

Abstract
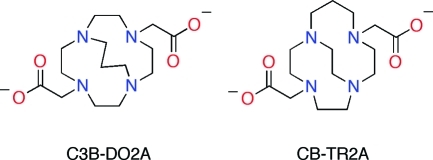
Ethylene cross-bridged tetraamine macrocycles are useful chelators in coordination, catalytic, medicinal, and radiopharmaceutical chemistry. Springborg and co-workers developed trimethylene cross-bridged analogues, although their pendant-armed derivatives received little attention. We report here the synthesis of a bis-carboxymethyl pendant-armed cyclen with a trimethylene cross-bridge (C3B-DO2A) and its isomeric ethylene-cross-bridged homocyclen ligand (CB-TR2A) as well as their copper(II) complexes. The in vitro and in vivo properties of these complexes are compared with respect to their potential application as 64Cu-radiopharmaceuticals in positron emission tomography (PET imaging). The inertness of Cu-C3B-DO2A to decomplexation is remarkable, exceeding that of Cu-CB-TE2A. Electrochemical reduction of Cu-CB-TR2A is quasi-reversible, whereas that of Cu-C3B-DO2A is irreversible. The reaction conditions for preparing 64Cu-C3B-DO2A (microwaving at high temperature) are relatively harsh compared to 64Cu-CB-TR2A (basic ethanol). The in vivo behavior of the 64Cu complexes was evaluated in normal rats. Rapid and continual clearance of 64Cu-CB-TR2A through the blood, liver, and kidneys suggests relatively good in vivo stability, albeit inferior to 64Cu-CB-TE2A. Although 64Cu-C3B-DO2A clears continually, the initial uptake is high and only about half is excreted within 22 h, suggesting poor stability and transchelation of 64Cu to proteins in the blood and/or liver. These data suggest that in vitro inertness of a chelator complex may not always be a good indicator of in vivo stability.
Short abstract
Isomeric trimethylene and ethylene cross-bridged tetraamine ligands C3B-DO2A and CB-TR2A have been prepared and their Cu(II) complexes characterized and compared. Each chelator has been successfully labeled with Copper-64 and evaluated in an animal model for potential PET imaging application.
Introduction
The family of ethylene cross-bridged tetraamine macrocyclic ligands (Figure 1a) has become valuable as metal chelators in coordination, catalytic, as well as medicinal chemistry.1,2 Their pendant-armed derivatives have been especially useful in endowing corresponding metal complexes with remarkable kinetic inertness. One lead bifunctional chelator for copper radiometals has been the bis-carboxymethyl pendant-armed cross-bridged cyclam, CB-TE2A (Figure 1b). Its 64Cu-labeled bioconjugates have significantly improved in vivo behavior and superior inertness toward radiometal loss and are of interest for positron emission tomography (PET) imaging applications.2,3 We have been developing a second generation of cross-bridged ligands aimed at improving the kinetics of complexation and decreasing nontarget organ accumulation.(4) Springborg and co-workers previously reported an interesting series of trimethylene cross-bridged tetraazamacrocycles and their coordination chemistry (Figure 1c, 1d).1,5
Figure 1.

(a) Cross-bridged tetraazamacrocycles; (b) CB-TE2A; (c−d) Trimethylene cross-bridged tetraazamacrocycles.
A limited number of pendant-armed derivatives of these ligands have been investigated, though none with ionizable functional groups.6,7 We were intrigued whether such a trimethylene cross-bridged tetraamine derivative would have distinct in vitro or in vivo behavior compared to its most closely related ethylene cross-bridged analogues. We report here the synthesis and characterization of a dicarboxymethyl pendant-armed cyclen with such a cross-bridge, C3B-DO2A (1), as well as its isomeric ethylene cross-bridged homocyclen ligand, CB-TR2A (2) (Figure 2). Their copper(II) complexes have been prepared and fully characterized. Finally, 64Cu radiolabeling and animal biodistribution studies have been carried out for comparison with each other, and with the widely used chelator CB-TE2A, with the overall goal of developing the optimal 64Cu-cross-bridged complex for conjugation to biomolecules as potential PET imaging agents.
Figure 2.

Isomeric dicarboxymethyl pendant-armed cross-bridged ligands 1 and 2.
Results and Discussion
Synthesis
The ligand C3B-DO2A (1) was synthesized by N-alkylation of the parent trimethylene cross-bridged Springborg ligand (3, C3B-cyclen)(6) with t-butyl bromoacetate (Scheme 1) to give diester 4 as a hydrobromide salt. Deprotection of this product with excess trifluoroacetic acid (TFA) provided H2-C3B-DO2A, also as a hydrobromide (H2-1·HBr). The synthetic route used for preparation of CB-TR2A (2) is shown in Scheme 2. The approach from homocyclen to cross-bridged homocyclen has been communicated previously,(8) but we include synthetic details and characterization data herein.
Scheme 1. Synthesis of H2-C3B-DO2A (H2-1).

Scheme 2. Synthesis of H2-CB-TR2A (H2-2).

Condensation of homocyclen with aqueous glyoxal in MeCN leads to a mixture of two constitutionally isomeric cis-fused tetracyclic bisaminals, 5 (major) and 6 (minor), which were separated chromatographically.(9) The major isomer (5), the desired compound, was shown to be cis-fused by virtue of dynamic NMR spectra indicative of enantiomerization (selected variable temperature 13C{1H} NMR spectral data are included in the experimental details for compound 5).9,10 Highly regio- and stereoselective dibenzylation of 5 gives rac salt 7 through benzylation on the most sterically available nitrogen lone pairs. Double reductive ring expansion with NaBH4 gives dibenzyl cross-bridged homocyclen 8, which is debenzylated to cross-bridged homocyclen 9 by hydrogenolysis. In analogy to our synthesis of CB-TE2A3 and C3B-DO2A (1) (vide supra), 9 was di-N-alkylated with t-butyl bromoacetate to give bis-ester-armed derivative 10, which was deprotected with TFA to give the desired ligand as H2-CB-TR2A (H2-2) as a TFA salt.
The respective copper(II) complexes of 1 and 2 were prepared by refluxing Cu(ClO4)2 overnight with the ligand in a 95% ethanol solution that was adjusted to pH 8. Isolated crude complexes were recrystallized from methanol/diethyl ether to give crystalline products.
Ligand and Complex Spectroscopic Studies
The 1H and 13C{1H} NMR spectral data for C3B-DO2A (1) and CB-TR2A (2) are detailed in the Experimental Section. The NMR spectra of H2-1 exhibit the expected time-averaged 2-fold symmetry in solution. The 13C{1H} NMR spectrum exhibits 6 resonances and the 1H NMR spectrum shows the expected singlet for the enantiotopic hydrogens of the acetate methylenes (N-CH2-CO2H). On the other hand, H2-2 (TFA salt) is completely asymmetric as shown by NMR spectroscopy. All of the carbon resonances are unique and every methylene hydrogen pair is diastereotopic. The two distinct N-CH2-CO2H resonances appear as AB and AX multiplets.
The solid-state IR spectra of Cu-C3B-DO2A (11) and Cu-CB-TR2A (12) showed carboxylate stretching bands at 1616 and 1609 cm−1, respectively, both consistent with coordinated pendant arms. Their d-d electronic spectral maxima in aqueous solutions are at 614 nm (85 M−1 cm−1) and 608 nm (32 M−1 cm−1) respectively, also typical of six-coordinate Cu(II) with N4O2 coordination spheres.
