

Abstract
This paper describes the fabrication and the performance of microfluidic paper-based electrochemical sensing devices (we call the microfluidic paper-based electrochemical devices, μPEDs). The μPEDs comprise paper-based microfluidic channels patterned by photolithography or wax printing, and electrodes screen-printed from conducting inks (e.g., carbon or Ag/AgCl). We demonstrated that the μPEDs are capable of quantifying the concentrations of various analytes (e.g., heavy-metal ions and glucose) in aqueous solutions. This low-cost analytical device should be useful for applications in public health, environmental monitoring, and the developing world.
Introduction
This article describes the fabrication and the performance of paper-based electrochemical sensing devices. We call these devices μPEDs (microfluidic paper-based electrochemical devices). The μPEDs are capable of quantifying the concentrations of various analytes in aqueous solutions, including biological fluids such as urine, serum and blood. The μPEDs comprise microfluidic channels, fabricated from patterned paper (either chromatography paper or a polyester–cellulose blend), and electrodes that were screen-printed on chromatography paper or polyester film (Fig. 1). Three electrodes (a working electrode, a counter electrode, and a reference electrode) were made from carbon ink or Ag/AgCl ink.
Fig. 1.
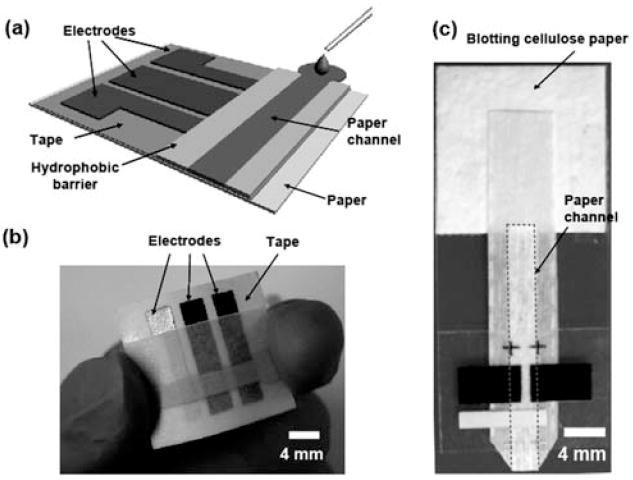
(a) Schematic of a paper-based electrochemical sensing device. The sensor comprises three electrodes printed on a piece of paper substrate (or plastic) and a paper channel. The paper channel was in conformal contact with the electrodes, and was held in place by double-sided adhesive tape surrounding the electrodes. A photograph of a paper-based electrochemical sensing device for the analysis of glucose (b), and a hydrodynamic paper-based electrochemical sensing device for the measurement of heavy-metal ions (c). The device consists of two printed carbon electrodes as the working and counter electrodes, and a printed Ag/AgCl electrode as the pseudo-reference electrode. The paper channel was fabricated by patterning SU-8 as a hydrophobic barrier for aqueous solution. The paper channel in (b) was colored with red ink to enhance imaging. The dashed line in (c) indicates the edge of the paper channel. The scale bar is 4 mm.
This work was carried out independently of a similar and complementary study by Dungchai et al.,1 in which μPEDs were fabricated and used for electrochemical detection of glucose, lactate, and uric acid in biological samples. Using μPEDs, we have demonstrated the detection and quantification of glucose in artificial urine using a chronoamperometric analysis based on glucose oxidase. The detection of glucose using the device showed high sensitivity and accuracy over the full range of clinically relevant concentrations of glucose in urine. The detection limit of glucose in the current μPED is about 0.22 mM (corresponding to 4 mg mL−1), which is lower than the approximately 1.0 mM specified by conventional glucometers, and 0.5 mM obtained by a colorimetric detection method reported previously.2,3 We estimated the sensitivity of the glucose analysis to be 0.43 μA mM−1 mm−2. We further demonstrated the use of μPEDs in the selective analysis of Pb(ii) in an aqueous solution containing a mixture of Pb(ii) and Zn(ii). The continuous wicking of the sample solution across the electrodes dramatically enhanced the efficiency of the deposition of metals during anodic stripping voltammetry, and improved the sensitivity and reliability of the detection. The measurement of Pb(ii) showed a limit of detection of 1.0 ppb. This value is much lower than the WHO (World Health Organization) guideline value (<10 ppb) for the safe level of lead in drinking water.4
We have recently developed methods for fabricating microfluidic devices from patterned paper, and demonstrated that paper-based microfluidic devices can be used for colorimetric detection of glucose and protein in artificial urine.2,5-7 Low-cost, paper-based diagnostic and analytical devices are attractive for use in developing countries, in the field, or in home health-care settings.8,9 Although colorimetric assays are the best choice for some applications, electrochemical sensing provides a more versatile and quantitative methodology for others.10 The μPEDs described in this article provide a simple, fast, and low-cost (~2 cents per device) platform for the quantitative detection of analytes (biological and inorganic species) in aqueous solutions, including body fluids. This type of detection has five advantages: (i) it is light-weight, portable, single-use and disposable; (ii) it is flexible and foldable; (iii) it has excellent reproducibility with high sensitivity and accuracy; (iv) it does not require professional medical personnel or complicated instruments; and (v) it allows for the integration of high-density detection systems into a small device.
