

Abstract
In a recent issue of Molecular Cell, Zheng et al. (2009) describe a surprising set of findings that highlight an unexpected negative regulation of FAK by oncogenic Ras and its consequences for cancer cell migration and invasion.
Ras is one of the best-known signaling proteins implicated in the initiation and progression of cancer. It is a GTP-binding/GTPase that functions as a molecular switch downstream from a number of growth factor receptors. Its role as a signal transducer for growth factor-signaling has been especially well worked out for members of the EGF receptor (EGFR) family such as EGFR itself and the Neu/ErbB2 tyrosine kinase which activate Ras following their autophosphorylation and recruitment of the adaptor protein Grb2 and the Ras-guanine nucleotide exchange factor (GEF), Son-of-sevenless. The activation of Ras and its ensuing stimulation of downstream signaling targets and effector proteins, in particular ERK, PI-3K and Ral-GDS, have been implicated in malignant transformation, and consequently, mutations in Ras that block its GTP-hydrolytic activity and cause it to be irreversibly activated, have been identified in a significant percentage of human tumors.
While the signaling of Ras to its downstream effector proteins has been shown to stimulate cell-cycle progression and cellular proliferation, Ras activation has also been demonstrated to be important for tumor cell migration, invasion, and metastasis. Presumably, such an involvement would require some form of interplay with integrins and other proteins implicated in cell adhesion and migration. One protein in particular that has often been implicated in cell migration, as well as being linked to tumor cell invasiveness and metastasis, is the integrin-activated, focal adhesion kinase (FAK). This is perhaps best seen in fibroblasts derived from FAK knockout mice, as these cells show increased numbers of large, stable focal adhesions, as well as a diminished ability to migrate on fibronectin-coated dishes and in response to growth factors and chemoattractants (Ilić et al., 1995, Zhao and Guan, 2009). In addition, EGFRs, which activate Ras as well as FAK, have been reported to signal through FAK to stimulate cell migration (Sieg et al., 2000). Thus, one might naturally assume that oncogenic Ras and FAK cooperate to promote tumor cell migration and invasive activity.
However, recent findings by Zheng et al. (2009), show that apparently things are not that straightforward in fibroblasts transformed by oncogenic Ras. In fact, these studies point to an unexpected twist regarding the interplay between Ras and FAK by showing that activated Ras causes the dephosphorylation of Tyr397 of FAK, a FAK-autophosphorylation site that leads to the recruitment of the c-Src tyrosine kinase and is essential for its signaling functions. The dephosphorylation of FAK appears to be the outcome of a Ras-dependent signaling pathway that consists of the small GTPase Cdc42, its upstream activator/GEF Fgd1 (Facial genital dysplasia-1), its downstream effector Pak1 (p21-activated kinase), MEK1 and ERK (Figure 1). The activation of MEK1 by PAK1, and subsequently ERK by MEK1, is suggested to result in the phosphorylation of FAK at Ser910. This enables the binding of PIN1 (Protein interacting with NIMA (never in mitosis A)-1), a peptidyl-prolyl cis-trans isomerase, which in turn leads to the recruitment of a protein tyrosine phosphatase, PTP-PEST, resulting in the dephosphorylation of Tyr397 of FAK. Remarkably, the inhibition of FAK appears to correlate with the ability of oncogenic Ras to induce cell migration, invasion, and metastasis.
Figure 1.
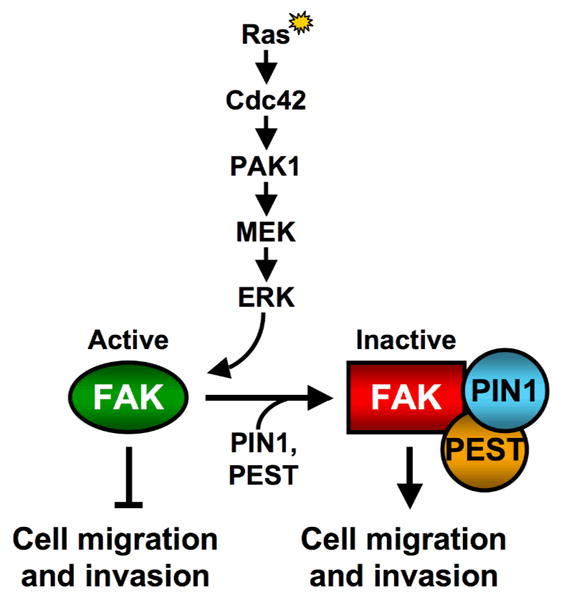
Scheme for the Ras-mediated inactivation of FAK. Activated Ras, signaling through Cdc42, PAK1 and MEK, leads to an ERK-induced phosphorylation of FAK at serine 910. This phosphorylation event triggers the binding of PIN1 to FAK which in turn leads to the recruitment of PTP-PEST to FAK. PTP-PEST then de-phosphorylates tyrosine 397 on FAK, a FAK-autophosphorylation site that is crucial for its signaling functions. Surprisingly, it is the resulting inactivation of FAK that appears to be necessary for Ras-stimulated cell migration and invasion.