Electrochemical and Acid Inertness Studies
Cyclic voltammetry of Cu-C3B-DO2A (11) in 0.1 N sodium acetate revealed an irreversible reduction wave with a peak potential at −1.05 V (Ag/AgCl, scan rate 200 mV/s) with a large copper stripping peak in the return scan. Under the same conditions, Cu-CB-TR2A (12) exhibited a quasi-reversible reduction wave at −0.95 V with a peak-to-peak separation of 138 mV. Our previous results indicated that Cu(II)-complexes of ethylene cross-bridged cyclam and its derivatives typically have quasi-reversible reductions while cyclen analogues display irreversible reductions.11,12 Thus, electrochemically Cu-C3B-DO2A (11) behaves as expected for a cross-bridged cyclen complex while the isomeric homocyclen-based complex Cu-CB-TR2A (12) is more similar to cross-bridged cyclam complexes. The possible relevance of Cu(II) reduction potentials to in vivo radio-copper loss from tetraazamacrocyclic chelators and their bioconjugates has been postulated.(12) It was hypothesized that Cu(II) reduction potentials lower than about −0.6 V (Ag/AgCl) may be necessary to avoid reduction by common bioreductants. If indeed so, neither of these complexes should be susceptible to in vivo Cu(II) reduction.
Acid inertness data of Cu(II)-tetraamine complexes provide a convenient measure of their resistance toward demetalation in aqueous solution. We have found acid inertness half-lives obtained under pseudo first-order conditions to be useful first predictors for in vivo viability of 64Cu-labeled chelator complexes.(2) In particular, Cu-CB-TE2A was shown to have a half-life of more than 6 d even in 5 M HCl at 90 °C.(11) Studies in 5 M HCl solutions at 30 °C revealed that Cu-C3B-DO2A (11) is indefinitely inert while Cu-CB-TR2A (12) dissociated with a half−life of 10.8(4) h. Remarkably, the Cu-C3B-DO2A (11) complex only demetalated in 12 M HCl at 90 °C with a half-life of 1.1(1) d. To our knowledge, this represents the most acid-inert copper amine complex studied. Since Springborg reported the half-life of the parent trimethylene cross-bridged copper complex in 5 M HCl at 25 °C to be almost 6 days while we have found the isomeric Cu-CB-homocyclen complex to be significantly less inert (half-life only 4.8 h in 1 M HCl, 30 °C),5,13 the trimethylene bridge does appear to impart greater acid inertness. Here, attachment of the two enveloping carboxymethyl pendant arms further enhanced the acid inertness of Cu-C3B-DO2A (11) even beyond that of Cu-CB-TE2A.
X-ray Structural Data
A summary of crystal data for the two X-ray diffraction studies can be found in Table 1. While the quality of the Cu-CB-TR2A (12) crystal used was relatively poor, nonetheless three distinct six-coordinated distorted octahedral complexes with N4O2 coordination spheres were found. Each of these provides a pendant arm oxygen to bridge two of three sodium cations which together form a central core for this assembly. Two of these, complexes 12A and 12B are well-defined (Figure 3). Complex 12A has a [333]/[2233] ligand conformation while complex 12B adopts instead a [333]/[2323] conformation, the distinction mainly being the sign of the N−C−C−N torsion angle of the short bridge. Complex 12C is conformationally disordered but possesses the same basic Cu coordination sphere as 12A and 12B.
Table 1. Crystal Data for Cu-CB-TR2A (12) and Cu-C3B-DO2A (11)a.
| Cu-CB-TR2A | Cu-C3B-DO2A | |
|---|---|---|
| chemical formula | C45H78Cl3Cu3N12Na3O27 | C21H30CuF12N4O6 |
| molecular weight | 1585.13 | 726.03 |
| space group | P2(1)/c | P2(1)/n |
| color | blue | blue |
| a (Å) | 20.2068(19) | 12.2973(7) |
| b (Å) | 24.613(2) | 13.3899(8) |
| c (Å) | 13.0914(12) | 17.5444(10) |
| α (deg) | 90.00 | 90.00 |
| β (deg) | 105.8090(10) | 106.581(3) |
| γ (deg) | 90.00 | 90.00 |
| V (Å3) | 6264.6(10) | 2768.7(3) |
| Z | 4 | 4 |
| Dcacld (g/cm3) | 1.681 | 1.742 |
| T (K) | 173(2) | 100(2) |
| μ (mm−1) | 1.252 (Mo Kα) | 2.293 (Cu Kα) |
| unique data, Rint | 13599 | 5164 |
| parameters/restraints | 804/40 | 399/0 |
| R1 (I > 2σ(I)) | 0.0784 | 0.0497 |
| R1 (all data) | 0.1396 | 0.0558 |
| wR2 (all data) | 0.2026 | 0.1299 |
For full details, see the Supporting Information.
Figure 3.
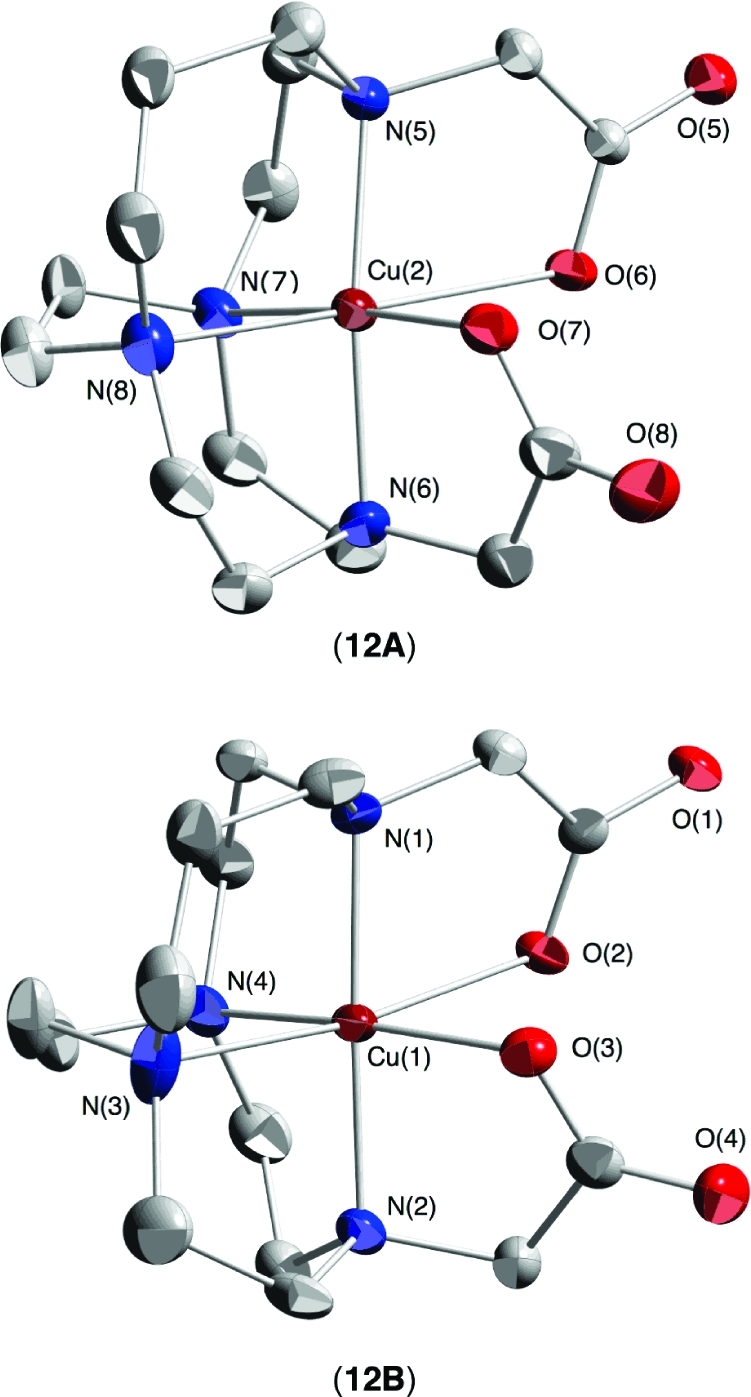
X-ray structure of 12: views of complexes 12A and 12B (50% thermal ellipsoids; hydrogens omitted for clarity).
