

Abstract
Of the 20 ribosomally coded amino acid residues, lysine is the most frequently post-translationally modified, which has important functional and regulatory consequences. Here we report the identification and verification of a previously unreported form of protein post-translational modification (PTM): lysine succinylation. The succinyllysine residue was initially identified by mass spectrometry and protein sequence alignment. The identified succinyllysine peptides derived from in vivo proteins were verified by western blot analysis, in vivo labeling with isotopic succinate, MS/MS and HPLC coelution of their synthetic counterparts. We further show that lysine succinylation is evolutionarily conserved and that this PTM responds to different physiological conditions. Our study also implies that succinyl-CoA might be a cofactor for lysine succinylation. Given the apparent high abundance of lysine succinylation and the significant structural changes induced by this PTM, it is expected that lysine succinylation has important cellular functions.
Protein post-translational modifications are one of the most efficient biological mechanisms for expanding the genetic code and for regulating cellular physiology1,2. The remarkable complexity of PTM networks is exemplified by modifications at the side chain of lysine, one of the three basic residues critical for protein structure and function. Lysine residues in proteins can be subjected to a variety of PTMs, including methylation, acetylation, biotinylation, ubiquitination, ubiquitin-like modifications, propionylation and butyrylation, the last two of which were recently identified by us3,4. Extensive studies in the past few decades have revealed that most, if not all, of these lysine PTMs are important in cellular physiology and pathology5–8.
The method of choice for mapping a PTM site uses the molecular weight of the peptide and its fragments, which can be determined by mass spectrometry. The PTM induces both a structural change and a mass shift to its substrate residue. For example, lysine acetylation and lysine dimethylation lead to mass increases of 42.0106 and 28.0313 daltons (Da), respectively. To map PTM sites, one or a few PTMs of interest are typically prespecified during the protein sequence alignment of MS/MS data9. Recent advances in nonrestrictive sequence alignment make it possible to identify PTMs without prior specification of mass shifts that are induced by undescribed PTMs10,11, enabling identification of new PTMs.
Here we report the identification and verification of a previously unreported form of PTM: lysine succinylation. The lysine-succinylated peptide FTEGAFSuccKDWGYQLAR of Escherichia coli isocitrate dehydrogenase was initially identified on the basis of a mass shift of 100.0186 Da at the lysine residue by HPLC-MS/MS analysis, sequence alignment using PTMap and manual verification. Four succinyllysine (1) peptide candidates identified from three proteins (isocitrate dehydrogenase, serine hydroxymethyltransferase and glyceraldehyde-3-phosphate dehydrogenase A (GAPDH)) were then comprehensively verified by four independent methods: western blot analysis, in vivo labeling with isotopic succinate, MS/MS and HPLC coelution of their corresponding synthetic peptides. In addition, we also show, by MS/MS and HPLC coelution of methylmalonyllysine (the succinyllysine isomer), that the detected mass shift of 100.0186 Da is caused by succinylation rather than methylmalonylation. Mutagenesis analysis of the succinylated lysine residues of isocitrate dehydrogenase indicated the importance of these sites for keeping the protein's enzymatic activity. By carrying out affinity purification using an anti-succinyllysine antibody, we identified 69 succinyllysine sites among 14 E. coli proteins. The results conclusively established that lysine succinylation is a naturally occurring lysine modification.
RESULTS
100.0186 Da mass shift in isocitrate dehydrogenase
Isocitrate dehydrogenase, a citric acid cycle (TCA cycle) protein, catalyzes the third step of the cycle: the conversion of isocitrate to α-ketoglutarate and CO2. This step is the rate-limiting step in the TCA cycle. Our earlier proteomics studies on lysine-acetylated proteins showed that this protein is lysine acetylated not only in E. coli cells but also in mouse mitochondria12,13.
To examine other possible PTMs in the protein, we purified His-tagged isocitrate dehydrogenase from E. coli K-12 using Ni-NTA agarose beads (Supplementary Methods). The isolated protein was resolved in SDS-PAGE gel, excised from the gel (Supplementary Fig. 1) and in-gel digested for HPLC-MS/MS analysis. We used an algorithm that enables the identification of all possible PTMs, called PTMap, to analyze the resulting MS/MS data.
Notably, we identified a tryptic peptide, FTEGAFKDWGYQLAR, as having a mass shift of 100.0186 Da (precursor ion mass at m/z 944.9487) localized at the lysine residue Lys242. The accurate mass shift was used to deduce the possible PTM. On the basis of the annotation from Unimod (http://www.unimod.org/)—an online protein modification database for mass spectrometry—the most likely structure for this mass shift is a succinyl group or its isomer, a methylmalonyl group (Fig. 1).
