

Abstract
Advances in cardiovascular imaging increasingly afford unique insights into heritable myocardial disease. As clinical presentation of genetic cardiomyopathies may range from nonspecific symptoms to sudden cardiac death, accurate diagnosis has implications for individual patients as well as related family members. The initial consideration of genetic cardiomyopathy may occur in the imaging laboratory, where one must recognize the patient with arrhythmogenic right ventricular cardiomyopathy (ARVC) among the many with ventricular arrhythmia referred to define myocardial substrate. Accurate diagnosis of the patient presenting with dyspnea and palpitations whose first-degree relatives have lamin A/C cardiomyopathy may warrant genetic testing1, 2 plus imaging of diastolic function and myocardial fibrosis3. As advances in cardiac imaging afford detection of subclinical structural and functional changes, the imaging specialist must be attuned to signatures of specific genetic disorders. With increased availability of both advanced imaging as well as genotyping techniques, this review seeks to provide cardiovascular imaging specialists and clinicians with the contemporary information needed for more precise diagnosis and treatment of heritable myocardial disease. A companion paper in this series covers imaging phenotype and genotype considerations in hypertrophic cardiomyopathy (HCM). This review details clinical features, imaging phenotype and current genetic understanding for two of the most common non-HCM conditions that prompt myocardial imaging - dilated cardiomyopathy (DCM) and arrhythmogenic right ventricular cardiomyopathy (ARVC). While all modalities are considered herein, considerable focus is given to CMR with its unique capabilities for myocardial tissue characterization.
Keywords: imaging, cardiomyopathy, genetics
I. Dilated Cardiomyopathy
DCM has a prevalence of at least 1 in 25004 and an incidence of 7 per 100,000/year5. The condition was classically defined as ‘idiopathic’ when a single member in a family was affected without a known cause and as ‘familial’ when the DCM phenotype was present in two or more related family members.6, 7 However, substantial work over the past two decades has confirmed that genetic factors are the underlying cause of both idiopathic and familial forms, and that a careful examination of relatives of an index case often reveals other affected family members and a familial pattern of disease. These systematic studies of idiopathic and familial cases have shown that dilated cardiomyopathies may be confined to ventricular enlargement and systolic dysfunction, or occur in the setting of extracardiac features such as skeletal myopathy and elevated serum creatine kinase levels (muscular dystrophy-associated cardiomyopathies are not included in this review). Consideration of a primary genetic disorder presumes that other secondary causes such as metabolic disorders, acute inflammatory conditions, valvular heart disease, toxins and ischemic heart disease have been excluded. Notably, presumed secondary DCM may occur in the setting of genetic predisposition8.
The incidence of DCM is increasing in part due to advances in diagnostics and increased awareness among physicians. In the early stages of disease, minimal symptoms may be present and diagnosis delayed, a situation that often becomes apparent when other ostensibly ‘healthy’ family members of a patient are evaluated and additional cases ascertained. Many cases of DCM have an apparent genetic origin with 30% to 50% of cases suspicious for a primary genetic etiology.9–11 These estimates are complicated by the fact that DCM is classified as a ‘mixed’ cardiomyopathy in deference to the broad list of genetic and exogenous causes in major classification guidelines.12 Although tempting to apply these guidelines as a mere roadmap to finding the ‘singular’ cause of a patient’s DCM, it is likely that both genetic and non-genetic factors interact to cause many instances of disease. In spite of the probable heterogeneity of etiology in DCM, the high risk of disease to biological relatives provides a compelling reason to assess each case of cardiomyopathy for the possibility of a primary genetic cause.
DCM: Imaging Phenotype
The phenotype of DCM is defined principally by cardiac enlargement and impaired systolic function6, 7, 12. Echocardiography readily detects both. Similar features can be recognized by contrast x-ray ventriculography or nuclear imaging. For instance, DCM may be diagnosed in the patient whose symptoms are initially ascribed to ischemic heart disease and undergoes stress nuclear scinitigraphy that shows a dilated, hypocontractile LV with no ischemia. Variability in cutoff values for abnormal chamber size across modalities, age, gender and indices of body size should be taken into account when assessing for cardiac enlargement. Recognizing abnormal myocardial relaxation via mitral inflow and tissue Doppler velocities is particularly important, since some genetic conditions classified as DCM such as lamin A/C cardiomyopathy predominantly affect diastolic function in the initial stages of disease. While many other conditions such as hypertensive heart disease may also manifest as diastolic dysfunction, these echo-Doppler findings warrant consideration of potential genetic etiologies when recognized in the context of a family history of cardiomyopathy or clinical markers of high risk (e.g. malignant ventricular arrhythmia). While more precise etiologic determination may be limited, echo-Doppler provides valuable information on the degree of pulmonary hypertension and LV filling pressures with prognostic implications13.
Left ventricular noncompaction (LVNC) may present as a distinct genetic cardiomyopathy14, but also may represent a phenotypic feature in a spectrum of other heritable cardiomyopathies15.
Clues to a specific genetic cause may come from techniques like cardiac magnetic resonance (CMR). An appropriate protocol to evaluate the patient with DCM of unknown etiology should include three important techniques for myocardial characterization: T2* quantification16, T2-weighted imaging or T2 mapping17, and late gadolinium enhancement (LGE)18. Briefly, T2* is an MR relaxation parameter whose value is influenced by tissue iron aggregates. The introduction of T2*-based screening of patients with thalassemia, a genetic disease associated with myocardial siderosis due to transfusion-related iron overload, has dramatically reduced mortality in this population. Notably, patients with sickle cell disease may develop hepatic siderosis but our laboratory and others19, 20 have not found significant myocardial overload in these patients despite lifelong exogenous iron overload, suggesting that additional yet-undefined genetic factors may influence myocardial siderosis. A normal myocardial T2* exceeds 20 ms at 1.5 Tesla; a diffusely shortened myocardial T2* in a patient presenting with cardiomyopathy without secondary causes such as chronic transfusions warrants consideration of hereditary hemochromatosis (HH). T2* screening of large HH cohorts has not been reported to provide a contemporary estimate of cardiac involvement, though histopathologic detection at autopsy examination after sudden cardiac death suggest it may be underrecognized21.
T2 increases with tissue water content, and may identify regions of myocardial inflammation or edema22. The magnetic resonance parameter T2 was recently reported to be increased in patients with dystrophin-associated cardiomyopathy23.
LGE is the essential CMR technique for myocardial characterization in DCM, providing both diagnostic and prognostic value24. LGE imaging leverages contrast-induced T1-shortening to distinguish between necrotic/fibrotic and normal myocardium. While findings such as mid-myocardial fibrosis may be nonspecific, they reliably distinguish DCM from infiltrative and ischemic cardiomyopathies. Notably, relying on angiography alone to exclude coronary artery disease as the cause of DCM could potentially misclassify up to 13% of cases18. Similarly, both LGE findings of nonischemic cardiomyopathy may coexist with infarct scar, that should prompt the interpreting team to consider nonischemic cardiomyopathy superimposed on ischemic heart disease. Genotypic evidence supporting DCM as an end-stage phenotype of HCM25 underscores the importance of considering a genetic cardiomyopathy when appropriate phenotypic findings are detected by cardiac imaging (Fig. 1). LGE-positivity in a patient with ventricular arrhythmia as well as a concerning family history for heritable disease may warrant genetic testing; however, recognition that patients with genetic cardiomyopathies may be LGE-negative at presentation underscores variability in phenotype and opportunities for imaging advances to better define signatures of genetic myocardial disease.
Figure 1.