Noteworthy are the distinct internal “axial” N−Cu−N angles of 191.3(2)° and 183.2(2)° respectively, with each cation sunken into the ligand cavity but with the [333]/[2233] ligand conformation engulfing the cation more. Corresponding “equatorial” N−Cu−N angles are similar at 81.3(2)° and 80.2(2)° respectively. Jahn−Teller elongations in both structures are along select N−Cu−O axes; N(8)−Cu(2)−O(6) in 12A and N(3)−Cu(1)−O(2) in 12B. Copper−nitrogen bond distances in both structures range from 1.96 to 2.24 Å while copper−oxygen distances are between 1.96 and 2.51 Å (Table 2).
Table 2. Selected Bond Data for Cu-CB-TR2A (12) and Cu-C3B-DO2A (11)a.
| Cu-CB-TR2A |
|||||
|---|---|---|---|---|---|
| A | B | Cu-C3B-DO2A | |||
| Cu−N Bond Lengths (Å) | |||||
| Cu(2)−N(5) | 1.975(5) | Cu(1)−N(1) | 1.968(5) | Cu(1)−N(1) | 2.201(3) |
| Cu(2)−N(6) | 1.988(5) | Cu(1)−N(2) | 1.956(6) | Cu(1)−N(2) | 2.003(3) |
| Cu(2)−N(7) | 2.051(5) | Cu(1)−N(3) | 2.244(6) | Cu(1)−N(3) | 2.013(3) |
| Cu(2)−N(8) | 2.228(6) | Cu(1)−N(4) | 2.041(5) | Cu(1)−N(4) | 2.002(3) |
| Cu−O Bond Lengths (Å) | |||||
| Cu(2)−O(6) | 2.513(6) | Cu(1)−O(2) | 2.501(5) | Cu(1)−O(1) | 2.003(2) |
| Cu(2)−O(7) | 1.960(4) | Cu(1)−O(3) | 1.981(4) | Cu(1)−O(3) | 2.449(2) |
| N−Cu−N Angles Inside Ligand Cleft (deg) | |||||
| N(5)−Cu(2)−N(6) | 191.3(2) | N(1)−Cu(1)−N(2) | 183.2(2) | N(2)−Cu(1)−N(3) | 173.6(1) |
| N(7)−Cu(2)−N(8) | 81.3(2) | N(3)−Cu(1)−N(4) | 80.2(2) | N(1)−Cu(1)−N(4) | 97.5(1) |
Estimated standard deviations in parentheses. For full details, see the Supporting Information.
The X-ray structure of Cu-C3B-DO2A (11) is shown in Figure 4. Its good metal−ligand fit is indicated by observed internal “axial” and “equatorial” N−Cu−N angles of 173.6(1)° and 97.5(1)° respectively. Similar numbers were found in a Cu(II) complex of the parent trimethylene cross-bridged cyclen (170.5° and 99.0°).(5) The first angle indicates that the cation is protruding slightly from the ligand cavity while the latter value is substantially larger than those found in reported ethylene cross-bridged ligand Cu(II) complexes (typically around 80°) as a result of its trimethylene cross-bridging.14,15 Its Jahn−Teller elongation is along the N(1)−Cu(1)−O(3) axis which deviates substantially from linearity at 159°. The copper−nitrogen bond distances range from 2.0 to 2.2 Å, similar to those found in Cu-CB-TR2A (12), while the Cu−O distances are 2.00 and 2.45 Å. In light of these observations, no significant structural insight can be gleaned to account for the very disparate acid inertness between these two complexes.
Figure 4.
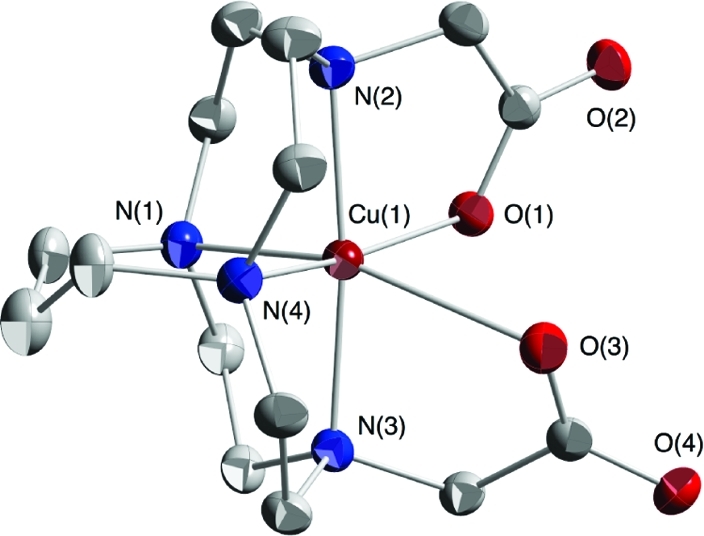
X-ray structure of complex 11 (50% thermal ellipsoids; hydrogens omitted for clarity).
Radiochemistry
Consistent with the extremely high acid inertness of its Cu-complex, radiolabeling of C3B-DO2A was found to be very challenging and ultimately required rather harsh conditions for successful labeling. Radiolabeling was attempted under a variety of aqueous conditions, varying concentration (0.5−1 μg/μL), buffer (0.1−1.25 M NH4OAc, 0.1−0.5 M NH4-citrate, and 0.1−0.5 Na2HPO4), pH (4.5−8), and temperature (25−95 °C). Reactions were monitored over time to assess the rate of complexation. All reactions had very disappointing yields that rarely exceeded 50% within 2 h, even at high temperature. At longer time points (3−18 h) the yields were generally better; however, they rarely were >70%. Furthermore, instability of the 64Cu-C3B-DO2A complex was observed in the form of secondary peak formation. Although in our hands, radiolysis has not been commonly observed for small molecules, reactions were carried out in the presence of two different radiolytic scavengers with no significant benefit. Carrier-added reactions did not improve radiolabeling yields.
After many failed attempts at radiolabeling 1 with high yields under aqueous conditions, the standard conditions for radiolabeling CB-TE2A were attempted,(16) by dissolving the compound (0.5−3 mg) in ethanol (50−100 mL), preincubating it in the presence of an excess of Cs2CO3 and then heating the reaction at 95 °C with 100−130 μCi of 64Cu. Unfortunately, the yields were still relatively low (30−70% after 1 h) and did not exceed 85% even after several hours. Replacing Cs2CO3 with Li2CO3 resulted in extremely low yields (3−6%) at all time points (1−18 h). Again, carrier-added reactions did not generate any improvement in radiolabeling yield.
In an attempt to speed up the reaction kinetics, a microwave reactor was employed. In all reactions 1.0 μg C3B-DO2A was incubated with ∼100 μCi 64Cu in a total volume of 300 μL. Many different reactions were performed where solvent, pH, temperature, and reaction time were varied, and it was found that the shortest reaction that consistently provided a greater than 97% radiochemical purity consisted of microwave heating a solution of 1 in Milli-Q water at pH = 3 to 100 °C for 2 h.
In contrast, CB-TR2A was successfully labeled with 64Cu by the method employed for CB-TE2A(16) (radiochemical purity ≥98%) at room temperature in basic ethanolic solution. A single peak corresponding to 64Cu-CB-TR2A was confirmed by radio-TLC.