Figure 1. Illustration of chemical structures of lysine, acetyllysine, succinyllysine and methylmalonyllysine residues.
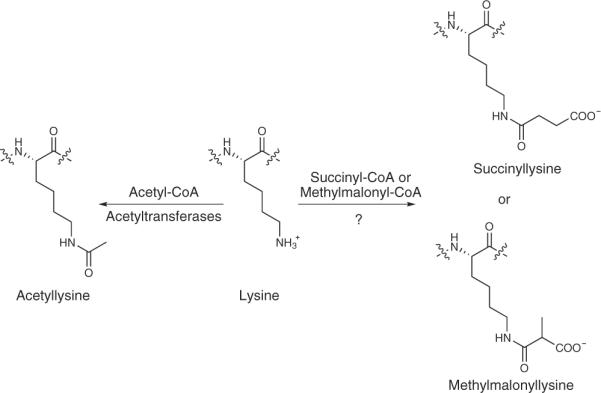
The enzymatic reaction for lysine acetylation and the hypothesized mechanism for lysine succinylation are indicated.
Verification of a lysine-succinylated peptide candidate
Identical peptides have the same MS/MS patterns and coelute in HPLC, which is the gold standard for confirming peptide identification. To verify that the PTM with a mass shift of 100.0186 Da represented a PTM induced by succinylation, we synthesized a succinyllysine peptide bearing the same sequence as the in vivo one. We analyzed both the in vivo–derived peptide and its synthetic counterpart by HRMS. MS/MS of the in vivo peptide matched perfectly with that of the synthetic counterpart (Fig. 2a; MS/MS spectrum with the detailed peak assignment is included in Supplementary Fig. 2).
Figure 2. Mass spectrometric identification and verification of a lysine-succinylated peptide from isocitrate dehydrogenase.

(a) MS/MS spectra of a doubly charged tryptic peptide (FTEGAFKDWGYQlAR) from isocitrate dehydrogenase (top), the synthetic succinyllysine peptide corresponding to the in vivo peptide sequence (middle) and the synthetic methylmalonyllysine peptide bearing the same peptide sequence (bottom). Insets show the precursor ion masses. The neutral loss of the Co2 group is shown in red. (b) Extracted ion chromatograms (XICs) of the in vivo-derived isocitrate dehydrogenase peptide (top), the synthetic succinyllysine peptide bearing the same peptide sequence (middle) and a mixture of the peptide from in vivo-derived isocitrate dehydrogenase tryptic peptide and its synthetic counterpart (bottom). (c) The HPlC coelution profile of FTEGAFSuccKDWGYQlAR (◆) and FTEGAFMeMalKDWGYQlAR (▼), showing the different retention times of these two peptides.
In contrast, the MS/MS spectrum from a peptide containing a methylmalonyllysine residue, FTEGAFMeMalKDWGYQLAR, showed a notably different pattern (Fig. 2a; its MS/MS spectrum with the detailed peak assignment is shown in Supplementary Fig. 3) from that of the same peptide bearing a succinyllysine residue (either in vivo or synthetic version). The peptide containing a methylmalonyllysine, but not a succinyllysine, showed significant neutral loss of CO2 peaks (−43.9898 Da for singly charged ions or −21.9949 Da for doubly charged ions). The resulting MS/MS result can be used as additional evidence to distinguish the lysine-succinylated peptide from lysine-methylmalonylated peptide.
The coelution experiment using HPLC-MS/MS demonstrated that the in vivo peptide, the synthetic succinyllysine peptide and a mixture of the two had similar retention times, and the mixture showed a single coeluted peak (Fig. 2b), further suggesting that the observed 100.0186 Da mass shift was derived from lysine succinylation. In addition, the synthetic methylmalonyllysine peptide showed a longer retention time than its corresponding lysine-succinylated peptide in the reversed-phase HPLC coelution chromatogram (Fig. 2c), indicating that the mass shift is not caused by methylmalonylation.
Confirmation of lysine succinylation using western blot
To further verify the lysine succinylation in isocitrate dehydrogenase, we developed a pan-anti-succinyllysine antibody using a method previously described14. The specificity of the antibody was confirmed by dot-spot assay (Fig. 3a).
Figure 3. Verification of lysine succinylation by western blot analysis.