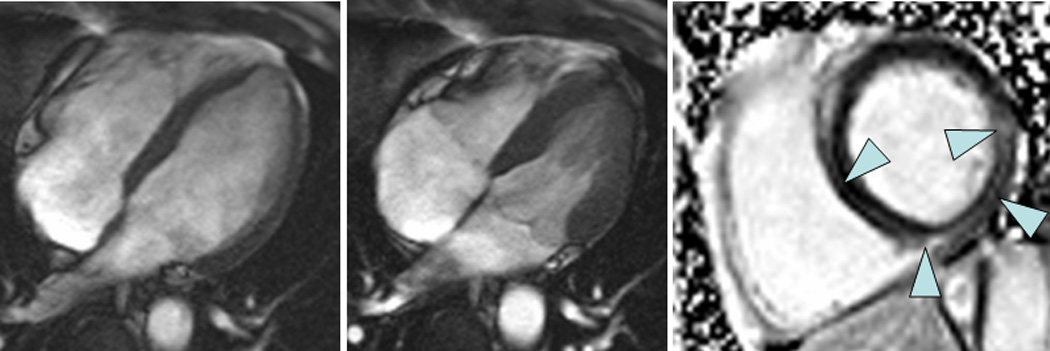
A 52 year-old male with ventricular arrhythmia by ambulatory ECG monitoring whose family history included a grandparent with sudden cardiac death at a young age underwent echocardiography that showed normal LV systolic function and mitral valve prolapse. CMR was performed and confirmed normal systolic function (left panel: end-diastole, middle panel: end-systole) but revealed midmyocardial fibrosis by LGE imaging (right panel). The patient underwent genetic testing via a 23-gene panel for dilated cardiomyopathy (GeneDx, Gaithersberg, MD) that revealed a mutation in myosin binding protein C.
DCM: Current Status of Genetic Testing
DCM is characterized by high genetic heterogeneity – over twenty-five different genes have been linked to the DCM phenotype (Table 1)1, 26–46. Early work identified genes predominantly responsible for coding cytoskeletal proteins, and a ‘cytoskeletal hypothesis’ implicating dysfunction of structural networks was proposed (Fig. 2).47, 48 More recent data has revealed that perturbations in proteins beyond the cytoskeleton can lead to DCM, and the idea of a ‘final common pathway’ now extends to sarcomeric, ion channel, nuclear lamina, and desmosomal proteins. Accurate prevalence estimates for pathogenesis of each gene have been difficult to obtain, in part because most studies have been in cohorts of modest size (<200 families) with each individual gene accounting for often <2% of cases in a given study. An exception to this has been the lamin A/C (LMNA) gene that currently represents the most commonly recognizable cause of DCM, particularly if accompanied by conduction system disease49, accounting for up to 10% of cases.50, 51
Table 1.
Genetic Causes of Dilated Cardiomyopathy*
| Author (ref) | Phenotype | Frequency (%) |
Chromosomal location |
LOCUS | OMIM | Gene Symbol |
Gene | Location |
|---|---|---|---|---|---|---|---|---|
| Durand et al.45 | Autosomal dominant | 56 | 1q32 | CMD1D | 191045 | TNNT2 | Cardiac troponin T | Sarcomere |
| Mogensen et al.44 | FDC | 3p21.1 | 191040 | TNNC1 | Cardiac troponin C | Sarcomere | ||
| Gerull et al.46 | 2q31 | CMD1G | 188840 | TTN | Titin | Sarcomere | ||
| Li et al.43 | 2q35 | CMD1I | 125660 | DES | Desmin | Cytoskeleton | ||
| 6q12-q16 | CMD1K | 172405 | ? | ? | ||||
| Schmitt et al.42 | (with mitral prolapse) | 6q22.1 | CMD1P | 609909 | PLN | Phospholamban | Calcium | |
| 9q13 | CMD1B | 600884 | ? | ? | ||||
| 9131 | CMD1X | 611615 | ? | ? | ||||
| Olson et al.41 | 10q21-q23 | CMD1W | 193065 | VCL | Metavinculin | Cytoskeleton | ||
| Duboscq-Bidot et al.40 | 10q22.1 | CMD | 608517 | MYPN | Myopalladin | Sarcomere | ||
| Vatta et al.39 | 10q23.3 | CMD1C | 601493 | ZASP/LDB3 | ZASP/ LIM domain binding 3 | Sarcomere | ||
| Daehmlow et al.38 | 11p11 | CMD1M | 600958 | MYBPC3 | Myosin-binding protein C | Sarcomere | ||
| Knoll et al.37 | 11p15.1 | CMD1A | 600824 | CSRP3 | Cysteine-glycine-rich protein | Sarcomere | ||
| Bienengraeber et al.36 | 12p12.1 | CMD1O | 601439 | ABCC9 | 3 | Ion channel | ||
| Kamisago et al.35 | 14q11.2-13 | CMD1A | 160760 | MYH7 | cardiac KATP channel | Sarcomere | ||
| Li et al.34 | 14q24.3 | CMD1U | 104311 | PSEN1 | Cardiac β-myosin heavy chain | Nuclear mem | ||
| Olson et al.33 | 15q14 | 102540 | ACTC | Presenilin 1 | Sarcomere | |||
| Olson et al.32 | 15q22.1 | CMD1N | 191010 | TPM1 | Cardiac actin | Sarcomere | ||
| Valle et al.31 | 17q12 | 604488 | TCAP | α-tropomyosin | Sarcomere | |||
| Tinin-cap (telethonin) | ||||||||
| Murphy et al.30 | Autosomal recessive | 16 | 19q13.42 | 191044 | TNNI3 | Cardiac troponin I | Sarcomere | |
| FDC | unknown | 212110 | ||||||
| Gold et al.29 | X-linked DC | 10 | Xp21 | XLCM | 300377 | DMD | Dystrophin | Cytoskeleton |
| Fatkin et al.1 | Autosomal dominant | 7.7 | 1q11-q23 | LGMD1 | 150330 | LMNA | Lamin A/C | Nucleoskeleton |
| Tsubata et al.28 | FDC with skeletal muscle disease |
5q33-34 | B | 601411 | SGCD | δ-sarcoglycan | Cytoskeleton | |
| Barresi et al.27 | 4q11 | LGMD2 | 600900 | SGCB | β-sarcoglycan | Cytoskeleton | ||
| 6q23 | F | 602067 | ||||||
| LGMD2 | ||||||||
| E | ||||||||
| CMD1F | ||||||||
| Fatkin et al.1 | Autosomal dominant | 2.6 | 1q1-q1 | CMD1A | 150330 | LMNA | Lamin A/C | Nucleoskeleton |
| FDC with conduction defects |
2q14-q22 | CMD1H | 604288 | |||||
| McNair et al.26 | 3p22.2 | CMD1E | 600163 | SCN5A | Na channel, voltage-gated, type V, αpolypeptide |
Ion channel |
organized by phenotype and sequentially by chromosome location
Figure 2.
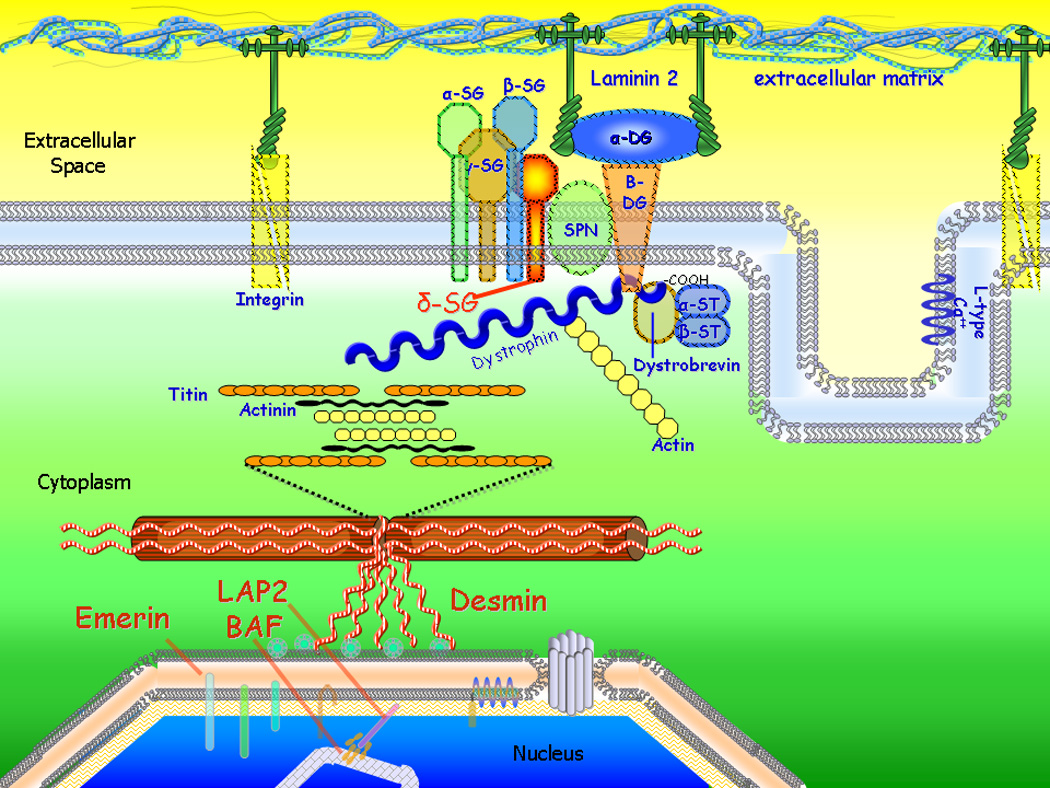
Proteins of the cytoskeletal network. Mutations in many cytoskeletal genes cause dilated cardiomyopathy (Table 1) supporting the ‘cytoskeletal hypothesis’. BAF=barrier to autointegration factor; DG=dystroglycan; LAP2=lamina-associated polypeptide 2; L-type Ca=L-type calcium channel; SG=sarcoglycan (alpha, beta, gamma, and delta isoforms shown); SPN=sarcospan; ST=syntrophin.