Biodistribution Studies
The biodistribution of 64Cu-C3B-DO2A and 64Cu-CB-TR2A were determined in normal, juvenile Lewis rats to examine their in vivo properties. The blood, liver, and kidney clearance of these agents are plotted in Figure 5 along with previously published data of 64Cu-CB-DO2A and 64Cu-CB-TE2A for comparison.(18) Although 64Cu-CB-TR2A demonstrated rapid clearance from the blood suggesting that it is stable for the limited time it remains in the bloodstream, significantly more activity was observed in the blood at 24 h for 64Cu-CB-TR2A compared to 64Cu-CB-TE2A (64Cu-CB-TE2A vs 64Cu-CB-TR2A (%ID/g; p value): 0.003 ± 0.0006 vs 0.01 ± 0.002; p = 0.003). Levels of 64Cu-CB-TR2A remaining in the blood at 24 h were comparable to 64Cu-CB-DO2A (0.01 ± 0.002 vs 0.011 ± 0.004).
Figure 5.
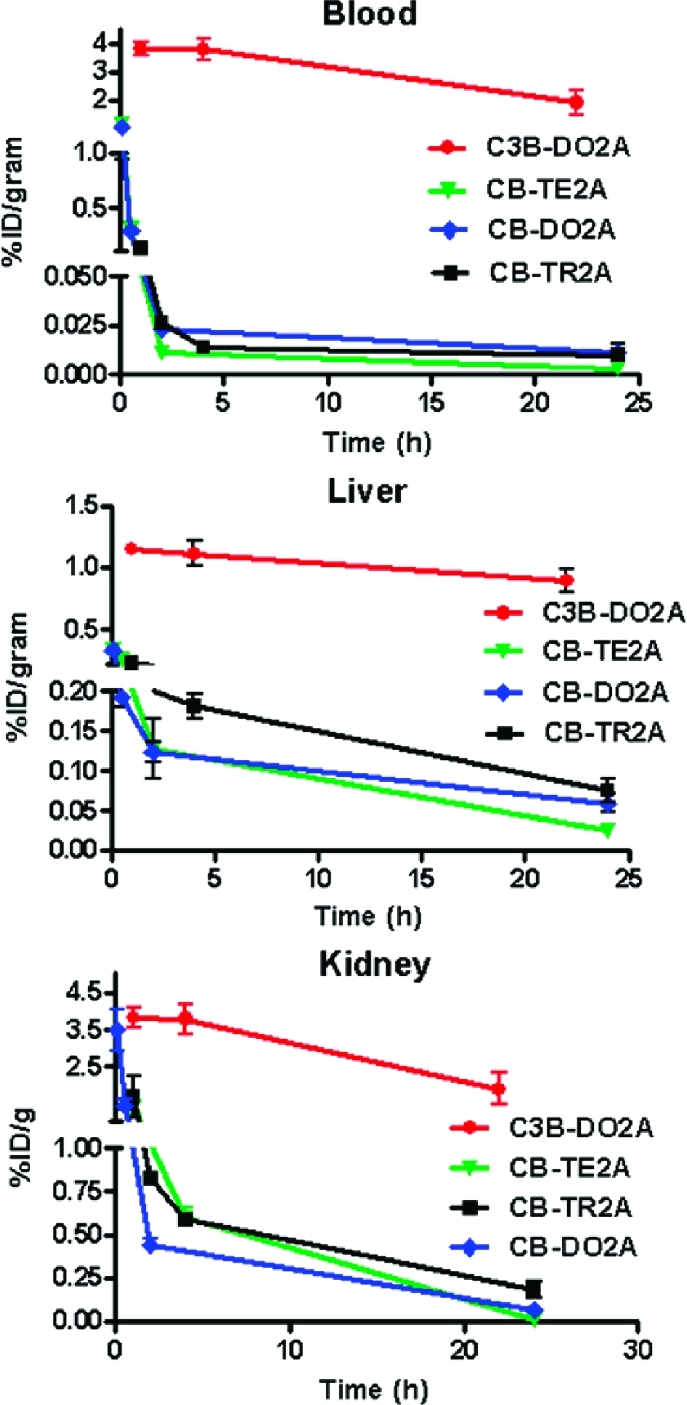
Biodistribution of 64Cu-11 and 64Cu-12 compared to published data for 64Cu-CB-DO2A, 64Cu-CB-TE2A.
Additionally, more tissue associated radioactivity was observed in the kidneys of rats receiving 64Cu-CB-TR2A at 24 h p.i. compared to the other two CB chelators (64Cu-CB-TR2A vs 64Cu-CB-TE2A vs 64Cu-CB-DO2A: 0.18 ± 0.05, 0.012 ± 0.003 and 0.064 ± 0.007%ID/g, respectively; p < 0.0001). Similarly, there was significantly greater accumulation in the liver of mice receiving 64Cu-CB-TR2A compared to 64Cu-CB-DO2A or 64Cu-CB-TE2A (0.075 ± 0.014, 0.024 ± 0.002, and 0.056 ± 0.012%ID/g, respectively; p = 0.0003). While the increased 64Cu in the liver from 64Cu-CB-TR2A may be attributed to hepatobilary clearance, transchelation of copper to liver proteins is also a likely possibility.(17) Boswell et al. reported the transchelation of radio-copper to liver proteins such as superoxide dismutase and metallotheinein after injection of 64Cu labeled CB-chelators.(18) At this time it is unclear why CB-TR2A was less stable in vivo than CB-DO2A despite its comparatively low reduction potential and its relative kinetic acid inertness. 64Cu-C3B-DO2A exhibited very slow clearance from the liver compared to CB-TR2A, CB-DO2A, and CB-TE2A (percent reduction at 22 or 24 h PI: 22.2, 66.4, 69.5 and 90.2% clearance, respectively). Kidney clearance showed a similar pattern out to 22 or 24 h PI (64Cu-C3B-DO2A, 50.4%; 64Cu-CB-TR2A, 89.2%; 64Cu-CB-DO2A, 95.6%; and 64Cu-CB-TE2A, 99.1%). The poor bioclearance of 64Cu-C3B-DO2A compared to the other radiolabeled cross-bridged chelators is surprising. Previously, in vitro assays of Cu(II)-complex acid inertness and resistance to Cu(II)/Cu(I) reduction have provided useful indicators of their in vivo stability and bioclearance behavior.1,12 The very slow hepatic clearance of 64Cu-C3B-DO2A observed here suggests significant radiometal loss in the liver from what was predicted to be a very inert complex. Close examination of the X-ray structure of Cu-C3B-DO2A revealed that the trimethylene cross-bridge led to a larger equatorial N−Cu−N angle of 98° instead of the 80−88° typical for its ethylene-bridged analogues but no other noteworthy differences in Cu-chelator bonding parameters. The possibility that an incompletely formed, perhaps out-of-cavity, 64Cu complex being the actual radiolabeled product can account for the observed radiocopper loss in vivo. However, the very harsh radiolabeling conditions (2 h, microwave 100 °C, pH 3) mitigate against such a kinetic product. Other contributing factors affecting in vivo clearance including lipophilicity, susceptibility to metabolic degradation, or formation of easily reduced ternary complexes as a consequence of this trimethylene cross-bridge may be implicated but will require additional SAR studies for validation.(19)
Summary
The trimethylene cross-bridged chelator C3B-DO2A (1) and its ethylene cross-bridged isomer CB-TR2A (2) have been synthesized and characterized as were their copper(II) complexes. Both complexes have distorted octahedral N4O2 coordination spheres and should be resistant to biological reduction. While Cu-CB-TR2A (12) is reasonably resistant to acid decomplexation, Cu-C3B-DO2A (11) is remarkably inert. At the same time, successful radio-copper labeling of CB3B-DO2A (11) required relatively harsh conditions. Contrary to the highly inert in vitro behavior of Cu-C3B-DO2A (11), biodistribution studies of 64Cu-C3B-DO2A revealed very poor in vivo clearance data, significantly inferior to those of 64Cu-CB-TE2A and even 64Cu-CB-DO2A.