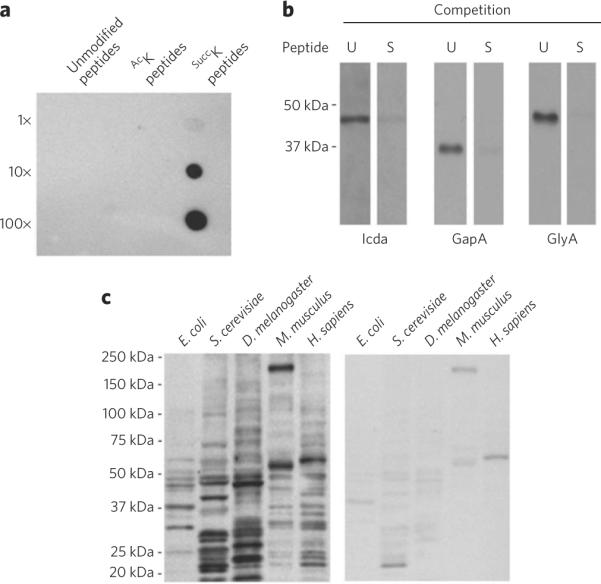
(a) Specificity of anti-SuccK antibody using dot-spot assays. Peptide libraries bearing a fixed unmodified lysine, acetyllysine or succinyllysine were spotted on nitrocellulose membrane with 10-fold dilutions. The 13-residue randomized peptide libraries have a fixed lysine residue at the seventh position: lane 1, unmodified lysine; lane 2, AcK; lane 3, SuccK. (b) Western blot analysis of recombinant E. coli isocitrate dehydrogenase (Icda), GADPH (GapA) and serine hydroxymethyltransferase (GlyA) competed with a lysine-succinylated (S) or an unmodified (U) peptide library. The same amounts of sample were loaded in both lanes for each protein. (c) Western blot analysis of lysine succinylation competed with a lysine succinylated (right) or an unmodified (left) peptide library in protein whole-cell lysates of E. coli (K-12 MG1655), S. cerevisiae (strain BY4741), D. melanogaster (S2 cells), M. musculus (C3H10 (T1/2) cells) and H. sapiens (Hela cells).
We performed western blot analysis using the purified E. coli isocitrate dehydrogenase with the anti-succinyllysine antibody. Through western blotting, a specific band was detected whose signal could be competed away by a peptide library bearing a fixed succinyllysine (Fig. 3b). This experiment provides additional evidence of lysine succinylation in isocitrate dehydrogenase.
In addition to isocitrate dehydrogenase, we also carried out western blot analysis in protein whole-cell lysates from E. coli, Saccharomyces cerevisiae, Drosophila melanogaster, Mus musculus and Homo sapiens cells (Fig. 3c). The results showed that signals derived from the full spectrum of cellular proteins can be detected by the antibody and can be efficiently competed away using a peptide library bearing a fixed succinyllysine. This experiment suggests that there are many protein substrates for lysine succinylation among these cells and that this PTM is evolutionarily conserved.
Taken together, the experiments described above conclusively verify that the mass shift of 100.0186 Da was caused by lysine succinylation and not by lysine methylmalonylation, shown by three independent methods: MS/MS, coelution in HPLC and western blot analysis.
Lysine succinylation in other two E. coli proteins
As succinyllysine signals were detected in a wide range of cellular proteins by western blot analysis of bacterial whole-cell lysates, we asked whether lysine succinylation is present in other E. coli proteins. To address this issue, we isolated two additional proteins, serine hydroxymethyltransferase and GAPDH (Supplementary Fig. 1). Lysine succinylation was detected in both proteins by western blot analysis (Fig. 3b). The isolated proteins were in-gel digested and analyzed by nano-HPLC-MS/MS. We detected succinyllysine residues in peptides GGSEELYSuccKK (Fig. 4a; its MS/MS spectrum with the detailed peak assignment is included in Supplementary Fig. 4) and NLTGSuccKEADAALGR (Supplementary Fig. 5a) from serine hydroxymethyltransferase and GASQNIIPSSTGAASuccKAVGK (Supplementary Fig. 6) from GAPDH. The identification of these peptides was confirmed by MS/MS and coelution in HPLC using their corresponding synthetic succinyllysine peptides (Fig. 4b and Supplementary Figs. 5 and 6). Similar to the peptide identified from isocitrate dehydrogenase, the synthetic methylmalonyllysine peptide, GGSEELYMeMalKK, was more retentive on the C12 column than its corresponding succinyllysine counterpart (Fig. 4c). Additionally, the neutral loss of CO2 peaks was only observed in methylmalonyllysine peptide (Fig. 4a). These unique features can be used to distinguish succinyllysine peptides from methylmalonyllysine peptides.