The broad genetic heterogeneity of DCM genes initially delayed the development of clinical genetic testing. Many cytoskeletal proteins are large; the large size of these genes made genetic testing costly, and utilization of such testing outside of large research centers was limited. More recently, several laboratories have developed cardiomyopathy panels including over twenty genes offered in a single panel. Testing is available in the United States and Europe and is probably less available in other regions of the world, although even in the United States testing may not always be covered by commercial insurance carriers.
DCM Benefits/Limitations of Testing
For many patients, the greatest benefit of genetic testing comes in evaluating family members at risk of developing the DCM phenotype. For the index patient with evident DCM, genetic testing is not needed to confirm the diagnosis, though it may help determine if the disease is primarily due to a genetic defect vs. another etiology. At-risk family members who have borderline changes on echocardiography may be considered for early treatment to prevent or delay progressive cardiac dysfunction, although studies supporting this approach in true genetic cases are lacking. It should be noted that mutations in LMNA may be more malignant than mutations in other DCM genes as LMNA mutation carriers appear to be at elevated risk for sudden death and more rapid or severe course of heart failure.
Testing should generally be undertaken after formal genetic counseling and a discussion of the benefits and limitations of testing in the context of the individual patient as well as the overall family structure. Since current genetic testing panels fail to identify a pathogenic mutation in up to 50% of cases, patients should be counseled on this important limitation of current testing. Private mutations are common making predictions of genotype to phenotype unreliable, with the possible exception of LMNA mutations which are expected to be more severe. Variants of unknown significance can be encountered and may be difficult to interpret even after additional individuals in the family undergo testing. It has been recommended that strong consideration be given for referral to centers with experience in cardiomyopathy genetics if genetic testing is to be undertaken.52
DCM: Family Screening
In deference to the large role of genetic factors in DCM, recommendations for collecting a detailed family history and offering genetic counseling have been proposed.52 The most common inheritance pattern is autosomal dominant showing multi-generational involvement, equal number of affected males and females, and male-to-male transmission. Other inheritance patterns, while less common, have been described and indeed the specific pattern of inheritance within a family can be used to guide genetic counseling and testing. In addition to evaluating a complete and accurate family history, direct clinical testing of first-degree relatives by objective measures such as electrocardiography and echocardiography are important to identify latent cases. A review of records of deceased individuals can also be critical in uncovering past cases in a family that were not recognized as manifesting the phenotype.
II. Arrhythmogenic Right Ventricular Cardiomyopathy (ARVC)
ARVC is an inherited cardiomyopathy characterized by fibrofatty replacement of right ventricular (RV) myocardium leading to RV failure and arrhythmias53, 54. Prevalence estimates in the general population range from 1:1000 to 1:500055, 56. It often affects young men who have an athletic lifestyle. Presenting symptoms range from palpitations to exertional syncope and sudden cardiac death54. Arrhythmias in ARVC most frequently originate from the right ventricle and have a left bundle branch block (LBBB) morphology. The disease often affects the RV outflow tract, the base of the RV and the RV apex, collectively termed the ‘triangle of dysplasia’. Early-stage patterns of RV involvement are poorly understood, making it difficult to diagnose early disease by imaging. Major and minor diagnostic criteria have been proposed that encompass structural, electrophysiologic and histopathologic variables57, 58. Identification of abnormalities in RV structure and function constitutes an important part of the diagnosis of ARVC and accounts for a major or minor criterion based on the severity of the abnormality. The task force criteria, initially proposed in 1994, were recently revised to include quantitative data for RV functional evaluation, underscoring the importance of thorough assessment of the RV in suspected ARVC58.
ARVC is a familial disease in at least 50% of cases, usually transmitted as an autosomal dominant trait with variable penetrance59, 60. Reduced penetrance and variable expressivity, together with the availability of small families for clinical evaluation, might explain the underestimation of ARVC as heritable disease. Family history alone cannot replace the prospective evaluation of family members in establishing inheritance of ARVC. In the absence of definite knowledge of gene-carrier status, the major clinical challenge consists in differentiating mild or atypical manifestations in family members from the so-called ‘phenocopies’, i.e. non-hereditary diseases that can mimic ARVC such as idiopathic RV outflow tract tachycardia, myocarditis and sarcoidosis.
ARVC: Imaging Phenotype
Echocardiography is widely available and is often the first imaging modality to assess cardiac structure and function in known or suspected ARVC (Fig. 3, Data Supplement 1). Three-dimensional echocardiography has been shown to accurately quantify RV size and systolic function compared to CMR61. Inherent limitations imposed by acoustic window with ultrasound-based cardiac imaging in some patients may preclude visualization of the segmental RV abnormalities that constitute phenotypic hallmarks of ARVC. X-ray right ventriculography is invasive and has fallen out of favor due to the availability of noninvasive imaging techniques. The modified ARVC/D Task Force Criteria62 provide detailed cutoffs regarding abnormal RV size and wall motion; briefly, RVEF ≤40% by CMR or regional akinesia, dyskinesia or aneurysm by 2D echo, CMR or RV angiography constitute major criteria for ARVC.
Figure 3.