Experimental design
Choice of materials
We used paper as the substrate for electrochemical detection. We used conductive carbon ink and Ag/AgCl ink to fabricate the electrodes used in the work.11 The conductive ink electrodes have four advantages: (i) they are inexpensive; (ii) they are robust; (iii) they are easy to fabricate; and (iv) they are widely used in both industrial and university research.12 We evaluated the quality of the μPED-based electrochemical cells by cyclic voltammetric measurements, using ferrocene carboxylic acid as a standard, because in bulk solution the electrochemical reaction is reversible under ambient conditions at room temperature.
Fabrication and design of the device
We fabricated a three-electrode sensor by screen-printing carbon ink (or Ag/AgCl ink) onto a piece of paper or polyester film. The printing process is fast, simple, and inexpensive, and suitable for mass production. Paper-based microfluidic channels were fabricated by patterning chromatography (Whatman 1 Chr) or polyester–cellulose blend paper (VWR®Spec-Wip) by photolithography2,5-7 or wax printing.13 We chose these papers in this work because they do not deform on the surface of electrodes when they are wetted by fluids. In order to achieve conformal contact between paper channels and electrodes, the patterned paper was taped onto the substrate that supported the electrodes using double-side adhesive tapes (Fig. 1).
Experimental
Chemical reagents
Carbon ink (E3456) and Ag/AgCl ink (AGCL-675C) were purchased from Ercon Inc (Wareham, MA) and Conductive Compound (Hudson, NH), respectively. Glucose oxidase (136 300 U mg−1, Aspergillus niger), glucose, and potassium ferricyanide were purchased from Aldrich and used as received. Stock solutions of β-d-glucose were prepared in a PBS buffer (pH 7.0) and allowed to mutarotate overnight before use. Atomic absorption standard solutions of Pb(ii) (104 mg L−1), Zn(ii) (103 mg L−1), and bismuth(iii) (103 mg L−1) were obtained from Aldrich, USA, and diluted as required. 0.1 M acetate buffer (pH 4.5) was used as a supporting electrolyte.
Apparatus
All chronoamperometric measurements were performed with a bipotentiostat (PINE Instrument Company, Model AFCBP1). A modular electrochemical system AUTOLAB equipped with PGSTAT12 was used in combination with GPES software (Eco Chemie) for the anodic stripping voltammetric measurements of heavy-metal ions.
Fabrication of the devices
Electrodes
We fabricated a three-electrode sensor by screen-printing carbon ink (or Ag/AgCl ink for a reference electrode) on a piece of paper or polyester film. We generated a stencil for printing by designing patterns of electrodes using Adobe Free-hand®, followed by cutting the pattern into double-sided adhesive tape using a laser-cutter (VersaLASER VLS3.50, Universal Laser Systems Inc.). We taped the stencil on top of a paper or plastic substrate, and filled the openings of the stencil with ink. We baked the electrodes on a hotplate at 100 °C for 30 min. After the ink dried, we removed the protective backing layer of the tape, and left the adhesive layer on the substrate for the assembly of a paper channel on the top of electrodes. The thickness of the electrodes was approximately 100 μm. A typical working and counter electrode had dimensions of 1.5 cm in length and 4 mm in width, and a typical reference electrode had dimensions of 1.5 cm in length and 3 mm in width.
Microfluidic channels
Paper-based microfluidic channels were fabricated by patterning chromatography paper (Whatman 1 Chr) or polyester–cellulose blend paper (VWR®Spec-Wip) by photolithography2,5-7 or wax printing.13 Briefly, we soaked a piece of paper with SU-8 2010 photoresist, baked it at 95 °C for 5 min to remove solvents, and photoexposed it to UV light for 10 s through a photomask. The unpolymerized photoresist was removed by soaking the paper in acetone and washing three times with isopropanol. The polymers patterned on the paper form hydrophobic barriers to confine liquids in the microchannel. The paper microfluidic channel had dimensions of 4 mm in width and 100 μm in height (determined by the thickness of the paper), Fig. 1. The paper-based channels were assembled onto the electrodes using double-sided adhesive tapes. The contact area between a paper channel and a working electrode was 4 mm by 4 mm.
Chronoamperometric measurements
Chronoamperometric experiments were performed using a 500 mV step potential (vs. a carbon pseudo-reference electrode) to generate the calibration curve; these experiments used glucose with concentrations ranged from 0 to 22.2 mM (corresponding to 400 mg dL−1). Solutions (glucose oxidase 250 U mL−1, K3[Fe(CN)6] 600 mM, KCl 1.0 M in pH 7.0 PBS buffer) containing glucose with different concentrations were measured (each sample was examined eight times). We spotted the solution of enzyme on top of the paper microchannel. The solution of enzyme was distributed evenly in the paper channel due to the capillary wicking. After the solution dried, the enzyme was uniformly absorbed in the paper. When a solution of analytes was added into the microchannel, the solution distributed and mixed well with the pre-loaded enzyme. In another set of experiments, we premixed the enzyme solution with glucose samples before the chronoamperometric measurements. We did not observe obvious difference between the two methods. Paper can also be easily modified to immobilize enzymes if necessary. All measurements were conducted at room temperature under ambient conditions. We used a carbon working electrode (on a plastic substrate) with a surface area of 16 mm2 in contact with the fluid for the detection of glucose in the bulk solution. We averaged the eight measurements of current readout and calculated the corresponding standard deviation.