Experimental Section
General Procedures
1H and 13C{1H}-NMR spectra were acquired on a Varian Mercury 400 MHz spectrometer operating at 399.75 and 100.51 MHz respectively, on a Varian INOVA 500 MHz spectrometer operating at 500 and 125.67 MHz respectively, or on a Bruker spectrometer operating at 360 and 90.56 MHz respectively or on a Varian Gemini operating at 300 and 75 MHz respectively. Variable temperature dynamic 13C{1H} NMR spectra of 5 were acquired on a JEOL FX-90Q spectrometer operating at 22.5 MHz. IR spectra were recorded using KBr pellets on a Nicolet MX-1 FT-IR Spectrophotometer. ESI-MS was performed on a Thermofinnigan LCQ Mass Spectrometer coupled to a Picoview electrospray source or on a Waters Micromass ZQ 4000. FAB-MS data were acquired on the JEOL JMS-AX505HA Mass Spectrometer at the University of Notre Dame. Electronic spectra were measured using a Cary 219 spectrophotometer. Elemental analyses were performed at Atlantic Microlab Inc. Norcross, GA.
All solvents were reagent grade and from commercial sources and were used without further purification. All metal complexation reactions were performed in standard Schlenk glassware under a nitrogen atmosphere. Recrystallizations were conducted in closed containers without precautions to exclude either air or moisture. Bulk solvent removal was by rotary evaporation under reduced pressure, and trace solvent removal from solids was by vacuum pump evacuation.
Caution! Perchlorate salts of metal complexes containing organic ligands as solids or in organic solvents are potentially explosive. Although no problems were encountered by us, cautious handling of only small amounts of these compounds should be the rule.
Ligand Synthesis
C3B-cyclen (3) was synthesized according to a procedure previously published by Springborg et al. in 1995.(20) Briefly, cyclen (1,4,7,10-tetraazacyclododecane) was treated with 2 equiv of p-toluenesulfonyl chloride (TsCl) in pyridine: the only product obtained was cyclen-1,7-bis-p-toluenesulfonamide. The 3-carbon cross bridge was then introduced by alkylation of the 2 secondary nitrogens with 1 equiv of 1,3-propanediol di-p-toluenesulfonate. The N-protecting tosyl groups were removed from the crude product in refluxing HBr/CH3COOH (3 days), and the crude product was recrystallized to give pure 3·3HBr.
4,10-Bis-(carbo-tert-butoxymethyl)-1,4,7,10-tetraazabicyclo[5.5.3]pentadecane Hydrobromide (4·HBr)
The compound C3B-cyclen·3HBr (3·3HBr, 0.45 g, 0.99 mmol) was dissolved in MeCN (25 mL). Na2CO3 (0.45 g, 4.2 mmol) and tert-butyl bromoacetate (0.34 mL, 2.3 mmol) were then added, and the reaction was heated to 60 °C for 3 days. After cooling, solids were removed by filtration, and the filtrate was evaporated under reduced pressure. Residual solvent was removed under high vacuum, and the product was triturated with diethyl ether to give 4·HBr (0.42 g) in 81% yield as the residual solid. This material was taken on without further purification. 1H NMR (DMSO-d6, 300 MHz): δ 1.32 (s, 18H, C(CH3)3), 1.68 (m, 2H, NCH2CH2CH2N), 2.65−3.05 (m, 20H, NCH2CH2N and NCH2CH2CH2N), 3.38 (s, 4H, NCH2C(O)), 12.80 (br s, inside N-H). 13C{1H} NMR (DMSO-d6, 75 MHz): δ 21.4, 28.5, 50.2, 53.3, 54.1, 55.8, 81.0, 171.4. MS (ESI+): m/z 441.1 (MH+), 385.1 (MH+ − 1tBu), 329.1 (MH+ − 2tBu).
4,10-Bis-(carboxymethyl)-1,4,7,10-tetrazazabicyclo[5.5.3]pentadecane Hydrobromide (H2-1·HBr)
The compound 4·HBr (0.47 g, 0.90 mmol) was dissolved in a 1:1 mixture of dichloromethane (DCM, 15 mL) and trifluoroacetic acid (TFA, 15 mL), and the reaction mixture was stirred at room temperature under argon for 3 days. DCM and TFA were then evaporated off, the remaining oil was dissolved in ethanol, and the desired product slowly precipitated with diethyl ether. After removal of trace solvents under high vacuum, H2-1·HBr (0.34 g) was obtained as a slightly off-white solid in a 92% yield. 1H NMR (D2O, 300 MHz): δ 1.82 (m, 2H, NCH2CH2CH2N), 2.95−3.25 (m, 20H, NCH2CH2N and NCH2CH2CH2N), 3.61 (s, 4H, NCH2C(O)). 13C{1H} NMR (D2O, 75 MHz): δ 18.2, 51.7, 53.8, 53.9, 60.2, 173.1. MS (ESI+): m/z 329.1 (MH+). Elem. analysis: calcd for C15H28N4O4·HBr: C 44.02, H 7.14, N 13.69; found: C 43.94, H 6.92, N 13.30.
cis-Decahydro-5H-2a,4a,7a,9a-tetraazacyclopenta[cd]phenalene (5)
Aqueous glyoxal (3.7 mL 40% (w/w); 25.1 mmol) was added dropwise over 1 h under nitrogen to a stirred solution of 1,4,7,10-tetraazacyclotridecane (homocyclen) (3.42 g, 18.36 mmol) in MeCN (100 mL). The reaction mixture was then heated to 60 °C for 2.5 h. Solvent was removed under reduced pressure to give a yellow oil, which was dissolved in CHCl3 and dried (Na2SO4). After concentration of the solution, the crude product was purified by flash chromatography (basic alumina, 10% EtOH/Et2O) to give the product as a clear oil (Rf 0.29) which crystallized upon storage at −10 °C. (1.91 g, 50%): mp 28−30 °C; 1H NMR (360 MHz, CDCl3) δ 1.20−1.32 (dm, 1H, J = 8.1 Hz) 1.90−3.70 (br m, 19H) 2.88 (d, J = 2.8 Hz, CH) 3.37 (d, J = 2.8 Hz, CH) (Note: the 1H NMR spectrum exhibited severe dynamic exchange broadening.); 13C{1H} NMR (22.5 MHz, CDCl3, −39 °C) δ 19.79 (CH2CH2CH2), (Note: three pairs of peaks (labeled A, B, and C) each broadened upon warming and coalesced to a singlet by 55 °C; see below) 45.51 and 53.18 (A), 49.35 and 51.82 (B), 51.82 and 54.76 (C), 49.93, 75.39 (CH), 77.74 (CH); 13C{1H} NMR (22.5 MHz, CDCl3, 55 °C) δ 20.35, (Note: the following peaks (labeled A, B, and C) correspond to three pairs of coalesced resonances.) 49.82 (br) (A), 50.97 (B), 53.83 (C), 50.41, 76.13 (CH), 78.17 (CH); MS, m/z 208 (M+); IR (CCl4) 2930, 2883, 2842, 2795, 2790, 1320, 1278, 1161, 1130, 1112, 1095 cm−1. Anal. Calcd for C11H20N4: C, 63.43; H, 9.68; N, 26.90. Found: C, 63.17; H, 9.98; N, 27.02. A minor component (0.20 g, 5%), was also isolated during the flash chromatography (Rf 0.07): 1H NMR (360 MHz, CDCl3) δ 1.40 (dtt, 1H, J = 15.0, 5.6, 2.0 Hz) 2.02 (dtt, 1H, J = 15.0, 10.7, 2.9 Hz) 2.45−2.85 (m, 8H) 2.92−3.08 (m, 3H) 3.13 (ddd, 2H, J = 13.7, 5.7, 2.6 Hz) 3.27−3.40 (m, 2H) 3.59 (s, 2H, CHCH); 13C NMR (90.56 MHz, CDCl3) δ 25.11, 49.33, 51.31, 51.63, 52.31, 80.94. These spectra are consistent with the constitutionally isomeric cis-fused tetracyclic bisaminal 6.