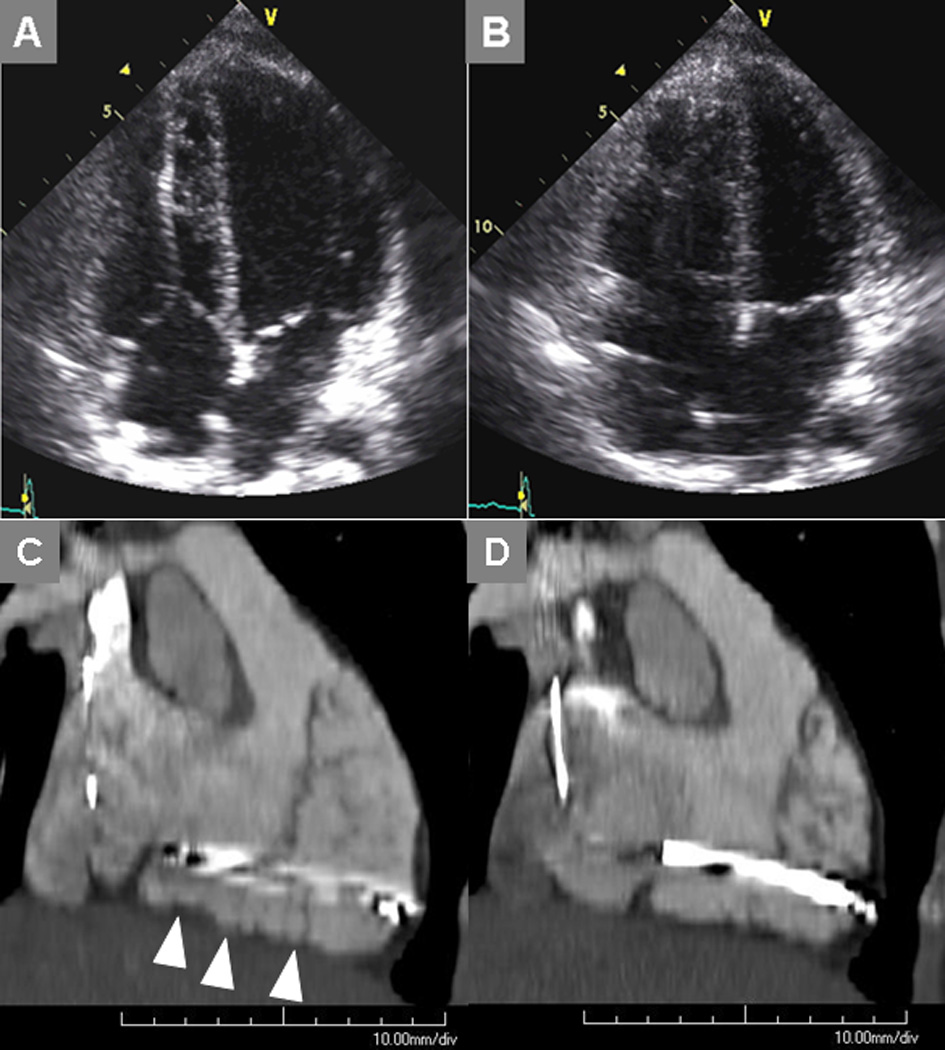
Transthoracic echocardiogram at end-diastole (A) and end-systole (B) indicates RV dysfunction in a 32 year-old female who underwent defibrillator placement shortly after surviving sudden cardiac death. She was then referred for cardiac computed tomography to assess for possible ARVC. Maximum intensity projection image in an oblique sagittal plane demonstrates the scalloped RV myocardium (arrowheads) and dyskinetic segments when comparing images reconstructed at end-diastole (C) vs. end-systole.
Cardiovascular computed tomography (CT) may sometimes be used to diagnose ARVC, particularly in the setting of contraindication to magnetic resonance (Fig. 3). While CT-based recognition of intramyocardial fat holds appeal63, cine reconstructions (Data Supplement 2) are essential to assess regional RV wall motion given the challenges (even with CT’s high spatial resolution) of defining fibrofatty replacement in a thin, diseased RV.
CMR is uniquely suited to evaluate ARVC: it not only provides excellent functional information for the RV, but also can provide tissue characterization to depict fibrosis and fatty infiltration in the RV64, 65. CMR provides accurate quantitative assessment of RV size and global and regional RV systolic function, important parts of the revised task force criteria. Limitations inherent to CMR remain presence of MR-incompatible devices or foreign bodies, severe claustrophobia and advanced renal disease that precludes use of the powerful late gadolinium enhancement technique for myocardial characterization.
CMR findings in ARVC include fat infiltration of the myocardium (Fig. 4), global and regional RV dysfunction, and myocardial fibrosis. Dark blood imaging may demonstrate replacement of ventricular myocardium with hyperintense fat signal, which infrequently appears as a signal void on a corresponding fat-suppressed image. The incidence of fat infiltration in ARVC ranges from 60%–100% in the literature, likely related to differences in patient selection66. Fat infiltration often affects the basal right ventricle, RV outflow tract, and the RV anterior wall close to the tricuspid inlet. Relying on intramyocardial fat visualization to make the diagnosis is problematic due to often abundant epicardial fat, underscoring the need to carefully distinguish between abnormal fat infiltrating the RV myocardium and fat in the atrio-ventricular groove. Fat suppression helps distinguish epicardial fat from the unaffected RV wall, though failure to distinguish normal epicardial fat from pathologic RV infiltration may result in misdiagnosis/over diagnosis of ARVC67. ARVC should be kept distinct from both fatty infiltration of the right ventricle and adipositas cordis. It is well known that a certain amount of intramyocardial fat is present in the right ventricular antero-lateral and apical regions even in the normal heart, and that intramyocardial and epicardial fat increase with increasing body weight and with aging68, 69, though prevalence is unknown70–72. However, both the fibro-fatty and fatty variants of ARVC show, besides fatty replacement of the right ventricular myocardium, degenerative changes of the myocytes and interstitial fibrosis, with or without extensive replacement-type fibrosis. As such, the suggestion of RV intramyocardial fat by dark blood imaging should prompt closer attention to segmental RV function and late gadolinium enhancement in the corresponding location to reduce ‘false-positive’ imaging-based diagnosis.
Figure 4.
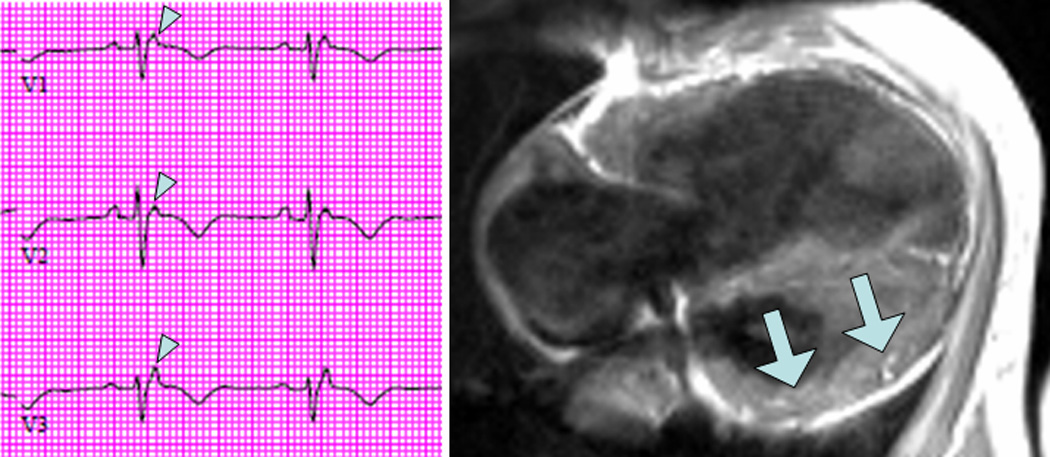
Electrocardiogram from a patient who presented with fatigue demonstrates classic epsilon waves of ARVC (left, arrowheads). Dark blood cardiac magnetic resonance image shows extensive fatty infiltration that also involves the LV myocardium (right, arrows).
Among CMR criteria, global and regional function are most useful in the diagnosis and are very reproducible73. RV regional dysfunction often precedes global dysfunction and affects the triangle of dysplasia. Regional functional changes include focal hypokinesis, dyskinesis and aneurysms (Data Supplement 2). By the time of diagnosis, the majority of probands with ARVC have global RV dysfunction. Reproducible CMR-derived measures of RV volumes and function, with published nomograms58, are invaluable in the longitudinal evaluation of patients with borderline abnormalities and can be used to assign major or minor criteria for ARVC.
Evaluation of plakophillin-2 (PKP2) mutation-positive asymptomatic first-degree relatives revealed minor crinkling contractions in the RV base that resembled an accordion74. This sign was seen with a high prevalence in mutation-positive relatives and none of the first-degree relatives who did not carry the pathologic mutation. Reproducibility of this finding has not been systematically assessed, and the diagnostic and prognostic significance remains unknown.
LGE imaging can non-invasively demonstrate RV fibrosis (Fig. 5), and is an essential component of the CMR examination of patients with suspected ARVC64. The extent of RV myocardial fibrosis correlates with the degree of RV dysfunction, and predicts inducibility of ventricular arrhythmias64. LGE also assists in distinguishing phenocopies of ARVC like sarcoidosis which occasionally results in isolated cardiac involvement75. Multiple, patchy regions of LV hyperenhancement favors a diagnosis of sarcoidosis76 and may also be seen in myocarditis. Notably, fat infiltration is distinctly absent in both conditions.
Figure 5.