Anodic stripping voltammetry
Stripping voltammetric measurements were performed by in situ deposition of the bismuth (500 μg L−1) and the target metal ions with concentrations ranged from 0 to 100 ppb (μg L−1). Non-deaerated solutions were used for all measurements, and each sample was measured eight times. All measurements were carried out using the square-wave anodic stripping voltammetry (SWASV) with a frequency of 20 Hz, a potential step of 5 mV, and an amplitude of 25 mV. SWASV experiments comprised an electrochemical deposition step at −1.2 V for 120 s, an equilibration period of 30 s, and a square-wave voltammetric stripping scan usually from −1.2 to −0.5 V. Before each measurement a pre-conditioning step (for cleaning of the electrode) at a potential of +0.5 V was applied for 60 s.
Detection method and principle
Glucose
We detected glucose in the μPEDs by chronoamperometric method. The reactions for the glucose detection were:
| (1) |
| (2) |
| (3) |
In the first step, glucose oxidase catalyzed the oxidation of glucose to gluconic acid with concomitant reduction of Fe(iii) to Fe(ii) (eqn (1)); the Fe(CN)64− ions generated were detected chronoamperometrically (eqn (2)). The corresponding cathodic reaction was described in eqn (3).
Metals
We used square-wave anodic stripping voltammetry for the measurements of heavy-metal ions in the μPEDs. Our measurements of trace metals relied on the simultaneous (in situ) plating of bismuth and target metals onto screen-printed carbon electrodes, which formed alloys followed by anodic stripping of metals from the electrode.
Results and discussion
Electrochemical characterization of paper-based electrochemical devices
We used ferrocene carboxylic acid as a model redox-active compound to characterize electrochemical behavior of μPEDs (Fig. 2a). The peak shape of the CVs shows a typical reversible (Nernstian) electrochemical reaction in which the rate of reaction is governed by the diffusion of the electroactive species to the surface of a planar electrode.14 The difference in potential between the peaks of the reduction (Epc) and oxidation (Epa) curves is 0.068 V (a value that is close to the theoretical value of 0.059 V for the ferrocene redox pair) for all scan rates between 50 and 500 mV s−1, and the peak current ratio (ipa/ipc) is equal to 1.0.14 This reversible behavior indicates that no side reactions take place, and that, as expected, the kinetics of electron transfer is sufficiently rapid to maintain the surface concentrations of redox-active species at the values required by the Nernst equation.
Fig. 2.
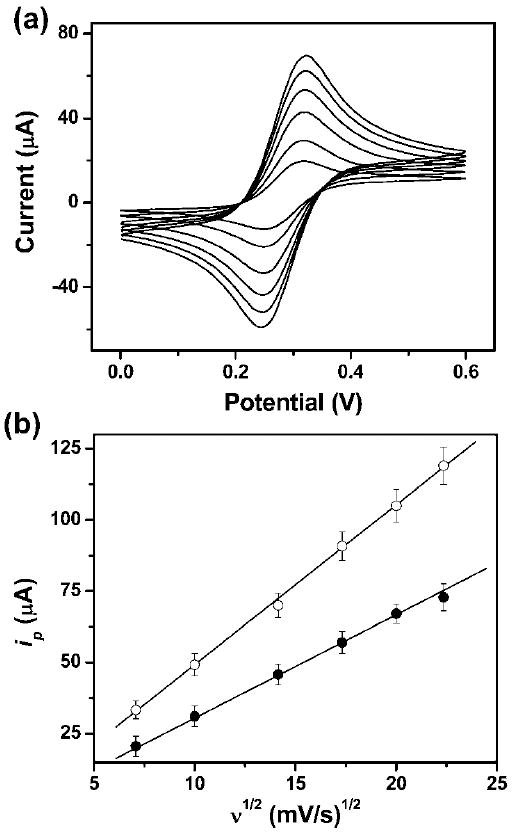
(a) Cyclic voltammograms of 2.0 mM ferrocene carboxylic acid in 0.5 M KCl aqueous solution (pH = 7.0) in a μPED at various scan rates (ascending along y-axis): 50, 100, 200, 300, 400, and 500 mV s−1. We used a 4 mm by 4 mm carbon-printed electrode as the working electrode, and a printed Ag/AgCl electrode as the reference electrode. (b) The plot of anodic peak current vs. the square root of the scan rate (ν1/2) for CV experiments conducted on a paper device (●) and in a bulk solution (○). The solid lines represent a linear fit to (●) with regression equation: y = −3.6 + 3.5x (R2 = 0.998, n = 8), and a linear fit to (○) with regression equation: y = −7.1 + 5.6x (R2 = 0.999, n = 8).