Figure 4. Mass spectrometric identification and verification of a lysine-succinylated peptide from serine hydroxymethyltransferase.

(a) MS/MS spectra of a doubly charged tryptic peptide (GGSEElYKK) from serine hydromethyltransferase (top), the synthetic succinyllysine peptide corresponding to the in vivo peptide sequence (middle) and the synthetic methylmalonyllysine peptide bearing the same peptide sequence (bottom). Insets show the precursor ion masses. The neutral loss of Co2 group was shown in red. (b) XICs of the in vivo-derived serine hydroxymethyltransferase peptide (top), the synthetic succinyllysine peptide bearing the same peptide sequence (middle) and a mixture of the peptide from in vivo-derived serine hydroxymethyltransferase tryptic peptide and its synthetic counterpart (bottom). (c) The HPlC coelution profile of GGSEElYSuccKK (◆) and GGSEElYMeMalKK (▼), showing the different retention times of these two peptides.
In vivo isotopic succinate labeling
Sodium acetate was used to label lysine acetylation to confirm its in vivo existence15. We rationalized that sodium succinate (2) may be used by cells in a similar fashion as sodium acetate for lysine acetylation. After entering the cells, sodium succinate can be converted into succinyl-CoA by succinyl-CoA synthetase16.
Indeed, sodium succinate significantly enhanced the global profile of lysine succinylation (Fig. 5a). To test whether the isotopic sodium succinate, 2,2,3,3-D4-succinate, can be used to label lysine succinylation, we treated E. coli cells with the compound for 40 min. After labeling, we extracted whole-cell lysate and digested the protein with trypsin. The resulting tryptic peptides were subjected to affinity enrichment using anti-succinyllysine antibody, and the enriched peptides were analyzed by HPLC-MS/MS analysis. The study identified succinyllysine-containing peptides bearing either H4- or D4-succinyl group (Fig. 5b and Supplementary Table 1) with a roughly equal ratio (in terms of the numbers of MS/MS spectra identified by Mascot algorithm). This result suggests that lysine succinylation can be in vivo labeled by isotopic succinate. Additionally, we identified 12 new sites in these three proteins with this approach (Supplementary Table 1 and Supplementary Data Set 1).
Figure 5. Stimulation of lysine succinylation by sodium succinate and in vivo isotopic succinate labeling.
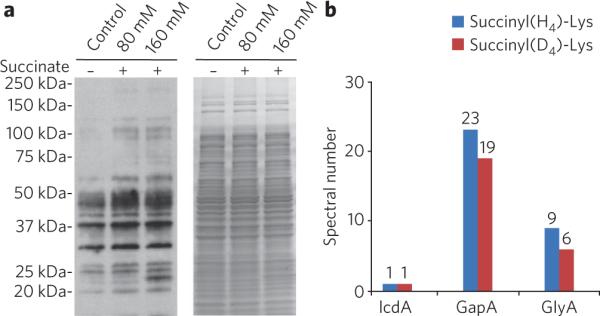
(a) Stimulation of lysine succinylation in response to succinate. Western blot analysis (left) of whole-cell lysates from untreated (control), 80 mM and 160 mM sodium succinate treated (for 4 h) E. coli cells. Coomassie blue gel shows equal loading amounts (right). (b) MS/MS spectral numbers of H4- and D4-labeled succinyllysine peptides identified from E. coli that were treated with 160 mM 2,2,3,3-D4-succinate. Results were based on Mascot sequence alignment using ion score cutoff 30 and manually verified.
Sequence alignment and mutagenesis analysis
We carried out sequence alignment and studied possible roles of the identified succinylated lysine residues on the basis of the structures of the three proteins. Of the two succinylated lysine residues (Lys100 and Lys242) in isocitrate dehydrogenase, Lys100 is evolutionarily conserved among all six species examined in this study, whereas Lys242 is not (Fig. 6a). We analyzed the possible interaction of the two residues with other important residues for the enzyme's functions (for example, binding to the cofactors or substrate) (Fig. 6b and Supplementary Fig. 7). Neither Lys100 nor Lys242 is known to be directly involved in substrate binding and catalysis. The unmodified Lys242 residue may also form a salt bridge with nearby residues such as Glu238 and Asp279. Succinylated Lys100 (with a negative charge after the modification) could potentially form a salt bridge with nearby residue Arg119 (within 4 Å), which is one of the residues responsible for the substrate binding. These results suggest that lysine succinylation may affect its enzymatic activity. Similar analysis was carried out for serine hydroxymethyltransferase and GAPDH (Supplementary Figs. 8 and 9).