(9bα,9cα)-Decahydro-2a,7a-bis(phenylmethyl)-5H-4a,9a-diaza-2a,7a-diazoniacyclopenta[cd]phenalene Dibromide (7)
To a stirred solution of purified homocyclen-glyoxal adduct 5 (1.49 g, 7.17 mmol) in MeCN (35 mL) at room temperature was added benzyl bromide (12.40 g, 72.50 mmol), all in one portion. The reaction mixture was stirrred at room temperature for 3 weeks. Precipitate was collected by suction filtration, washed with MeCN (2 × 30 mL), then CH2Cl2 (3 × 15 mL). Residual solvent was removed under vacuum. This gave product as white solid (3.60 g, 90%): mp 222−225 °C (dec); 1H NMR (360 MHz, D2O, MeCN secondary ref set at 2.06 ppm) δ 1.86−1.97 (dm, 1H, J = 15.0 Hz, H6-eq) 2.15−2.33 (m, 1H, H6-ax) 2.78 (td, 1H, J = 12.2, 2.9 Hz) 3.09−3.92 (m, 13H) 4.21−4.39 (m, 2H), 4.78 and 5.09 (AB, 2H, J = 13.3 Hz, CH2Ph) 4.87 and 5.18 (AB, 2H, J = 13.0 Hz, CH2Ph), 4.95 and 5.12 (AB, 2H, J = 1.1 Hz, CHCH), 7.30−7.90 (m, 10H, Ph); 13C{1H} NMR (90.56 MHz, D2O, MeCN secondary ref set at 1.70 ppm) δ 19.40 (CH2CH2CH2), 43.37, 46.34, 47.68, 48.94, 51.25, 52.84, 61.05, 61.16, 61.37, 63.34, 76.81 (CH), 78.82 (CH), 125.56 (1C), 126.62 (1C), 130.28 (2C), 130.52 (2C), 132.18 (2C), 133.18 (2C), 134.10 (2C); IR (KBr) 3062, 3050, 3026, 3018, 2996, 2957, 2915, 2886, 2864, 2844, 2821, 2807, 1480, 1456, 1437, 1283, 1212, 1146, 1110, 1055, 769, 711, 706 cm−1. Anal. Calcd for C25H34N4Br2·0.5H2O: C, 53.68; H, 6.31; N, 10.02. Found: C, 53.94; H, 6.10; N, 9.97.
4,11-Bis(phenylmethyl)-1,4,8,11-tetraazabicyclo[6.5.2]pentadecane (8)
NaBH4 (13.10 g, 0.340 mol) was added in small portions over 20 min to a stirred solution of dibenzyl tetracyclic bisaminal dibromide salt 7 (3.05 g, 5.55 mmol) in 95% EtOH (75 mL). The reaction mixture was stirred at room temperature for 10 days. Excess NaBH4 was then decomposed by slow addition of 150 mL 3 M HCl. Evaporation of solvent under reduced pressure gave a white solid which was dissolved in H2O (100 mL), adjusted to pH 14 with solid KOH (with cooling), and extracted with benzene (6 × 50 mL). The combined extracts were dried (Na2SO4), and solvent was removed under reduced pressure to yield 1.44 g (quant) of product as a viscous oil; 1H NMR (360 MHz, C6D6) δ 1.37−1.60 (m, 2H, CH2CH2CH2), 2.26−2.49 (m, 5H), 2.53−2.73 (m, 7H), 2.78 (ddd, 1H, J = 13.7, 4.3, 2.6 Hz), 2.82−3.04 (m, 5H), 3.09−3.21 (m, 1H), 3.26 and 3.64 (AB, 2H, J = 13.4 Hz, CH2Ph), 3.60 and 3.62 (AB, 2H, J = 14.4 Hz, CH2Ph), 3.88 (ddd, 1H, J = 12.7, 8.2, 4.9 Hz), 7.07−7.41 (m, 10H, CH2Ph); 13C{1H} NMR (90.56 MHz, C6D6) δ 28.59 (CH2CH2CH2), 52.39, 53.78, 55.43, 55.84, 56.36, 56.43, 57.04, 57.93, 59.04, 59.48, 60.82, 60.99, 126.93, 126.98, 128.41, 128.42, 129.08, 129.39, 140.92, 141.03; MS, m/z 392 (M+); IR (neat) 3077, 3053, 3020, 2919, 2863, 2800, 2784, 2745, 2721, 2702, 1448, 1364, 1110, 1046, 1025, 729, 696 cm−1; HRMS exact mass calcd for C25H37N4 393.3018, found 393.3012. The 13C and 1H NMR spectra showed the material to be >98% purity.
1,4,8,11-Tetraazabicyclo[6.5.2]pentadecane (9)
Hydrogenolysis of 8 was carried out in a glass apparatus designed for the exclusion of O2 and for measurement of H2 uptake with maintenance of constant pressure. 10% Pd/C (0.20 g) and glacial HOAc (60 mL) were added to a 125 mL hydrogenation flask which was connected to the apparatus. The system was evacuated (water aspirator) and flushed with nitrogen four times. The system was then evacuated, filled with hydrogen, and catalyst was equilibrated under H2 (767 mmHg) for 1.5 h. To this was added a solution of 8 (1.11 g, 2.83 mmol) in glacial HOAc (5 mL). The mixture was stirred for 19 h under H2 (767 mmHg). (The theoretical H2 uptake for this reaction was 138 mL. The observed uptake was 144 mL.) The apparatus was then evacuated and flushed with nitrogen four times, the reaction flask was removed from the apparatus, the contents were filtered through Celite, and the catalyst and Celite were washed with glacial HOAc (2 × 5 mL). The combined filtrate and washings were concentrated under reduced pressure to give a light yellow oil which was dissolved in H2O (50 mL), adjusted to pH 14 with KOH, and extracted with benzene (5 × 50 mL). The combined extracts were dried (Na2SO4), and solvent was removed under reduced pressure to give an oil. After kugelrohr distillation (90−120 °C air bath temperature, 0.03 mmHg), the product oil solidified upon standing to give 0.494 g (82% crude yield) of solid product. This material contained minor impurities and was therefore further purified. The solid was dissolved in absolute EtOH (5 mL) containing conc HCl (1 mL). A hydrochloride salt crystallized, was filtered, washed with cold (0−5 °C) absolute EtOH (2 × 1 mL), and air-dried. This solid was dissolved in H2O (10 mL), adjusted to pH 14 with solid KOH (with cooling), and extracted with benzene (6 × 15 mL). The combined extracts were dried (Na2SO4), and solvent was removed under reduced pressure to give an oil. This oil was kugelrohr distilled (100 °C air bath temperature, 0.03 mmHg) and subsequently solidified upon standing to give 0.283 g (47%) of pure product as a white solid: mp 28−30 °C; 1H NMR (360 MHz, C6D6) δ 1.26−1.51 (m, 2H CH2CH2CH2), 2.10−2.90 (m, 21H), 3.30 (ddd, 1H, J = 12.1, 7.5, 2.4 Hz); 13C{1H} NMR (90.56 MHz, C6D6) δ 27.14 (CH2CH2CH2), 47.68, 48.56, 48.89, 49.02, 52.85, 53.27, 53.59, 54.20, 55.97, 59.41; MS, m/z 212 (M+); IR (KBr) 3474, 2948, 2929, 2907, 2894, 2845, 2699, 1577, 1464, 1399, 1356, 1342, 812, 612, 523 cm−1; HRMS exact mass calcd for C11H25N4 213.2079, found 213.2078.