Fig. 2b shows that the anodic peak current, ip, is linearly proportional to the square root of the scan rate (ν1/2) in both the bulk solution and the μPED. The value of diffusion coefficient evaluated by analyzing the slope observed in bulk solution was 4.3 × 10−6 cm2 s−1 (see ESI†), which is fairly close to the reported value of 5.7 × 10−6 cm2 s−1.15 The current readout (Fig. 2b) measured using the paper device is about 30% lower than that measured in bulk solution. We presume that this difference is due to the fact that 30–40% of the volume in diffusional contact with the electrodes is occupied by the cellulose fiber of the paper. These results containing the redox-active species do not slow the rate of mass-limited charge transfer relative to that in solution.
Chronoamperometric analysis of glucose in urine
We demonstrated the use of our device in the analysis of a clinically relevant analyte in a simulant body fluid of glucose in artificial urine using chronoamperometry. Chronoamperometry offers a better signal-to-noise ratio than other electrochemical techniques in this kind of experiment, and the use of a thin slab of fluids mechanically clamped to the electrodes is more resistant to vibration than analysis in a larger volume of solution.14 The chronoamperometric measurement of current—reflecting charge transfer to/from the redox-active species as a function of time at constant applied voltages—begins with an initially large capacitive current. Upon the decay of the initial capacitive current within 1–2 s, Faradaic current (the current that is proportional to the concentration of the analyte) dominates. The current, i, decays as t−1/2 as described by the Cottrell equation14 (eqn (4))
| (4) |
where n is the number of electrons, t is the time, F is Faraday’s constant, A is the area of the electrode, D is the diffusion coefficient of analytes, and C is the initial concentration of the reactants.
The μPED confines fluids in the paper channel, inhibits the convective movement of fluids, and thus facilitates the chronoamperometric measurements by minimizing the disturbances of the stationary boundary layer in the vicinity of electrodes due to vibration, thermal or density-based convection, and other disturbing sources. Fig. 3a shows a representative chronoamperometric response of the measurements of glucose using a μPED. Over the range of concentrations of glucose examined (0–22.2 mM), all the response curves reached a steady state 2 s after the step potential (also see the Cottrell plot in ESI†). Fig. 3b shows a calibration curve for the detection of glucose. When the concentration of glucose is in the range of 0–22.2 mM, the current is linearly proportional to the glucose concentration in the artificial urine.
Fig. 3.
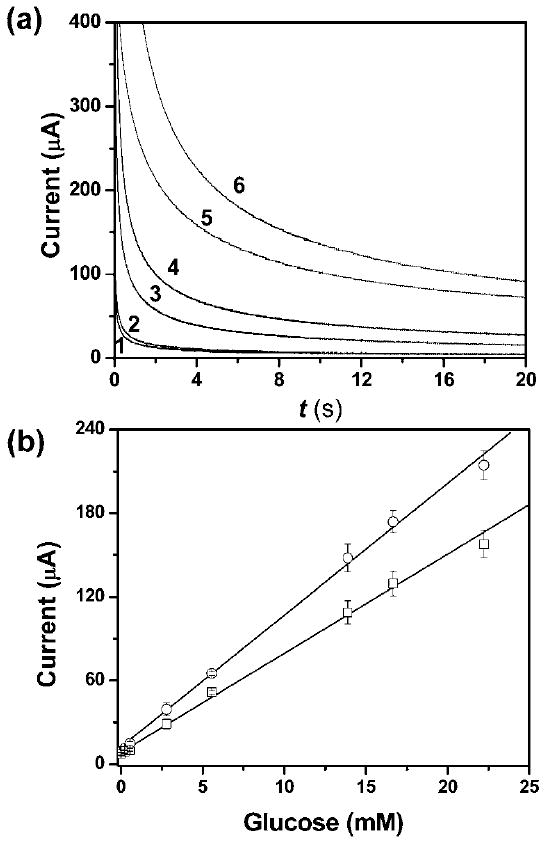
(a) Representative chronoamperometric curves for glucose concentrations (mM): 0 (1), 0.2 (2), 2.8 (3), 5.6 (4), 13.9 (5) and 22.2 (6) in the μPEDs. (b) Calibration plots of current as a function of the concentration of glucose for the detection of glucose in the μPEDs (□) and in bulk solutions (○). We used three carbon electrodes as working, counter, and pseudo-reference electrodes, respectively. The working electrodes had a surface area of 16 mm2 in contact with the solution in both the μPEDs and the bulk solutions. The distance between electrodes was 1.0 mm. A 500 mV step potential (vs. a carbon pseudo-reference electrode) was used to generate the calibration curve. The solid line represents a linear fit to (□) with regression equation: y = 9.5 + 6.9x (R2 = 0.995, n = 8), and a linear fit to (○) with regression equation: y = 12.1 + 9.4x (R2 = 0.997, n = 8).