Figure 6. Sequence alignment and mutagenesis analysis of isocitrate dehydrogenase.

(a) ClustalW (2.0.12) alignment of isocitrate dehydrogenase homologs from H. sapiens (GenInfo Identifier (GI): 5031777), M. musculus (GI: 148693874), D. melanogaster (GI: 24643268), C. elegans (GI: 71986051), S. cerevisiae (GI: 6324709) and E. coli (GI: 170080787). Conserved sites are shaded with gray and black. Conserved and nonconserved succinyllysine residues are indicated by red and blue triangles, respectively. The positions are labeled corresponding to the E. coli sequence. (b) localization of the succinyllysines and functional sites in isocitrate dehydrogenase. The three-dimensional structure was obtained from the Molecular Modeling Database (MMDB) (MMDB ID 49631) and viewed by Cn3D (v4.1). Succinylated and known functionally important sites (Uniprot ID P08200) are indicated by red and yellow arrows, respectively. The superscripts SB, NB, MB, CAT and SUCC on the labeled residues refer to: substrate binding, NADP binding, metal binding, catalytic sites and succinylation, respectively. (c) Purity of the wild-type and mutated proteins shown by SDS-PAGE gel. (d) Enzymatic activities of wild-type (control), K242R, K242E (top), K100E and K100R (bottom) isocitrate dehydrogenase mutants.
To analyze possible functional consequence of lysine succinylation, we carried out mutagenesis experiments. The modified lysine residues, Lys242 and Lys100, were mutated to either arginine (K-to-R mutant), which conserved the positive charge, or glutamate (K-to-E mutant), which mimicked lysine succinylation. Primers that were used for site-specific mutagenesis are shown in Supplementary Table 2. Each mutant protein and its wide-type counterpart were expressed and then purified up to >95% purity (Fig. 6c). The isolated proteins were used to assay enzymatic activity17. Our results showed that mutation of Lys242 to either glutamate or arginine led to a decrease of the enzymatic activity and that the K-to-E mutant had a more significant activity drop than the K-to-R mutant (Fig. 6d). In a parallel experiment for the analysis of Lys100 mutants, both mutants almost completely abolished the enzymatic activity (Fig. 6d). We performed circular dichroism spectroscopic analysis on purified the proteins to test conformational changes of these mutants. The mutants (K100R and K100E) folded properly but showed some changes in the alpha helices. In particular, the signal attributable to the alpha helices increased considerably in the K100R mutant, whereas it decreased slightly in the K100E mutant (Supplementary Fig. 10). All these results suggest that both Lys242 and Lys100 are important for the activity of isocitrate dehydrogenase and that lysine succinylation is likely to inhibit its enzymatic function.
Lysine succinylation in other substrate proteins
To identify additional succinyllysine peptides in E. coli, peptide immunoprecipitation was carried out using anti-succinyllysine antibody and HPLC-MS/MS analysis. We identified 69 succinyllysine sites among 14 substrate proteins (Supplementary Table 3 and Supplementary Data Set 2). Many of these proteins are involved in the regulation of energy metabolism and translation, two biological processes that are highly connected with cellular energy status, implying a possible role for lysine succinylation in energy metabolism.
In addition to E. coli proteins, we also analyzed affinity- purified GAPDH protein of S. cerevisiae by MS. The study identified one peptide succinylated at Lys225 (VLPELQGSuccKLTGMAFR) (Supplementary Fig. 11). This succinylation site is evolutionarily conserved between the yeast protein and its E. coli counterpart (Lys225, Supplementary Fig. 9). Additionally, we analyzed the lysine succinylation profiles among different cancer cell lines by western blot analysis. The results showed differential lysine succinylation patterns among the cell lines, which implies a dynamic nature for lysine succinylation in mammalian cells (Supplementary Fig. 12).
DISCUSSION
Lysine residues are key to the formation of spatial structures of proteins and the regulation protein functions. Lysine is one of the three ribosomally coded amino acid residues with positively charged side chains at physiological pH. Its side chain can be involved in manifold noncovalent interactions in a protein, including van der Waals interactions, hydrogen bonds and electrostatic interactions with negatively charged residues. For example, salt bridge formation between lysine residues and acidic residues is thought to be important in forming the leucine zipper structure18. The lysine residue plays a key role in general acid-base catalyzed enzymatic reactions in which proton transfer is required19. Neutralization of the basic side chain of lysine could lead to significant effects on protein function, as has been described for lysine acetylation, ubiquitination and methylation13,20. Given the importance of lysine in protein folding and function, it is expected that lysine succinylation is likely to have a significant impact on the substrate proteins.