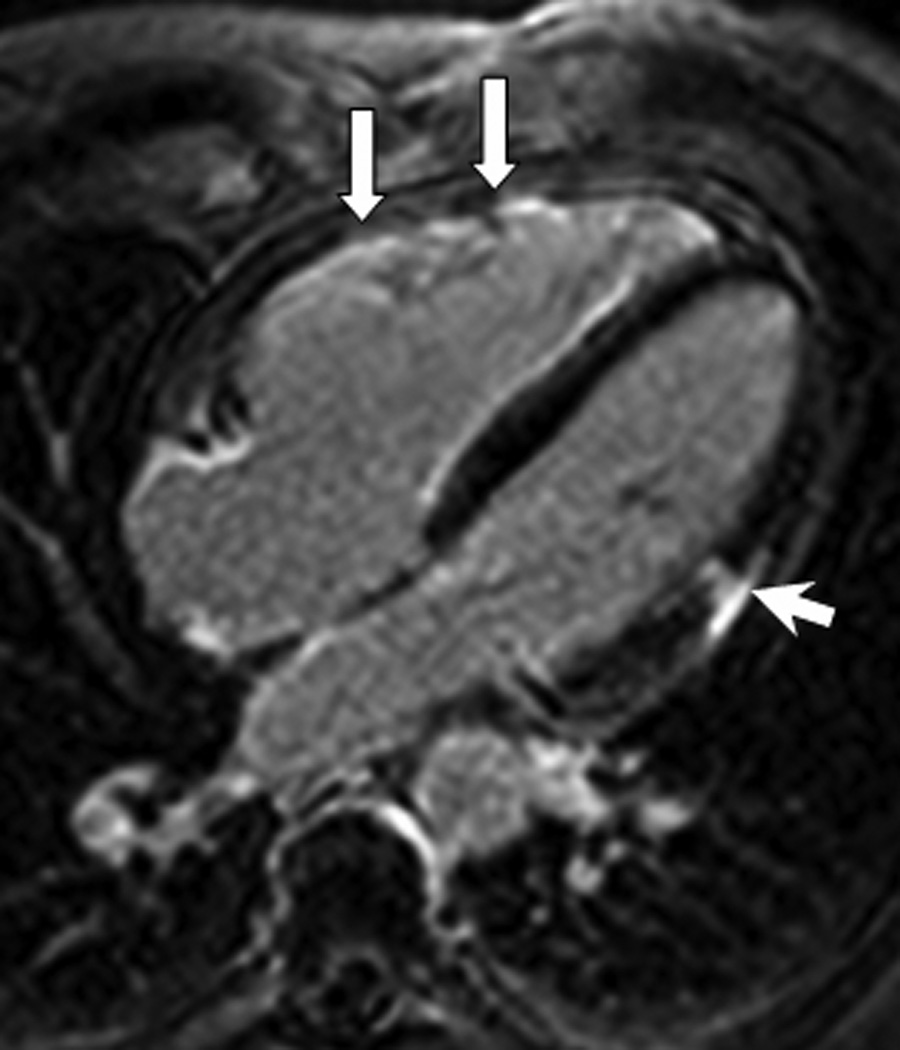
LGE image in the horizontal long-axis plane shows diffuse hyperenhancement of the right ventricular myocardium suggestive of fibrosis (arrows). Also seen is an area of focal hyperenhancement in the lateral LV myocardium (arrowhead).
Recent evidence suggests that ARVC is a biventricular cardiomyopathy; the extent and severity of LV involvement may be related to the underlying genotype, and can appear early in the disease course77. Histopathologic data suggest an inflammatory component in left-dominant arrhythmogenic cardiomyopathy; further studies are needed to define T2 imaging’s potential utility in delineating this feature of the disease. In PKP2-related ARVC, the most common mutation in the Unites States, LV fat infiltration is seen in up to 25% of these patients and most commonly affects the postero-lateral LV epicardium74. Recently, tagged cine CMR has revealed regional LV dysfunction in the postero-lateral LV wall in patients with early ARVC even in the presence of normal global function78. Mid-myocardial hyperenhancement by LGE, which may be seen in DCM, and sub-epicardial hyperenhancement have been reported in ARVC, particularly in desmoplakin (DSP) mutation carriers79. This underscores limitations in defining the underlying genetic abnormality by imaging phenotype alone. Individual patient assessment continues to require aggregate data assessment – history, examination, serologies, electrocardiography, imaging – in making the correct diagnosis.
ARVC: Current Status of Genetics
Since the discovery of the first ARVC locus in 199480, multiple disease loci have been mapped but the disease-causing genes remained elusive (Table 2)46, 81–89. The genetic cause of the recessive variant Naxos syndrome was elucidated first, as it is a highly penetrant disease with a clear-cut cutaneous phenotype89. Notably, epidermal cells in the palms and soles as well as cardiomyocytes are exposed to high shear stress and share components of the mechanical junctional apparatus (desmosome and fascia adherens) that is responsible for cell-to-cell adhesion. Proteins from three separate families assemble (Fig. 6)56 to form desmosomal cadherins (desmoglein and desmocollin), armadillo proteins (plakoglobin and plakophillin), and plakins (desmoplakin).
Table 2.
Genetic Causes of Arrhythmogenic Right Ventricular Cardiomyopathy
| Author (ref) | Gene | Encoded Protein | Chromosome locus |
OMIM* | Mode of inheritance | Comment |
|---|---|---|---|---|---|---|
| McKoy et al.89 Asimaki et al.81 |
JUP | Plakoglobin | 17q21 | #173325 #601214 |
Autosomal dominant Autosomal recessive |
Naxos disease |
| Rampazzo et al.86 Norgett et al.84 |
DSP | Desmoplakin | 6p24 | #125647 #605676 |
Autosomal dominant Autosomal recessive |
Carvajal syndrome |
| Gerull et al.46 | PKP2 | Plakophilin-2 | 12p11 | #602861 | Autosomal dominant | |
| Pilichou et al.85 | DSG2 | Desmoglein-2 | 18q12 | #125671 | Autosomal dominant | |
| Syrris et al.87 | DSC2 | Desmocollin-2 | 18q12 | #125645 | Autosomal dominant | |
| Extradesmosomal genes | ||||||
| Tiso et al.88 | RYR2 | Ryr2 | 1q42-q43 | #180902 | Autosomal dominant | CPVT* |
| Beffagna et al.82 | TGF-β3 | TGF β3 | 14q23-q24 | #190230 | Autosomal dominant | |
| Merner et al.83 | TMEM43 | TMEM 43 | 3p25 | #612048 | Autosomal dominant | |
CPVT = catecholaminergic polymorphic ventricular tachycardia
Figure 6.
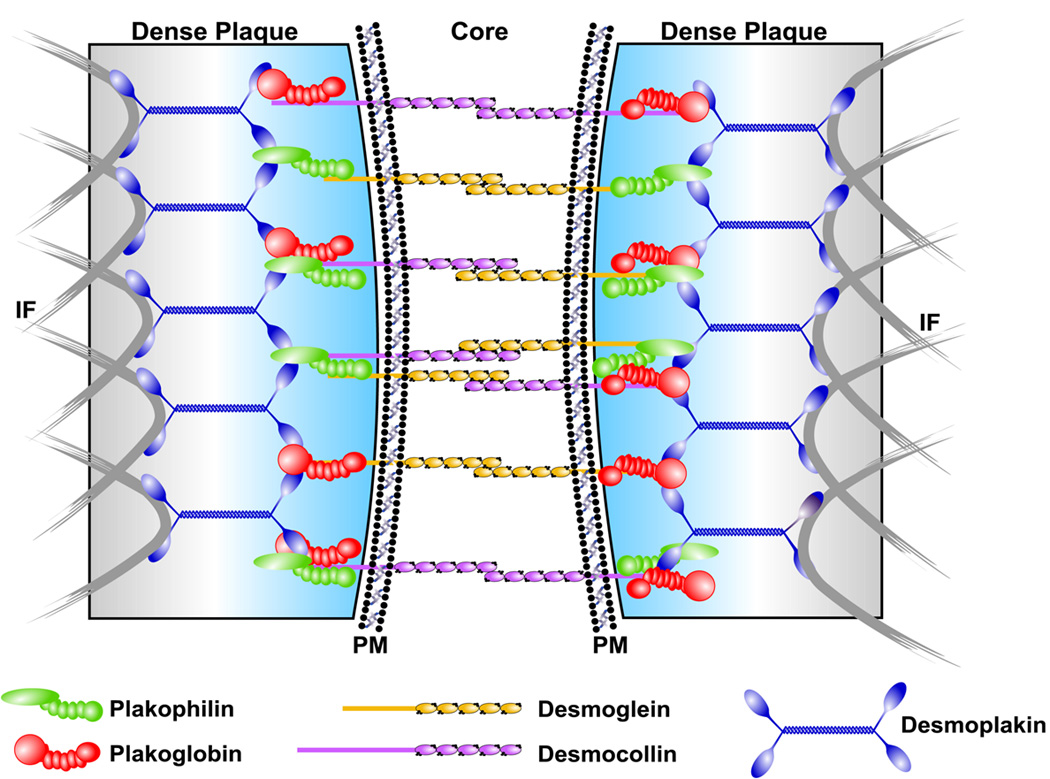
Components of the intercellular mechanical junction or desmosome between cardiomyocytes are shown. IF = intermediate filaments, PM = plasma membrane. Reproduced with permission from [56].