4,11-Bis-(carbo-tert-butoxymethyl)-1,4,8,11-tetraazabicyclo[6.5.2]pentadecane (10)
To a stirred solution of 9 (35.0 mg, 0.165 mmol) in MeCN (8 mL), sodium carbonate (50.0 mg, 0.472 mmol) was added in one portion, followed by tert-butyl bromoacetate (0.050 mL, 0.34 mmol) via syringe. The mixture was stirred for 3 days at room temperature. Solvent was removed under reduced pressure to yield crude product, which was subjected to flash chromatography (SiO2, CH2Cl2:MeOH = 10:1) to yield product as light yellow oil (40.0 mg, 0.0909 mmol, 55%): 1H NMR (500 MHz, C6D6) δ 1.30−1.46 (m, 2H), 1.40 (s, 18H), 2.25 (dt, 1H, J = 13.9, 2.9 Hz), 2.50−2.62 (m, 5H), 2.72−2.88 (m, 10H), 2.88−2.95 (m, 1H), 2.96−3.03 (m, 1H), 3.09 and 3.17 (AB, 2H, J = 16.4 Hz), 3.20 (br s, 2H), 3.24−3.31 (m, 1H), 3.59 (ddd, 1H, J = 12.5, 9.8, 4.9 Hz); 13C{1H} NMR (125.7 MHz, C6D6) δ 28.36, 28.60, 28.61, 51.24, 53.34, 55.08, 55.82, 56.86, 57.39, 57.52 (2C), 57.66, 57.74, 59.84, 60.22, 80.17, 80.22, 171.52, 172.08; IR(neat) 2976, 2924, 2852, 2800, 1734, 1719, 1457, 1367, 1152, 1124; HRFABMS, m/z (M + H)+ exact mass for C23H45N4O4: 441.3441; Found: 441.3456 (error +1.5 mmu/+0.3 ppm)
4,11-Bis-(carboxymethyl)-1,4,8,11-tetraazabicyclo[6.5.2]pentadecane (H2-2·3.7TFA)
A solution of 10 (96.0 mg, 0.218 mmol) in trifluoroacetic acid (3.4 mL, 0.046 mol) and CH2Cl2 (3.4 mL) was stirred under N2 for 2 days. The reaction mixture was then concentrated under aspirator pressure and residual solvent was removed under vacuum to give 0.1635 g (0.2179 mmol, quant) of H2-CB-TR2A (H2-2) as a brown oil with 3.7 equiv of TFA by mass. 1H NMR (MeCN-d3, 500 MHz) δ 1.69−1.76 (dm, 1H, J = 16.8 Hz), 2.14−2.28 (m, 1H), 2.82−3.52 (m, 22−23H), 3.53 and 3.89 (AX, 2H, J = 17.6 Hz, NCH2CO2), 3.86 and 4.05 (AB, 2H, J = 17.4 Hz, NCH2CO2), 11.64 (br s, 6H, includes all COOH peaks); 13C{1H} NMR (MeCN-d4, 125.68 MHz) δ 20.94, 48.58, 50.04, 50.40, 52.25, 53.36 (br), 53.85, 55.16 (br), 55.69, 56.37, 57.59, 58.21, 58.40, 117.08 (q, 1JCF= 283.2 Hz, F3CCOOH), 160.67 (q, 2JCF= 37.0 Hz, F3CCOOH), 170.88, 172.99. (1H NMR integration from 2.82 to 3.52 ppm was slightly high, likely because of H2O). This material was complexed with Cu(ClO4)2·6H2O without further purification (vide infra).
Complex Synthesis
Cu-C3B-DO2A (11)
An amount of 100 mg (0.203 mmol) of the C3B-DO2A (1) as a trifluoroacetate salt was combined with 75 mg (0.203 mmol) of Cu(ClO4)2·6H2O in 8 mL of 95% ethanol. The pH was adjusted to 8.5 with 0.1 N NaOH. This suspension was refluxed under nitrogen for 24 h to give a dark blue solution. After thorough removal of volatiles under reduced pressure, a dark-blue sticky solid was obtained. This was extracted with 4 mL of methanol and centrifuged. The supernatant was then placed in vials inside a glass container for diethylether diffusion. A total crop of 75 mg of dendritic dark-blue crystals formed (64% yield). Elem. analysis: calcd for C15CuH26N4O4·1.33NaClO4·1.33H2O: C 31.24, H 5.01, N 9.71, Cl 8.18%. Found: C 31.05, H 5.21, N 9.45, Cl 8.06%. Electronic spectrum (aq.) λmax 614 nm (ε = 84.6 M−1 cm−1). FT-IR (KBr) νCOO 1687, 1616 cm−1. X-ray crystals were grown from a hexafluoro-i-propanol solution upon diethylether diffusion.
Cu-CB-TR2A (12)
To a solution of 2·3.7 TFA (0.1635 g, 0.2179 mmol) in 95% EtOH (15 mL) was added Cu(ClO4)2·6H2O (136 mg, 0.366 mmol), and the pH of the reaction mixture was adjusted to 8 with 1 M NaOH. The reaction was then refluxed under N2 for approximately 23 h. The reaction mixture was centrifuged, and the supernatant collected and concentrated under aspirator pressure. Residual solvent was removed under vacuum to give 264 mg of a blue solid. This solid was dissolved in 95% EtOH (15 mL), and the blue solution was placed in a diethyl ether diffusion chamber. A powdery blue solid was collected to give 55.3 mg (44% yield) of purified product. Elem. analysis: calcd for Cu(C15H26N4O4)· 2H2O·1.2NaClO4: C, 31.45; H, 5.28; N, 9.78; Cl, 7.43. Found C, 31.49; H, 5.14, N, 9.68, Cl, 7.10; IR (KBr) 3536, 3490, 3430, 3394, 3364, 1610, 1485, 1437, 1397, 1320, 1098, 631 cm−1. UV−vis (H2O) λmax 608 (ε = 32.1 M−1 cm−1); Cyclic voltammetry conducted in 0.1 sodium acetate: indicated a quasi-reversible reduction with Eρ = −0.950 V (Ag/AgCl). A 0.13 M solution of Cu-CB-TR2A·2(H2O)·1.2(NaClO4) in 95% EtOH was placed in a diethylether diffusion chamber. Plate-like blue crystals were collected for X-ray structural analysis, which yielded an empirical formula of Cu-CB-TR2A·H2O·NaClO4.
X-ray Crystallography
Cu-CB-TR2A (11)
A blue plate crystal was mounted in a Nylon loop, and X-ray diffraction data collected on a Bruker D8 diffractometer equipped with an APEX CCD detector at 173 K. The monoclinic space group P21/c was uniquely assigned from systematic absences. The structure was solved by direct methods. Except as noted, non-hydrogen atoms were refined with anisotropic thermal parameters, and hydrogen atoms were included as idealized contributions. The structure was found to be extensively disordered with regard to both the macrocycle and the counterions. Satisfactory disorder models could be created for both, but extensive restraint was necessary because of limited resolution in the diffraction data. The perchlorate ions were refined as rigid tetrahedra, and several carbon atoms in the heterocycle were refined with isotropic thermal parameters. Additionally, the thermal parameters for C37 and its disorder partner, C37a were refined with the EADP command.