Lysine succinylation induces more substantial changes to a protein's chemical properties than do lysine methylation and acetylation, two PTMs known to have important cellular roles. At physiological pH (7.4), succinylation, acetylation and monomethylation at a lysine residue will change the charge status from +1 to −1, from +1 to 0 and not at all, respectively. Thus, the change of charge status at the lysine residue caused by succinylation is comparable to that caused by protein phosphorylation at serine, threonine or tyrosine residues (from 0 to −2 charges). In addition, succinylation adds a bigger structural moiety than acetylation or methylation. Accordingly, the more dramatic structural alteration resulting from lysine succinylation, as demonstrated in the mutagenesis experiment, is likely to lead to more significant changes in protein structure and function.
Enzymatic reactions of short-chain lysine acylations, such as acetylation, propionylation and butyrylation, use their corresponding high-energy CoAs to perform reactions. By analogy to these reactions, and in combination with our isotopic succinate labeling experiments and observations of differential succinylation among cancer cell lines (Supplementary Fig. 12), we therefore propose that succinyl-CoA is the cofactor of enzyme-mediated lysine succinylation. Succinyl-CoA is an important metabolic intermediate in a variety of metabolic pathways, including TCA cycle, porphyrin synthesis and catabolism of odd-chain fatty acids and some branched-chain amino acids. Homeostasis of succinyl-CoA is critical to normal cellular physiology. Mutations in the genes that are involved in succinyl-CoA metabolism, such as ketoglutarate dehydrogenase, succinyl-CoA-3-ketoacid-coenzyme A transferase and succinyl-CoA synthetase21–23, could lead to diseases.
In summary, we present evidence to conclusively establish lysine succinylation as a new PTM. Given the high abundance of lysine succinylation and its induced chemical changes, it is highly likely that lysine succinylation could have important cellular functions. This study provides a stepping stone for dissection of this PTM pathway and studies of its biological significance.
METHODS
Materials
Ni-NTA agarose beads were purchased from Qiagen; modified sequencing-grade trypsin from Promega; C18 ZipTips from Millipore Corporation; E. coli ASKA library (a complete set of E. coli from the K-12 Open Reading Frame Archive) obtained from H. Mori (Keio University); water and acetonitrile from Burdick & Jackson; protein A beads from GE Healthcare; trifluoroacetic acid (TFA), formic acid, dithiothreitol, trichloroacetic acid, acetone and iodoacetamide from Sigma-Aldrich. 2,2,3,3-D4-succinic acid was purchased from Cambridge Isotope Laboratories. All the peptides were synthesized by commercial customer synthesis using Fmoc-Lys(mono-tert-butyl succinate)-OH and Fmoc-Lys(monotert-butyl methylmalonate)-OH (HPLC analysis and purity were included in Supplementary Fig. 13).
Isotopic succinate labeling and affinity purification
The E. coli strain K-12 MG1655 was grown aerobically in LB medium at 37 °C with 250 r.p.m. shaking. For in vivo succinyl group labeling, the cultured cells were treated with 160 mM sodium 2,2,3,3-D4-succinate (pH 7.5) for 40 min before harvesting. After the collected cells were washed twice with ice-cold PBS, cell pellets were resuspended in a chilled lysis buffer (10 mM Tris-HCl, 100 mM NaCl, 1 mM EDTA, 0.5% (w/v) NP-40, pH 8.0) and then sonicated. The unbroken cells and debris were removed by centrifugation, and the supernatant was kept at −80 °C until use.
Affinity purification was performed using protein A agarose beads conjugated with 20 μl of anti-succinyllysine antibody at 4 °C for 6 h13. The beads were washed three times with 1 ml of NETN buffer (100mM NaCl, 1mM EDTA, 20mM Tris pH 8.0 and 0.5% (w/v) NP-40) and three times with ETN (50 mM Tris-HCl, pH 8.0, 100 mM NaCl, 1 mM EDTA). The bound peptides were eluted from the beads by washing three times with 100 μl of 0.1 M glycine (pH 2.5). The elutes were combined and dried in a SpeedVac.