A plakoglobin deletion was first found in Naxos disease89. This was followed by discovery of mutations in desmoplakin86, plakophilin-246, desmoglein-285, desmocollin-287, and plakoglobin81 in the dominant forms. A recessive mutation of desmoplakin has been reported in another cardiocutaneous disease Carvajal syndrome84. Thus, ARVC was found to be mainly a disease of the desmosome56, and plakophilin-2 is the most frequently identified gene90.
Extradesmosomal genes implicated in ARVC include the genes encoding cardiac ryanodine 2 receptor (RyR2)88, transforming growth factor β3 (TGF-β3)82, and transmembrane protein 43 (TMEM43)83.
Mutations in the cardiac RyR2 cause ARVC2, characterized by effort-induced polymorphic ventricular arrhythmias and sudden death at young age. The ARVC2 phenotype is more similar to catecholaminergic polymorphic ventricular tachycardia than to ARVC since affected individuals do not show the typical electrocardiographic features and structural abnormalities, and are limited to mild or absent right ventricular hypokinesis.
Mutations in the untranslated regions of transforming growth factor (TGF-β3) have been identified in one large family and an unrelated proband with ARVC1 linkage (locus 14q24.3)82. It has been demonstrated that this protein stimulates production of components of the extracellular matrix and modulates expression of desmosomal genes in vitro.
Finally, a missense mutation in the TMEM43 has been identified in the ARVC5 phenotype in the Newfoundland founder population83. Affected patients show right precordial R-wave reduction and ventricular extrasystoles on ECG, and have early LV involvement and a high incidence of sudden death. At present, definitive proof that TGF-β3 and TMEM43 contribute to ARVC is missing, and these extrademosomal genes are currently screened in just a few research laboratories. Comprehensive mutation screening of the five desmosomal (JUP, DSP, PKP2, DSG2, and DSC2) ARVC genes is routinely carried out by sequence analysis. This approach can detect rare variants in at least 30–60% of probands, according to different cohorts91.
Plakophilin-2, desmoplakin, and desmoglein-2 account for the majority of isolated variants, although a high variability in their prevalence has been reported in different cohorts of probands87, 92–96. For instance, the high prevalence of PKP2 mutations (70%) among ARVC families in the Netherlands can be ascribed to founder effects87. Preliminary genotype-phenotype correlations suggest that PKP2 ARVC patients usually present with the classic, right-dominant disease, while other series with a relatively higher prevalence of desmoplakin mutations consist of patients who show a more diverse phenotype, including the so-called left dominant ARVC79, 95, 97.
Finally, preliminary genotype-phenotype data suggest that disease severity is greater in double mutations carriers, further emphasising the need to screen all known disease-causing genes even after isolation of a pathogenic mutation98, 99.
ARVC: Benefit/Limitations of Genetic Testing
Candidates for genetic screening include both index cases and family members of gene-positive ARVC probands56, 90, 91.
Genetic analysis in the diagnosis of index cases
As a general rule, there is no role at present for routine genetic screening to confirm a definite clinical diagnosis. In fact, a positive result from genotyping is supportive but not always confirmatory of ARVC diagnosis, while a negative genetic screening is non-contributory. About 50% of ARVC probands do not carry a defect in a known desmosomal gene. On the other hand, the identification of a rare genetic variant raises the index of suspicion but cannot be diagnostic per se. The latter uncertainty is typical for missense mutations and is mainly ascribable to the marked allelic heterogeneity of the main desmosomal genes and to the high prevalence of private mutations.
When a rare genetic variant is identified in ARVC, there are two possibilities: a) the genetic variant has been previously reported as causally linked to ARVC, and in such cases the diagnosis can be confirmed, or b) mutation screening yields a novel genetic variant. In the latter (most frequent) situation, pathogenicity must be proven by traditional criteria as with other heritable cardiomyopathies: i-the absence of the variant in a significant number of healthy individuals; ii-clinical correlation within families i.e. cosegregation with the disease; iii-a change in amino acid polarity and/or size; iv-a change involving a conserved amino acid; v-localization of the variant within a functional protein domain; and vi-in vitro functional studies.
PKP2 mutation variants require careful interpretation87, 92, 94, 96. In fact, increasing evidence suggests that some PKP2 mutations labelled as “pathogenic” may not be causal since they have been subsequently identified in healthy controls. Recently, Xu et al.98 demonstrated that among 38 ARVC index cases carrying PKP2 variants, 9 were compound heterozygotes and 16 double heterozygotes, i.e. showing an additional mutation in another desmosomal gene. These findings suggest that many PKP2 mutations may have a contributory role rather than causal for ARVC development and this might be true also for other desmosomal gene variants.
Cascade genetic screening of family members
Predictive testing of relatives is the main current indication for genetic analysis in ARVC, as in other inherited cardiomyopathies91. However, its implementation suffers from most of the limitations of confirmatory testing in index cases. In fact, before using any novel genetic variant for predictive testing in family members, it is mandatory to prove its pathogenicity. Co-segregation with the phenotype is not always easy to demonstrate because of the reduced penetrance and the variable expressivity of ARVC. Conversely, functional studies for every novel genetic variant are not practically feasible. Also for these reasons, genetic counselling is mandatory in each patient undergoing genetic screening to emphasize that it is the allele, rather than the disease, that is inherited.
When the pathogenicity of the allele variant is unequivocal, cascade screening of family members is of utmost value. In fact, it allows the early identification of asymptomatic carriers (healthy carriers) who would require life-long clinical evaluation due to variable and age-related penetrance of ARVC. These subjects must be considered at-risk because the disease is progressive and can appear late during life, and frequent clinical evaluation is mandatory. Sports activity increases the risk of sudden death in subjects with ARVC/D by five-fold, since acute volume overload and stretching of the RV during effort as well as sympathetic stimulation are major triggers of ventricular arrhythmias. Detection of asymptomatic individuals affected by ARVC/D at pre-participation screening has been proven to be a lifesaving strategy. The clinically unaffected family member carrying a disease gene mutation (“healthy carrier”) must be considered potentially at risk because the disease is progressive and can appear late during life, and frequent clinical checkups are mandatory. According to recent guidelines, all competitive sports should be always forbidden considering the legal implications. Noncompetitive sports may be allowed, providing regular follow-up assessment.
Genetic testing that identifies non-carriers, who represent about 50% of those tested, allows them to be considered healthy–they do not need further cardiac screening for ARVC and can be reassured that they carry no risk of disease transmission to their children56, 91. Predictive diagnosis is usually proposed in all family members of a genotyped proband after the age of 10 years, which is the age at which cardiac screening is considered mandatory in ARVC59.