We tested the interference of the sensing device with bovine serum albumin (BSA) as a typical globular protein; serum albumins are present in highest concentrations in serum, and thus relevant to bioanalysis. We found that the presence of 40 μM BSA did not interfere with the measurement of glucose. This selectivity is due to the specificity of enzymatic oxidation of glucose; the BSA apparently does not foul the electrodes. Comparing the results of the detection of glucose in the μPEDs to those in bulk solutions (Fig. 3b), we noticed that the two methods showed comparable sensitivity and detection limits; the paper matrix does not interfere with the detection. The paper matrix in the μPEDs has, however, several advantages: (i) it stabilizes the geometry of the electrode; (ii) it reduces the effect of convection of liquids due to random motion, vibration and heating; (iii) it minimizes the total volume of solution required for analysis. The normal level of glucose in urine is 0.1–0.8 mM and 3.5–5.3 mM in whole blood.3 Our device should therefore be capable of measuring glucose in other biological fluids such as serum and blood (see measurement of glucose in blood plasma and whole blood in ESI†). The detection limit of glucose in the current μPED is about 0.22 mM (corresponding to 4 mg mL−1). This value is below the approximately 1.0 mM claimed in specifications of conventional glucometers, and 0.5 mM obtained by colorimetric detection method reported previously.2,3 We estimate the sensitivity of the glucose analysis to be 0.43 μA mM−1 mm−2. In principle, other species in real urine and blood may interfere when we use 500 mV potentials for the analysis of glucose. The potential can be reduced to around 300 mV for the measurements in biological fluids, thanks to the enzymatic selectivity of glucose oxidase. The paper-based device also has the potential to be integrated with various separation techniques such as paper chromatography to minimize interferences.
Anodic stripping voltammetric analysis of heavy-metal ions
Heavy-metal ions such as mercury, lead, and cadmium are toxic, non-biodegradable, and tend to accumulate in plants and animals.16,17 The pollution of heavy-metal ions in soil and water presents a global issue, and poses a severe threat to both the ecosystem and humans. Square-wave anodic stripping voltammetry (SWASV) is an ASV method frequently used for the measurement of trace heavy metals because it greatly reduces the background noise coming from the charging current during the potential scan.18,19 Conventional ASV measurements of heavy-metal ions are usually performed either by dipping electrodes in a sample solution under controlled stirring condition or by placing a sample droplet onto the electrodes.20-23 The former approach is not practical in field measurements due to the difficulty of synchronizing the stirring and ASV procedures.20 The latter one shows limited sensitivity of measurement because pre-accumulation of analytes in stripping is limited by diffusion. Additionally, in this case, new electrodes are usually required for each measurement since it is difficult to remove the residue of deposited metals in a stagnant drop of solution before the next cycle of ASV.
We have demonstrated the use of μPEDs in the selective measurement of Pb(ii) in an aqueous mixture of Pb(ii) and Zn(ii) using SWASV. We modified the design of the μPED by introducing a pad of cellulose blotting paper as a sink in the outlet of the paper channel (Fig. 1c) (see ESI†). The μPED allows the continuous wicking of fluids to pass across the electrodes, and facilitates the plating of metals, as well as the cleaning of electrodes. By tuning the size of the cellulose blotting paper, we optimized the wicking time of fluids in the paper channel of μPEDs so that the flow stopped before the system entered the equilibration step in the process of SWASV.24
Fig. 4 displays representative stripping voltammograms for the measurement of 25 ppb (μg L−1) Pb(ii) in acetate buffer solution in the presence of Zn(ii). The voltammograms in the hydrodynamic μPEDs, in which the fluid of the sample solution continuously wicked in the paper microchannel, showed a well-defined, sharp peak for Pb(ii) at ca. 780 mV vs. the Ag/AgCl reference electrode (C and D in Fig. 4). In contrast, under the same SWASV conditions, a stagnant solution of analytes, both in the μPEDs (without a pad of blotting paper as a sink) and in an experiment that placed a droplet of sample solution on the electrodes, resulted in a much weaker signal (A in Fig. 4) or a poorly defined response (B in Fig. 4). The hydrodynamic μPEDs thus exhibited a much higher sensitivity by a factor of five than the stagnant systems. We ascribe the enhanced sensitivity of the dynamic system to the high efficiency of the accumulation of metals on the electrodes by convection of flowing fluids in the porous matrix of paper over the electrodes, and to the large volume (~800 μL) of sample that flows across the surface of the electrodes.
Fig. 4.