HPLC-MS/MS analysis and database searching
The dried peptide samples were dissolved in 3-μl HPLC buffer A (0.1% formic acid in water, v/v) and loaded onto a home-made capillary column (10 cm length with 75 μm ID) packed with Jupiter C12 resin (4-μm particle size, 90-Å pore size, Phenomenex) connected to an Eksigent NanoLC-1Dplus HPLC system (Eksigent Technologies). Peptides were eluted with a 2-h gradient of 2% to 90% HPLC buffer B (0.1% formic acid in acetonitrile, v/v) in buffer A at a flow rate of 200 nl min−1. The eluted peptides were ionized and introduced into an LTQ Orbitrap Discovery mass spectrometer using a nanospray source. Survey full-scan MS spectra (from m/z 350–2,000) were acquired in the Orbitrap with resolution R = 30,000 at m/z 400. The ten most intense ions were sequentially isolated in the linear ion trap and subjected to collisionally activated dissociation (CAD) with a normalized energy of 35%. The exclusion duration for the data-dependant scan was 36 sec, the repeat count was 2, and the exclusion window was set at +2 Da and −1 Da.
Unrestrictive identification of PTMs was carried out by the PTMap program11 Mass tolerance for precursor ions was set at 0.01 Da and for MS/MS was set at ±0.5 Da. In addition, all the HPLC–MS/MS data were analyzed by Mascot (v2.1, Matrix Science). Peak lists were generated by extract_msn.exe software by ThermoFisher. Precursor mass tolerance for Mascot analysis was set at ±2 Da, and fragment mass tolerance was set at ±0.5 Da. All data were searched against the National Center for Biotechnology Information nonredundant E. coli database. Cysteine alkylation by iodoacetamide, methione oxidation and lysine succinylation (lysine +100.01604) were specified as variable modifications. All results were manually verified. Precursor ion mass errors of all the identified peptides were within 10 p.p.m.
Verification of lysine succinylation sites by HPLC–MS/MS
The in vivo peptide digest, its synthetic counterpart and their mixture for each lysine-succinylated peptide were subjected to nano-HPLC–MS/MS analysis. Extensive washing was carried out to avoid sample carryover before each sample injection. Full MS scans were acquired with resolution R = 30,000 at m/z 400, and targeted MS/MS spectra were acquired at a resolution of 7,500 at m/z 400.
Mutagenesis and activity assay
The isocitrate dehydrogenase clone (pCA24N-icdA) from the ASKA library was used as a template for PCR synthesis of mutants using the QuickChange Lightning site-directed mutagenesis kit (Agilent Technologies). Both the wild-type and the mutant proteins were expressed in E. coli BL21 (DE3) (Invitrogen) and purified. The enzymatic assays were carried out at 25 °C in 300-μl reactions containing 67 mM glycylglycine (pH 7.4), 0.44 mM DL-isocitrate, 1.0 mM β-nicotinamide adenine dinucleotide phosphate, 0.60 mM manganese chloride and 1 μg of isocitrate dehydrogenase. The production of NADPH was monitored at 340 nm with a Cary 300 UV–Vis spectrophotometer (Varian).
Supplementary Material
Acknowledgments
We would like to thank Z. Cheng and Z. Wang for their assistance during the course of this study. This work was supported by US National Institutes of Health grants to Y.Z.
Footnotes
Competing financial interests The authors declare no competing financial interests.
Additional information Supplementary information and chemical compound information is available online at http://www.nature.com/naturechemicalbiology/. Reprints and permissions information is available online at http://npg.nature.com/reprintsandpermissions/.