Summary and Future Directions
With increased understanding of the genetics of cardiomyopathy, active synthesis of clinical data and family history informs the interpretation of phenotypic information yielded by contemporary cardiovascular imaging. Such synthesis has implications for not only individual patients but also at-risk family members. Those involved in imaging have a responsibility to recognize phenotypic features that suggest a genetic cause (Table 3), just as clinicians and genetics specialists should recognize where imaging may be useful to refine diagnosis and prognosis. Much work remains to be done to identify specific imaging signatures that guide diagnosis toward particular genetic mechanisms of disease. Further insight is needed from histopathology in conjunction with genetic studies to define what, if any, phenotypic signatures correspond to specific genotypes; such insights will, in turn, inform refined imaging-based diagnosis in cardiomyopathy. For some mutations, it is unknown what long-term clinical significance is for currently asymptomatic mutation carriers. Even in the case of lamin A/C gene mutations there is considerable variability in symptom onset, severity, and rate of progression. Consensus on clinical screening, imaging, and frequency of assessments in asymptomatic mutation carriers is limited and currently not based on solid prospective longitudinal data. The fact that many mutations are unique, so called ‘private mutations’ restricted to one or a few families, will continue to limit efforts to provide broad recommendations. As recommended by a recent expert panel100, referral of patients and families with heritable cardiomyopathies to centers with genetic expertise should be strongly considered.
Table 3.
Phenotypic Clues Linking Imaging to Non-HCM Genetic Cardiomyopathies*
| Imaging Phenotype | Additional Clinical Clues† | Genetic Considerations |
|---|---|---|
| Dilated cardiomyopathy with diastolic dysfunction, atrial myopathy |
Conduction system disease | Lamin A/C |
| Dilated cardiomyopathy with circumferential or confluent midwall enhancement by LGE‡ |
Acute myocarditis-like presentation | Left-dominant arrhythmogenic cardiomyopathy |
| Right ventricular dilatation, segmental contraction abnormalities, aneurysms; fibrofatty changes in myocardium |
Left bundle branch-morphology ventricular arrhythmia |
Arrhythmogenic right ventricular cardiomyopathy |
This summary relates to DCM and ARVC, and does not include findings related to hypertrophic cardiomyopathy or genetic cardiomyopathies associated with muscular dystrophies or inborn errors of metabolism.
A valuable clinical clue pointing to genetic cardiomyopathy may be obtained from a meticulous family history.
LGE = late gadolinium enhancement imaging. Note that left-dominant arrhythmogenic cardiomoypathy may present with preserved LV size and systolic function as well.
The major obstacle for a widespread clinical use of genotyping has been the high costs of mutation screening by conventional direct sequencing. With increased availability of cost-effective tools, genotyping may become available at any center that performs family evaluation for inherited cardiomyopathies. In this setting, phenotype recognition by imaging that identifies disease in its earliest or concealed stages should prompt consideration of genotyping when clinical abnormalities are still subtle but individuals are already at risk of sudden death. Obstacles to widespread myocardial characterization and precise diagnosis with CMR may include variations across scanner platforms and interpreters; we advise that patients be referred to established CMR centers when considering this modality in evaluating genetic cardiomyopathies. More broadly, the shortcomings of current imaging to detect signatures of specific genotypes should encourage researchers to develop new imaging approaches based on our advancing understanding of the genetic and molecular bases of cardiomyopathies. As our understanding of the phenotypic spectrum and genetics of DCM and ARVC unfolds, longitudinal genotype-phenotype studies that take advantage of refined myocardial imaging and preclinical models will provide mechanistic insights to further improve our ability to diagnose and treat heritable cardiomyopathies.
Supplementary Material
Acknowledgements
The authors are indebted to Tam Tran, BS, for his assistance in manuscript preparation.
Sources of Funding
HL095563 & HL102450 (SVR), HL093350 (HT), Registry of Cardio-Cerebro-Vascular Mortality, Veneto Region, Venice, Italy (CB)
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosures
Dr. Raman receives research support from Siemens. Dr. Taylor receives research support from Genzyme Therapeutics.
REFERENCES
- 1.Fatkin D, MacRae C, Sasaki T, Wolff MR, Porcu M, Frenneaux M, Atherton J, Vidaillet HJ, Jr, Spudich S, De Girolami U, Seidman JG, Seidman C, Muntoni F, Muehle G, Johnson W, McDonough B. Missense mutations in the rod domain of the lamin A/C gene as causes of dilated cardiomyopathy and conduction-system disease. N Engl J Med. 1999;341:1715–1724. doi: 10.1056/NEJM199912023412302. [DOI] [PubMed] [Google Scholar]
- 2.Pan H, Richards AA, Zhu X, Joglar JA, Yin HL, Garg V. A novel mutation in LAMIN A/C is associated with isolated early-onset atrial fibrillation and progressive atrioventricular block followed by cardiomyopathy and sudden cardiac death. Heart Rhythm. 2009;6:707–710. doi: 10.1016/j.hrthm.2009.01.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Raman SV, Sparks EA, Baker PM, McCarthy B, Wooley CF. Mid-myocardial fibrosis by cardiac magnetic resonance in patients with lamin A/C cardiomyopathy: possible substrate for diastolic dysfunction. J Cardiovasc Magn Reson. 2007;9:907–913. doi: 10.1080/10976640701693733. [DOI] [PubMed] [Google Scholar]
- 4.Michels VV, Moll PP, Miller FA, Tajik AJ, Chu JS, Driscoll DJ, Burnett JC, Rodeheffer RJ, Chesebro JH, Tazelaar H. The frequency of familial dilated cardiomyopathy in a series of patients with idiopathic dilated cardiomyopathy. N Engl J Med. 1992;326:77–82. doi: 10.1056/NEJM199201093260201. [DOI] [PubMed] [Google Scholar]
- 5.Codd MB, Sugrue DD, Gersh BJ, Melton LJ. Epidemiology of idiopathic dilated and hypertrophic cardiomyopathy. A population-based study in Olmsted County, Minnesota, 1975–1984. Circulation. 1989;80:564–572. doi: 10.1161/01.cir.80.3.564. [DOI] [PubMed] [Google Scholar]
- 6.Richardson P, McKenna W, Bristow M, Maisch B, Mautner B, O'Connell J, Olsen E, Thiene G, Goodwin J, Gyarfas I, Martin I, Nordet P. Report of the 1995 World Health Organization/International Society and Federation of Cardiology Task Force on the Definition and Classification of cardiomyopathies. Circulation. 1996;93:841–842. doi: 10.1161/01.cir.93.5.841. [DOI] [PubMed] [Google Scholar]
- 7.Mestroni L, Maisch B, McKenna WJ, Schwartz K, Charron P, Rocco C, Tesson F, Richter A, Wilke A, Komajda M. Guidelines for the study of familial dilated cardiomyopathies. Eur Heart J. 1999;20:93–102. doi: 10.1053/euhj.1998.1145. [DOI] [PubMed] [Google Scholar]
- 8.Fernandez-Sola J, Nicolas JM, Oriola J, Sacanella E, Estruch R, Rubin E, Urbano-Marquez A. Angiotensin-converting enzyme gene polymorphism is associated with vulnerability to alcoholic cardiomyopathy. Ann Intern Med. 2002;137(5 Part 1):321–326. doi: 10.7326/0003-4819-137-5_part_1-200209030-00007. [DOI] [PubMed] [Google Scholar]