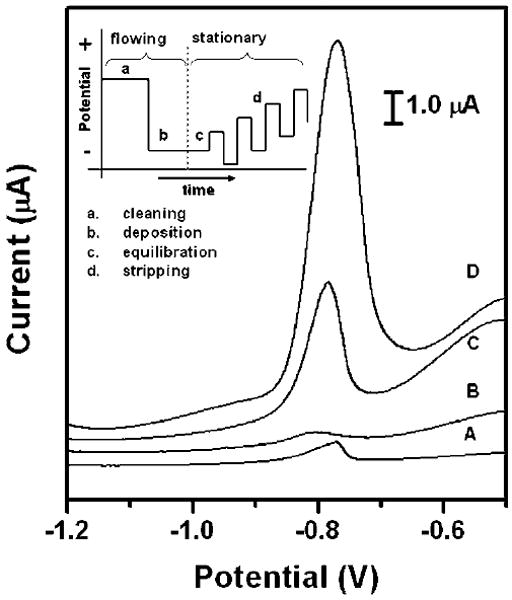
Square-wave anodic stripping voltammograms for 25 ppb solution of Pb(ii) in 0.1 M acetate buffer (pH 4.5) in the presence of 25 ppb Zn(ii) : (A) a 100 μL solution placed directly on the electrodes; (B) a 100 μL solution added to the stagnant μPEDs (without a pad of blotting paper as sink); (C and D) a solution of analytes continuously wicking the paper channel of the hydrodynamic μPEDs. The deposition time was 120 s (A, B, C) or 360 s (D). The SWASV was performed in the potential range of −1.2 to −0.5 V under optimized conditions: frequency, 20 Hz; amplitude, 25 mV; potential increment, 5 mV; equilibration time, 30 s. Deposition was performed at −1.2 V; ‘cleaning’ was performed at +0.5 V for 60 s. A bismuth(iii) concentration of 500 μg L−1 was chosen for the co-deposition of heavy-metal ions. We used screen-printed carbon electrodes as the working and counter electrodes, and a screen-printed Ag/AgCl electrode as the reference electrode. Inset shows the schematic of the four steps of the square-wave anodic stripping voltammetry.
In the hydrodynamic μPEDs, the peak current of the analysis of Pb (ii) dramatically increased with increasing the deposition time (Fig. 4). The peak current increased from 3.9 μA to 10.3 μA with the increase of deposition time from 120 s to 360 s. This increase was not obvious in the stagnant systems, since the deposition efficiency decays quickly with time due to the mass-transfer-limited reaction in the vicinity of the surface of electrodes. Moreover, we found that the stagnant μPEDs showed a more poorly defined signal, compared to the system with a drop of sample solution directly placed onto the electrodes (Fig. 4). We presume that this is because the cellulose matrix in the stagnant μPEDs inhibits convective movement of the solutions, and thus affected the stripping behavior of Pb(ii).
The stripping voltammograms for the analysis of Pb(ii) in the hydrodynamic μPEDs showed well-defined peaks and a strong signal over a wide range of concentrations of Pb(ii); this level of performance offers convenient quantification of low ppb levels of lead (Fig. 5a). The peak intensity increases proportionally with the concentration of Pb(ii), which yields a highly linear calibration plot with a slope of 0.17 μA ppb−1 for lead (correlation coefficient, 0.996) (Fig. 5b). The limit of detection of lead was estimated from the signal-to-noise characteristics of the data to be approximately 1.0 ppb (μg L−1); this value is even lower than 2.5 ppb obtained in conventional systems with controlled stirring.20-22,25,26 This value is also much lower than the 10 ppb (μg L−1) WHO guideline value for lead concentration in drinking water.4 We believe that even lower concentrations of lead could be detected if longer deposition periods were used. The sensitive measurement of Pb(ii) in the hydrodynamic μPEDs is highly reproducible, as indicated by the low relative standard deviation.
Fig. 5.
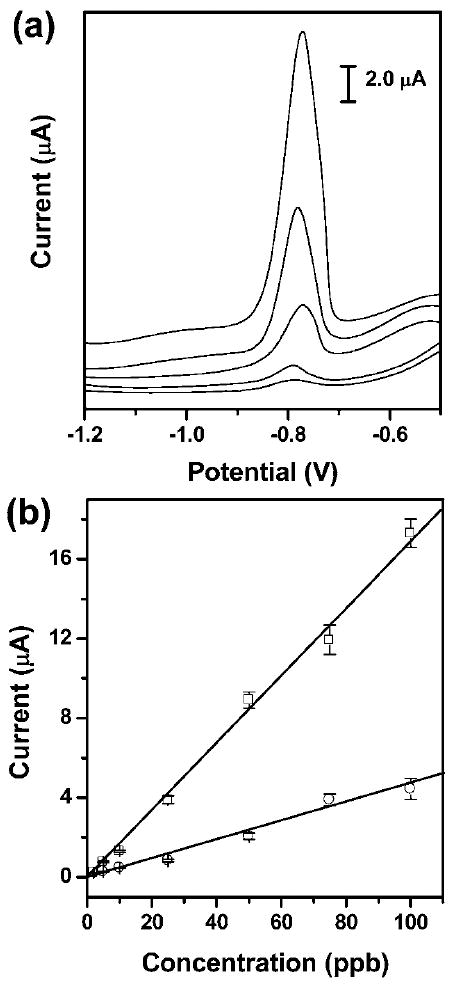
(a) Square-wave anodic stripping voltammograms for the analysis of trace Pb(ii) in 0.1 M acetate buffer (pH 4.5) in the presence of Zn(ii) (1: 1 molar ratio of Pb(ii) to Zn(ii)) in the μPEDs with a solution of analytes continuously wicking along the paper channel. The concentrations (ppb) of Pb(ii) (ascending along the y-axis) are 5, 10, 25, 50, and 100. The data are unsmoothed. (b) The resulting calibration plots for the analysis of trace Pb(ii): a 100 μL solution of analytes placed on the electrodes (○), and a solution of analytes continuously wicking along the paper channel in the μPEDs (□). Square-wave voltammetric stripping was performed in the potential range of −1.2 to −0.5 V under optimized conditions: frequency, 20 Hz; amplitude, 25 mV; potential increment, 5 mV; equilibration time, 30 s. Deposition was performed at −1.2 V for 120 s; ‘cleaning’ was performed at +0.5 V for 60 s. A bismuth(iii) concentration of 500 μg L−1 was chosen for the co-deposition of heavy-metal ions. We used screen-printed carbon electrodes as working and counter electrodes, and a screen-printed Ag/AgCl electrode as the reference. The solid lines represent a linear fit to (○) with regression equation: y = −0.08 + 0.05x (R2 = 0.978, n = 8), and a linear fit to (□) with regression equation: y = −0.21 + 0.17x (R2 = 0.996, n = 8).