Cu-C3B-DO2A (12)
A blue rod 0.15 × 0.10 × 0.10 mm in size was mounted on a Cryoloop with Paratone oil. Data were collected in a nitrogen gas stream at 100(2) K using φ and ω scans. Crystal-to-detector distance was 60 mm and exposure time was 10 s per frame using a scan width of 0.5°. Data collection was 99.9% complete to 67.00° in q. A total of 26635 reflections were collected covering the indices, −14 ≤ h ≤ 14, −16 ≤ k ≤ 16, −21 ≤ l ≤ 16. A total of 5166 reflections were found to be symmetry independent, with an Rint of 0.0299. Indexing and unit cell refinement indicated a primitive, monoclinic lattice. The space group was found to be P2(1)/n (No. 14). The data were integrated using the Bruker SAINT software program and scaled using the SADABS software program. Solution by direct methods (SIR-97) produced a complete heavy-atom phasing model consistent with the proposed structure. All non-hydrogen atoms were refined anisotropically by full-matrix least-squares (SHELXL-97). All hydrogen atoms were placed using a riding model. Their positions were constrained relative to their parent atom using the appropriate HFIX command in SHELXL-97.
Radiochemistry
The highest radiochemical purity with greatest consistency of yield was obtained using a microwave assisted reaction. A solution of 64CuCl2 (750 μCi in 50 μL Milli-Q water, pH 3) was added to 4.61 μg (14.0 nmol) C3B-DO2A dissolved in 250 μL of Milli-Q water (pH 3). The sealed vial containing the reaction mixture was placed in the microwave reactor, stirred for 2 min, and then heated at 100 °C for 2 h. Complex formation was confirmed by radio-TLC (Rf ∼ 0.8, C18 silica plates, eluent: methanol/10% ammonium acetate 7:3, Rf (64Cu-acetate = 0). Radiochemical purity was determined by HPLC and found to be ≥98% before use in animal studies (Complex retention time 10.55 min; Agilent C8 column; isocratic, 0.1% TFA in water; 0.5 mL/min).
Biodistribution Studies
Animal experiments were carried out in compliance with the Guidelines for the Care and Use of Research Animals established by Washington University’s Animal Studies Committee. Tissue distribution studies were performed in male Lewis rats (34−42 day old) after intravenous injection of the radiolabeled compound, 64Cu-C3B-DO2A (45 μCi; 0.9 nmol ligand) and 64Cu-CB-TR2A (100 μCi) via the tail vein (100−150 μL). Tissue biodistribution data were obtained at 1, 4, and 24 h post injection (PI). Animals were sacrificed at the appropriate time points, organs of interest were removed and weighed, and the radioactivity was measured in a gamma counter. The percent injected dose per gram (%ID/g) and percent injected dose per organ (%ID/organ) were calculated by comparison to a weighed and counted standard. Rats were allowed food and water ad libitum. Animals in the 24 h group for 64Cu-C3B-DO2A and 64Cu-NOTA were maintained in metabolism cages; urine and feces were collected.
All biodistribution data are presented as the mean ± standard deviation. Group comparisons were made using standard ANOVA methods. Post hoc testing of individual group differences was accomplished with the Bonferroni test. Groups with p < 0.05 were considered significantly different. GraphPad Prism software (version 5.02; San Diego, CA) was used for all statistical analyses.
Acknowledgments
We acknowledge financial support from NIH NCI-CA093395.
Supporting Information Available
Listings of 1H and 13C{1H} NMR spectra of synthetic intermediates and ligands. This material is available free of charge via the Internet at http://pubs.acs.org. X-ray crystallographic files in CIF format. These X-ray files have also been deposited with the Cambridge Crystallographic Data Centre (CCDC reference numbers 806127 and 806128) and are available from the Director, Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, U.K.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Springborg J. Dalton Trans. 2003, 9, 1653–1665. [Google Scholar]
- Anderson C., J.; Wadas T. J.; Wong E. H.; Weisman G. R. Q. J. Nucl. Med. Mol. Imaging 2008, 52, 185–192. [PMC free article] [PubMed] [Google Scholar]
- Sprague J. E.; Peng Y.; Sun X.; Weisman G. R.; Wong E. H.; Achilefu S.; Anderson C. J. Clin. Cancer Res. 2004, 10 (24), 8674–8682. [DOI] [PubMed] [Google Scholar]
- Stigers D. J.; Ferdani R.; Weisman G. R.; Wong E. H.; Anderson C. J.; Golen J. A.; Moore C.; Rheingold A. L. Dalton Trans. 2010, 39 (7), 1699–1701. [DOI] [PubMed] [Google Scholar]
- Springborg J.; Sotofte I. Acta Chem. Scand. 1997, 51 (3, Suppl.), 357–366. [Google Scholar]
- Springborg J.; Kofod P.; Olsen C. E.; Toftlund H.; Soetofte I. Acta Chem. Scand. 1995, 49 (8), 547–54. [Google Scholar]
- Springborg J.; Nielsen B.; Olsen C. E.; Sotofte I. Acta Chem. Scand. 1999, 53 (11), 985–991. [Google Scholar]
- Weisman G. R.; Wong E. H.; Hill D. C.; Rogers M. E.; Reed D. P.; Calabrese J. C. Chem. Commun. (Cambridge) 1996, 8, 947–948. [Google Scholar]
- Weisman G. R.; Johnson V.; Fiala R. E. Tetrahedron Lett. 1980, 21 (38), 3635–8. [Google Scholar]
- Kolinski R. A.; Riddell F. G. Tetrahedron Lett. 1981, 22 (23), 2217–20. [Google Scholar]
- Woodin K. S.; Heroux K. J.; Boswell C. A.; Wong E. H.; Weisman G. R.; Niu W.; Tomellini S. A.; Anderson C. J.; Zakharov L. N.; Rheingold A. L. Eur. J. Inorg. Chem. 2005, 23, 4829–4833. [Google Scholar]
- Heroux K. J.; Woodin K. S.; Tranchemontagne D. J.; Widger P. C. B.; Southwick E.; Wong E. H.; Weisman G. R.; Tomellini S. A.; Wadas T. J.; Anderson C. J.; Kassel S.; Golen J. A.; Rheingold A. L. Dalton Trans. 2007, 21, 2150–2162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odendaal A. Y. Ph.D. Thesis, University of New Hampshire, Durham, NH, 2009. [Google Scholar]
- Wong E. H.; Weisman G. R.; Hill D. C.; Reed D. P.; Rogers M. E.; Condon J. S.; Fagan M. A.; Calabrese J. C.; Lam K.-C.; Guzei I. A.; Rheingold A. L. J. Am. Chem. Soc. 2000, 122 (43), 10561–10572. [Google Scholar]
- Niu W.; Wong E. H.; Weisman G. R.; Zakharov L. N.; Incarvito C. D.; Rheingold A. L. Polyhedron 2004, 23 (6), 1019–1025. [Google Scholar]
- Wadas T. J.; Anderson C. J. Nat. Protoc. 2006, 1, 3062–3068. [DOI] [PubMed] [Google Scholar]
- Bass L. A.; Wang M.; Welch M. J.; Anderson C. J. Bioconjug. Chem. 2000, 11 (4), 527–32. [DOI] [PubMed] [Google Scholar]
- Boswell C. A.; Sun X.; Niu W.; Weisman G. R.; Wong E. H.; Rheingold A. L.; Anderson C. J. J. Med. Chem. 2004, 47 (6), 1465–1474. [DOI] [PubMed] [Google Scholar]
- Wadas T. J.; Wong E. H.; Weisman G. R.; Anderson C. J. Chem. Rev. 2010, 110 (5), 2858–2902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Springborg J.; Kofod P.; Olsen C. E.; Sotofte I. Acta Chem. Scand. 1995, 49, 547–554. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.