106:24–36. doi: 10.1111/j.1471-4159.2008.05385.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veasey SC, Valladares O, Fenik P, Kapfhamer D, Sanford L, Benington J, Bucan M. An automated system for recording and analysis of sleep in mice. Sleep. 2000;23:1025–1040. [PubMed] [Google Scholar]
- Veasey SC, Yeou-Jey H, Thayer P, Fenik P. Murine Multiple Sleep Latency Test: phenotyping sleep propensity in mice. Sleep. 2004;27:388–393. doi: 10.1093/sleep/27.3.388. [DOI] [PubMed] [Google Scholar]
- Williams RW, Rakic P. Three-dimensional counting: an accurate and direct method to estimate numbers of cells in sectioned material. J Comp Neurol. 1988;278:344–352. doi: 10.1002/cne.902780305. [DOI] [PubMed] [Google Scholar]
- Xu H, Chen M, Manivannan A, Lois N, Forrester JV. Age-dependent accumulation of lipofuscin in perivascular and subretinal microglia in experimental mice. Aging Cell. 2008;7:58–68. doi: 10.1111/j.1474-9726.2007.00351.x. [DOI] [PubMed] [Google Scholar]
- Zarow C, Lyness SA, Mortimer JA, Chui HC. Neuronal loss is greater in the locus coeruleus than nucleus basalis and substantia nigra in Alzheimer and Parkinson diseases. Arch Neurol. 2003;60:337–341. doi: 10.1001/archneur.60.3.337. [DOI] [PubMed] [Google Scholar]
- Zecca L, Stroppolo A, Gatti A, Tampellini D, Toscani M, Gallorini M, Giaveri G, Arosio P, Santambrogio P, Fariello RG, Karatekin E, Kleinman MH, Turro N, Hornykiewicz O, Zucca FA. The role of iron and copper molecules in the neuronal vulnerability of locus coeruleus and substantia nigra during aging. Proc Natl Acad Sci U S A. 2004;101:9843–9848. doi: 10.1073/pnas.0403495101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zee RS, Yoo CB, Pimentel DR, Perlman DH, Burgoyne JR, Hou X, McComb ME, Costello CE, Cohen RA, Bachschmid MM. Redox regulation of sirtuin-1 by S-glutathiolation. Antioxid Redox Signal. 2010;13:1023–1032. doi: 10.1089/ars.2010.3251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhan G, Shaheen F, Mackiewicz M, Fenik P, Veasey SC. Single cell laser dissection with molecular beacon polymerase chain reaction identifies 2A as the predominant serotonin receptor subtype in hypoglossal motoneurons. Neuroscience. 2002;113:145–154. doi: 10.1016/s0306-4522(02)00137-9. [DOI] [PubMed] [Google Scholar]
- Zhan G, Fenik P, Pratico D, Veasey SC. Inducible nitric oxide synthase in long-term intermittent hypoxia: hypersomnolence and brain injury. Am J Respir Crit Care Med. 2005;171:1414–1420. doi: 10.1164/rccm.200411-1564OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, Wang SY, Fleuriel C, Leprince D, Rocheleau JV, Piston DW, Goodman RH. Metabolic regulation of SIRT1 transcription via a HIC1:CtBP corepressor complex. Proc Natl Acad Sci U S A. 2007;104:829–833. doi: 10.1073/pnas.0610590104. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Zhu Y, Fenik P, Zhan G, Mazza E, Kelz M, Aston-Jones G, Veasey SC. Selective loss of catecholaminergic wake active neurons in a murine sleep apnea model. J Neurosci. 2007;27:10060–10071. doi: 10.1523/JNEUROSCI.0857-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]