We compared the performance of the hydrodynamic μPEDs for the analysis of lead with the stagnant system, in which a drop of sample solution was placed on electrodes (Fig. 5b). The stagnant system exhibits a much lower sensitivity of 0.05 μA ppb−1 for lead (correlation coefficient, 0.978), and a higher limit of detection of 4.3 ppb, than the dynamic measurement. Unlike the stagnant system, small perturbations (e.g., vibration, heating) did not interfere with the analysis of lead in the hydrodynamic μPEDs due to the stabilization of the flow of the sample solution by the paper matrix; this stabilization results in a high reliability and reproducibility of the measurements. The device is reusable by simply replacing the pad of blotting paper, since the continuous wicking removes dissolved analytes before the next cycle of the deposition of metals.
Conclusions
Paper provides a useful matrix for small electrochemical devices. It provides a thin, mechanically stabilized film of water or other fluids that deliver analytes to the surface of the electrodes. The fact that the cellulose matrix is mechanically rigid, and that the electrodes are in physical contact with the paper slab containing the analytes inhibits convective movement of the solutions after the complete wetting of the paper channel with fluids, and thus increases the accuracy of time-dependent measurements (chronoamperometry and chronopotentiometry). The continuous capillary wicking of fluids along the paper channel and across the electrode that is easily accomplished by introducing a cellulose blotting paper that acts as a sink provides an excellent platform for the measurements required for convection and hydrodynamic conditions (anodic stripping voltammetry, hydrodynamic amperometry and voltammetry).
Electrochemistry substantially increases the scope of options for detection using paper and fiber-based systems.1,27,28 We have demonstrated the chronoamperometric analysis of glucose, and the square-wave anodic stripping voltammetry measurements of heavy-metal ions using μPEDs. The resulting analysis of glucose is similar to that in bulk solution (the cellulose matrix occupies 30–40% of the volume of the space between the electrodes, and limiting differential currents are accordingly ~30% lower on paper than unstirred bulk solution). The measurements of heavy-metal ions in the hydrodynamic μPEDs have higher sensitivity and lower limit of detection (ca. 1.0 ppb) than conventional systems with controlled stirring (ca. 2.5 ppb).21,29 The μPEDs thus have three important advantages over conventional systems with controlled stirring: (i) they are simple, low-cost, and portable, (ii) they do not require extra instruments, such as stirring plates, and (iii) they are reusable, and are easily disposable after use.
The use of conductive inks to print electrodes is well suited for mass production using screen-printing technology. This methodology is suitable for further miniaturization, and to accommodate smaller lateral spacing between electrodes, and thus (with development) to higher density of detectors. The resulting paper-based electrochemical sensing devices combine the advantages of paper-based microfluidics—low-cost, small sample volumes, capillary wicking of fluids, facile multiplexed assays—with the advantages of electrochemical detection—high sensitivity, quantitative analysis, rapid response and applicability to a wide range of analytes. For the use in the field, it would be necessary to develop a portable electrochemical reader. The μPEDs described in the paper could easily be adapted to a portable reader. The development of this kind of devices is underway in this lab.
Supplementary Material
Acknowledgments
This work was supported by the Nano/Micro-electromechanical Systems Science and Technology Micro/Nano Fluidics Fundamentals Focus Center (Defense Advanced Research Projects Agency), the National Institutes of Environmental Health and Safety (016665), the Bill and Melinda Gates Foundation (51308), and a postdoctoral fellowship from the Natural Science and Engineering Research Council of Canada (to Z.N.). The Netherlands Organization for Scientific Research (NWO) is kindly acknowledged for the Rubicon grant (C.A.N.) supporting this research.
Footnotes
Electronic supplementary information (ESI) available: Geometry of the hydrodynamic μPED for the analysis of metals, estimation of diffusion coefficient, Cottrell plot for the analysis of glucose, chronoamperometric analysis of glucose in blood plasma and bovine blood, performance of gold electrodes on the paper-based devices.