References
- 1.Walsh CT, Garneau-Tsodikova S, Gatto GJ., Jr. Protein post-translational modifications: the chemistry of proteome diversifications. Angew. Chem. Int. Edn. Engl. 2005;44:7342–7372. doi: 10.1002/anie.200501023. [DOI] [PubMed] [Google Scholar]
- 2.Witze ES, Old WM, Resing KA, Ahn NG. Mapping protein post-translational modifications with mass spectrometry. Nat. Methods. 2007;4:798–806. doi: 10.1038/nmeth1100. [DOI] [PubMed] [Google Scholar]
- 3.Chen Y, et al. Lysine propionylation and butyrylation are novel post-translational modifications in histones. Mol. Cell. Proteomics. 2007;6:812–819. doi: 10.1074/mcp.M700021-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang K, Chen Y, Zhang Z, Zhao Y. Identification and verification of lysine propionylation and butyrylation in yeast core histones using PTMap software. J. Proteome Res. 2009;8:900–906. doi: 10.1021/pr8005155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hake SB, Xiao A, Allis CD. Linking the epigenetic `language' of covalent histone modifications to cancer. Br. J. Cancer. 2007;96(Suppl):R31–R39. [PubMed] [Google Scholar]
- 6.Martin C, Zhang Y. The diverse functions of histone lysine methylation. Nat. Rev. Mol. Cell Biol. 2005;6:838–849. doi: 10.1038/nrm1761. [DOI] [PubMed] [Google Scholar]
- 7.Kruse JP, Gu W. Modes of p53 regulation. Cell. 2009;137:609–622. doi: 10.1016/j.cell.2009.04.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hershko A, Ciechanover A. The ubiquitin system. Annu. Rev. Biochem. 1998;67:425–479. doi: 10.1146/annurev.biochem.67.1.425. [DOI] [PubMed] [Google Scholar]
- 9.Sadygov RG, Cociorva D, Yates JR., III Large-scale database searching using tandem mass spectra: looking up the answer in the back of the book. Nat. Methods. 2004;1:195–202. doi: 10.1038/nmeth725. [DOI] [PubMed] [Google Scholar]
- 10.Bandeira N, Tsur D, Frank A, Pevzner PA. Protein identification by spectral networks analysis. Proc. Natl. Acad. Sci. USA. 2007;104:6140–6145. doi: 10.1073/pnas.0701130104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen Y, Chen W, Cobb MH, Zhao Y. PTMap—a sequence alignment software for unrestricted, accurate, and full-spectrum identification of post-translational modification sites. Proc. Natl. Acad. Sci. USA. 2009;106:761–766. doi: 10.1073/pnas.0811739106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang J, et al. Lysine acetylation is a highly abundant and evolutionarily conserved modification in Escherichia coli. Mol. Cell. Proteomics. 2009;8:215–225. doi: 10.1074/mcp.M800187-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kim SC, et al. Substrate and functional diversity of lysine acetylation revealed by a proteomics survey. Mol. Cell. 2006;23:607–618. doi: 10.1016/j.molcel.2006.06.026. [DOI] [PubMed] [Google Scholar]
- 14.Cheng Z, et al. Molecular characterization of propionyllysines in non-histone proteins. Mol. Cell. Proteomics. 2009;8:45–52. doi: 10.1074/mcp.M800224-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Allis CD, Chicoine LG, Richman R, Schulman IG. Deposition-related histone acetylation in micronuclei of conjugating Tetrahymena. Proc. Natl. Acad. Sci. USA. 1985;82:8048–8052. doi: 10.1073/pnas.82.23.8048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.MacDonald MJ, et al. Perspective: emerging evidence for signaling roles of mitochondrial anaplerotic products in insulin secretion. Am. J. Physiol. Endocrinol. Metab. 2005;288:E1–E15. doi: 10.1152/ajpendo.00218.2004. [DOI] [PubMed] [Google Scholar]
- 17.Bergmeyer HU. Methods of Enzymatic Analysis. Academic Press; New York: 1974. pp. 624–627. [Google Scholar]
- 18.O'Shea EK, Klemm JD, Kim PS, Alber T. X-ray structure of the GCN4 leucine zipper, a two-stranded, parallel coiled coil. Science. 1991;254:539–544. doi: 10.1126/science.1948029. [DOI] [PubMed] [Google Scholar]
- 19.Nelson DL, Cox MM. Lehninger Principles of Biochemistry. W.H. Freeman and Company; New York: 2005. Chapter 6. Enzymes; pp. 200–202. [Google Scholar]
- 20.Norris KL, Lee JY, Yao TP. Acetylation goes global: the emergence of acetylation biology. Sci. Signal. 2009;2:76. doi: 10.1126/scisignal.297pe76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Berry GT, et al. Neonatal hypoglycaemia in severe succinyl-CoA: 3-oxoacid CoA-transferase deficiency. J. Inherit. Metab. Dis. 2001;24:587–595. doi: 10.1023/a:1012419911789. [DOI] [PubMed] [Google Scholar]
- 22.Bonnefont JP, et al. Alpha-ketoglutarate dehydrogenase deficiency presenting as congenital lactic acidosis. J. Pediatr. 1992;121:255–258. doi: 10.1016/s0022-3476(05)81199-0. [DOI] [PubMed] [Google Scholar]
- 23.Ostergaard E. Disorders caused by deficiency of succinate-CoA ligase. J. Inherit. Metab. Dis. 2008;31:226–229. doi: 10.1007/s10545-008-0828-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.