So how do the authors explain this apparent paradox? First, they point out that while FAK expression appears to be upregulated in a number of human cancers, there are some cancers where lower levels of FAK have been associated with higher rates of metastasis and poor patient prognosis (Zhao and Guan, 2009). Moreover, despite the various reports implicating FAK in the stimulation of cell migration, it has also been shown that under some conditions, FAK inhibits cell migration (Yano et al., 2004). This leads the authors to suggest that the overexpression of FAK in some tumors might promote cell survival under the stressful and adverse conditions encountered by tumor cells, primarily through a FAK-Src-signaling pathway, in which cases FAK activation would play a positive role in tumorigenesis. However, in cancers that are initiated by the actions of oncogenic Ras, which presumably do not require c-Src for their transformed phenotypes, the activation of the Cdc42-PAK1-MEK-ERK pathway leads to FAK dephosphorylation and inactivation, reduced cell adhesion and consequently increased migration and invasiveness. The authors further speculate that FAK activity may need to be dynamically regulated during tumorigenesis. In particular, when metastatic cancer cells re-adhere at their new sites of colonization, integrin-dependent activation of FAK would likely be important for the formation of adhesions.
All of this seems reasonable enough, at least at first glance. However, as is always the case with new and important findings, the more one looks at the model being presented, the more one finds questions that need answers. Perhaps the most obvious question, as raised by the authors, is how might FAK block migration such that its inactivation gives rise to increased cell motility? Based on studies by Yano et al. (2004), which show that FAK and its phosphosubstrate paxillin inhibit cell motility and promote N-cadherin-containing adhesions, one possibility is that FAK attenuates signaling by the small GTPase Rac1, a well-known regulator of cell migration. However, it is not obvious how this might occur. In fact, earlier reports had suggested that Rac1 is activated downstream of Ras and is essential for the transforming actions of oncogenic Ras (Khosravi-Far et al., 1995). There are also questions surrounding the role of Cdc42. While it has been demonstrated that like Rac1, activated Cdc42 is necessary for Ras-induced transformation (Qiu et al., 1997), the mechanism by which Cdc42 fulfills this requirement has been unclear. Expression of activated Cdc42 mutants in cells have been shown to activate JNK and p38, but not ERK1/2 (Minden et al., 1995). Another question involves the mechanism by which oncogenic Ras signals to the Cdc42-GEF Fgd1. Might other GEFs, such as Intersectin, which is capable of activating both Ras- and Cdc42-signaling pathways (Wang et al., 2005), or the Cool/Pix proteins that have been implicated in EGFR signaling as well as in cell migration (Feng et al., 2006), also in some circumstances help to mediate an interplay between Ras and FAK?
Despite these questions, the work by Zheng et al. highlights a mechanism for regulating FAK that has important consequences for Ras-promoted cell motility and invasion. Whether the ability of oncogenic Ras to inactivate FAK-signaling is necessary and sufficient to promote cell migration and invasive activity, or it represents one aspect of a dynamic regulation of FAK that involves distinct pathways responsible for its activation and inactivation, remains to be determined. However, what the work of Zheng et al. does show us is an important new regulatory aspect of invasion/metastasis that in the long run might offer novel modes of intervention against Ras-induced cancers.
References
- Feng Q, Baird D, Peng X, Wang J, Ly T, Guan JL, Cerione RA. Nat Cell Biol. 2006;8:945–956. doi: 10.1038/ncb1453. [DOI] [PubMed] [Google Scholar]
- Ilić D, Furuta Y, Kanazawa S, Takeda N, Sobue K, Nakatsuji N, Nomura S, Fujimoto J, Okada M, Yamamoto T. Nature. 1995;377:539–544. doi: 10.1038/377539a0. [DOI] [PubMed] [Google Scholar]
- Khosravi-Far R, Solski PA, Clark GJ, Kinch MS, Der CJ. Mol Cell Biol. 1995;15:6443–6453. doi: 10.1128/mcb.15.11.6443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minden A, Lin A, Claret FX, Abo A, Karin M. Cell. 1995;81:1147–1157. doi: 10.1016/s0092-8674(05)80019-4. [DOI] [PubMed] [Google Scholar]
- Qiu RG, Abo A, McCormick F, Symons M. Mol Cell Biol. 1997;17:3449–3458. doi: 10.1128/mcb.17.6.3449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sieg DJ, Hauck CR, Ilic D, Klingbeil CK, Schaefer E, Damsky CH, Schlaepfer DD. Nat Cell Biol. 2000;2:249–256. doi: 10.1038/35010517. [DOI] [PubMed] [Google Scholar]
- Wang JB, Wu WJ, Cerione RA. J Biol Chem. 2005;280:22883–22891. doi: 10.1074/jbc.M414375200. [DOI] [PubMed] [Google Scholar]
- Yano H, Mazaki Y, Kurokawa K, Hanks SK, Matsuda M, Sabe H. J Cell Biol. 2004;166:283–295. doi: 10.1083/jcb.200312013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Guan JL. Cancer Metastasis Rev. 2009;28:35–49. doi: 10.1007/s10555-008-9165-4. [DOI] [PubMed] [Google Scholar]
- Zheng Y, Xia Y, Hawke D, Halle M, Tremblay ML, Gao X, Zhou XZ, Aldape K, Cobb MH, Xie K, He J, Lu Z. Mol Cell. 2009;35:11–25. doi: 10.1016/j.molcel.2009.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]