References
- 1.Dungchai W, Chailapakul O, Henry CS. Anal Chem. 2009;81:5821–5826. doi: 10.1021/ac9007573. [DOI] [PubMed] [Google Scholar]
- 2.Martinez AW, Phillips ST, Carrilho E, Thomas SW, Sindi H, Whitesides GM. Anal Chem. 2008;80:3699–3707. doi: 10.1021/ac800112r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hones J, Muller P, Surridge N. Diabetes Technol Ther. 2008;10:S10–S26. [Google Scholar]
- 4.World Health Organization. Guidelines for Drinking-Water Quality. WHO, WHO Press; Switzerland: 2006. [Google Scholar]
- 5.Martinez AW, Phillips ST, Butte MJ, Whitesides GM. Angew Chem, Int Ed. 2007;46:1318–1320. doi: 10.1002/anie.200603817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Martinez AW, Phillips ST, Whitesides GM. Proc Natl Acad Sci U S A. 2008;105:19606–19611. doi: 10.1073/pnas.0810903105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Martinez AW, Phillips ST, Wiley BJ, Gupta M, Whitesides GM. Lab Chip. 2008;8:2146–2150. doi: 10.1039/b811135a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mabey D, Peeling RW, Ustianowski A, Perkins MD. Nat Rev Microbiol. 2004;2:231–240. doi: 10.1038/nrmicro841. [DOI] [PubMed] [Google Scholar]
- 9.Yager P, Domingo GJ, Gerdes J. Annu Rev Biomed Eng. 2008;10:107–144. doi: 10.1146/annurev.bioeng.10.061807.160524. [DOI] [PubMed] [Google Scholar]
- 10.Wang Y, Xu H, Zhang JM, Li G. Sensors. 2008;8:2043–2081. doi: 10.3390/s8042043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Other types of electrodes, for example, gold stripes coated on plastics are also suitable for the paper-based electrochemical device, and may be the best used for more specific applications. Paper devices with electrodes made from gold stripes showed excellent performance in cyclic voltammetry, but were less robust than the carbon-based electrodes. This problem is, however, simply due to the adhesion, which could be fixed by improved engineering.
- 12.Renedo OD, Alonso-Lomillo MA, Martinez MJA. Talanta. 2007;73:202–219. doi: 10.1016/j.talanta.2007.03.050. [DOI] [PubMed] [Google Scholar]
- 13.Carrilho E, Martinez AW, Whitesides GM. Anal Chem. 2009;81:7091–7095. doi: 10.1021/ac901071p. [DOI] [PubMed] [Google Scholar]
- 14.Bard AJ, Faulkner LR. Electrochemical Methods: Fundamentals and Applications. Wiley; New York: 2000. [Google Scholar]
- 15.Bartlett PN, Pratt KFE. J Electroanal Chem. 1995;397:53–60. [Google Scholar]
- 16.Patra M, Bhowmik N, Bandopadhyay B, Sharma A. Environ Exp Bot. 2004;52:199–223. [Google Scholar]
- 17.Porter SK, Scheckel KG, Impellitteri CA, Ryan JA. Crit Rev Environ Sci Technol. 2004;34:495–604. [Google Scholar]
- 18.Jena BK, Raj CR. Anal Chem. 2008;80:4836–4844. doi: 10.1021/ac071064w. [DOI] [PubMed] [Google Scholar]
- 19.Yantasee W, Lin Y, Hongsirikarn K, Fryxell GE, Addleman R, Timchalk C. Environ Health Perspect. 2007;115:1683–1690. doi: 10.1289/ehp.10190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang J, Lu JM, Kirgoz UA, Hocevar SB, Ogorevc B. Anal Chim Acta. 2001;434:29–34. [Google Scholar]
- 21.Rico MAG, Olivares-Marin M, Gil EP. Electroanalysis. 2008;20:2608–2613. [Google Scholar]
- 22.Honeychurch KC, Hart JP. TrAC, Trends Anal Chem. 2003;22:456–469. [Google Scholar]
- 23.Kadara RO, Tothill IE. Anal Bioanal Chem. 2004;378:770–775. doi: 10.1007/s00216-003-2351-x. [DOI] [PubMed] [Google Scholar]
- 24.We are aware that this device is sensitive to temperature due to temperature-dependent rate of wicking of flow. This problem, however, can be fixed by improved engineering.
- 25.Wang J, Lu JM, Hocevar SB, Farias PAM, Ogorevc B. Anal Chem. 2000;72:3218–3222. doi: 10.1021/ac000108x. [DOI] [PubMed] [Google Scholar]
- 26.Hwang GH, Han WK, Park JS, Kang SG. Sens Actuators, B. 2008;135:309–316. [Google Scholar]
- 27.Cui G, Kim SJ, Choi SH, Nam H, Cha GS, Paeng KJ. Anal Chem. 2000;72:1925–1929. doi: 10.1021/ac991213d. [DOI] [PubMed] [Google Scholar]
- 28.Cui G, Yoo JH, Yoo J, Lee SW, Nam H, Cha GS. Electroanalysis. 2001;13:224–228. [Google Scholar]
- 29.Honeychurch KC, Hart JP, Cowell DC. Electroanalysis. 2000;12:171–177. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.