- 9.Gregori D, Rocco C, Miocic S, Mestroni L. Estimating the frequency of familial dilated cardiomiopathy in the presence of misclassification errors. J. Appl. Statistics. 2001;28:53–62. [Google Scholar]
- 10.Grünig E, Tasman JA, Kucherer H, Franz W, Kubler W, Katus HA. Frequency and phenotypes of familial dilated cardiomyopathy. J Am Coll Cardiol. 1998;31:186–194. doi: 10.1016/s0735-1097(97)00434-8. [DOI] [PubMed] [Google Scholar]
- 11.Baig MK, Goldman JH, Caforio ALP, Coonar AS, Keeling PJ, McKenna WJ. Familial dilated cardiomyopathy: cardiac abnormalities are common in asymptomatic relatives and may represent early disease. J Am Coll Cardiol. 1998;31:195–201. doi: 10.1016/s0735-1097(97)00433-6. [DOI] [PubMed] [Google Scholar]
- 12.Maron BJ, Towbin JA, Thiene G, Antzelevitch C, Corrado D, Arnett D, Moss AJ, Seidman CE, Young JB. Contemporary definitions and classification of the cardiomyopathies: an American Heart Association Scientific Statement from the Council on Clinical Cardiology, Heart Failure and Transplantation Committee; Quality of Care and Outcomes Research and Functional Genomics and Translational Biology Interdisciplinary Working Groups; and Council on Epidemiology and Prevention. Circulation. 2006;113:1807–1816. doi: 10.1161/CIRCULATIONAHA.106.174287. [DOI] [PubMed] [Google Scholar]
- 13.Wood MJ, Picard MH. Utility of echocardiography in the evaluation of individuals with cardiomyopathy. Heart. 2004;90:707–712. doi: 10.1136/hrt.2003.024778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hoedemaekers YF, Caliskan K, Michels M, Frohn-Mulder I, van der Smagt JJ, Phefferkorn JE, Wessels MW, Ten Cate FJ, Sijbrands EJ, Dooijes D, Majoor-Krakauer DF. The importance of genetic counseling, DNA diagnostics and cardiologic family screening in left ventricular noncompaction cardiomyopathy. Circ Cardiovasc Genet. 2010;3:232–239. doi: 10.1161/CIRCGENETICS.109.903898. [DOI] [PubMed] [Google Scholar]
- 15.Biagini E, Ragni L, Ferlito M, Pasquale F, Lofiego C, Leone O, Rocchi G, Perugini E, Zagnoni S, Branzi A, Picchio FM, Rapezzi C. Different types of cardiomyopathy associated with isolated ventricular noncompaction. Am J Cardiol. 2006;98:821–824. doi: 10.1016/j.amjcard.2006.04.021. [DOI] [PubMed] [Google Scholar]
- 16.Anderson LJ, Holden S, Davis B, Prescott E, Charrier CC, Bunce NH, Firmin DN, Wonke B, Porter J, Walker JM, Pennell DJ. Cardiovascular T2-star (T2*) magnetic resonance for the early diagnosis of myocardial iron overload. Eur Heart J. 2001;22:2171–2179. doi: 10.1053/euhj.2001.2822. [DOI] [PubMed] [Google Scholar]
- 17.Giri S, Chung YC, Merchant A, Mihai G, Rajagopalan S, Raman SV, Simonetti OP. T2 quantification for improved detection of myocardial edema. J Cardiovasc Magn Reson. 2009;11:56. doi: 10.1186/1532-429X-11-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McCrohon JA, Moon JC, Prasad SK, McKenna WJ, Lorenz CH, Coats AJ, Pennell DJ. Differentiation of heart failure related to dilated cardiomyopathy and coronary artery disease using gadolinium-enhanced cardiovascular magnetic resonance. Circulation. 2003;108:54–59. doi: 10.1161/01.CIR.0000078641.19365.4C. [DOI] [PubMed] [Google Scholar]
- 19.Raman SV, Simonetti OP, Cataland SR, Kraut EH. Myocardial ischemia and right ventricular dysfunction in adult patients with sickle cell disease. Haematologica. 2006;91:1329–1335. [PubMed] [Google Scholar]
- 20.Wood JC, Tyszka JM, Carson S, Nelson MD, Coates TD. Myocardial iron loading in transfusion-dependent thalassemia and sickle cell disease. Blood. 2004;103:1934–1936. doi: 10.1182/blood-2003-06-1919. [DOI] [PubMed] [Google Scholar]
- 21.Klintschar M, Stiller D. Sudden cardiac death in hereditary hemochromatosis: an underestimated cause of death? Int J Legal Med. 2004;118:174–177. doi: 10.1007/s00414-004-0451-6. [DOI] [PubMed] [Google Scholar]
- 22.Abdel-Aty H, Simonetti O, Friedrich MG. T2-weighted cardiovascular magnetic resonance imaging. J Magn Reson Imaging. 2007;26:452–459. doi: 10.1002/jmri.21028. [DOI] [PubMed] [Google Scholar]
- 23.Wansapura JP, Hor KN, Mazur W, Fleck R, Hagenbuch S, Benson DW, Gottliebson WM. Left ventricular T2 distribution in Duchenne Muscular Dystrophy. J Cardiovasc Magn Reson. 2010;12:14. doi: 10.1186/1532-429X-12-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Assomull RG, Prasad SK, Lyne J, Smith G, Burman ED, Khan M, Sheppard MN, Poole-Wilson PA, Pennell DJ. Cardiovascular magnetic resonance, fibrosis, and prognosis in dilated cardiomyopathy. J Am Coll Cardiol. 2006;48:1977–1985. doi: 10.1016/j.jacc.2006.07.049. [DOI] [PubMed] [Google Scholar]
- 25.Regitz-Zagrosek V, Erdmann J, Wellnhofer E, Raible J, Fleck E. Novel mutation in the alpha-tropomyosin gene and transition from hypertrophic to hypocontractile dilated cardiomyopathy. Circulation. 2000;102:E112–E116. doi: 10.1161/01.cir.102.17.e112. [DOI] [PubMed] [Google Scholar]
- 26.McNair WP, Ku L, Taylor MR, Fain PR, Dao D, Wolfel E, Mestroni L. SCN5A mutation associated with dilated cardiomyopathy, conduction disorder, and arrhythmia. Circulation. 2004;110:2163–2167. doi: 10.1161/01.CIR.0000144458.58660.BB. [DOI] [PubMed] [Google Scholar]
- 27.Barresi R, Di Blasi C, Negri T, Brugnoni R, Vitali A, Felisari G, Salandi A, Daniel S, Cornelio F, Morandi L, Mora M. Disruption of heart sarcoglycan complex and severe cardiomyopathy caused by beta sarcoglycan mutations. J Med Genet. 2000;37:102–107. doi: 10.1136/jmg.37.2.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tsubata S, Bowles KR, Vatta M, Zintz C, Titus J, Muhonen L, Bowles NE, Towbin JA. Mutations in the human delta-sarcoglycan gene in familial and sporadic dilated cardiomyopathy. J Clin Invest. 2000;106:655–662. doi: 10.1172/JCI9224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gold R, Kress W, Meurers B, Meng G, Reichmann H, Muller CR. Becker muscular dystrophy: detection of unusual disease courses by combined approach to dystrophin analysis. Muscle Nerve. 1992;15:214–218. doi: 10.1002/mus.880150214. [DOI] [PubMed] [Google Scholar]
- 30.Murphy RT, Mogensen J, Shaw A, Kubo T, Hughes S, McKenna WJ. Novel mutation in cardiac troponin I in recessive idiopathic dilated cardiomyopathy. Lancet. 2004;363:371–372. doi: 10.1016/S0140-6736(04)15468-8. [DOI] [PubMed] [Google Scholar]
- 31.Valle G, Faulkner G, De Antoni A, Pacchioni B, Pallavicini A, Pandolfo D, Tiso N, Toppo S, Trevisan S, Lanfranchi G. Telethonin, a novel sarcomeric protein of heart and skeletal muscle. FEBS Lett. 1997;415:163–168. doi: 10.1016/s0014-5793(97)01108-3. [DOI] [PubMed] [Google Scholar]
- 32.Olson TM, Kishimoto NY, Whitby FG, Michels VV. Mutations that alter the surface charge of alpha-tropomyosin are associated with dilated cardiomyopathy. J Mol Cell Cardiol. 2001;33:723–732. doi: 10.1006/jmcc.2000.1339. [DOI] [PubMed] [Google Scholar]
- 33.Olson TM, Michels VV, Thibodeau SN, Tai YS, Keating MT. Actin mutations in dilated cardiomyopathy, a heritable form of heart failure. Science. 1998;280:750–752. doi: 10.1126/science.280.5364.750. [DOI] [PubMed] [Google Scholar]
- 34.Li D, Parks SB, Kushner JD, Nauman D, Burgess D, Ludwigsen S, Partain J, Nixon RR, Allen CN, Irwin RP, Jakobs PM, Litt M, Hershberger RE. Mutations of presenilin genes in dilated cardiomyopathy and heart failure. Am J Hum Genet. 2006;79:1030–1039. doi: 10.1086/509900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kamisago M, Sharma SD, DePalma SR, Solomon S, Sharma P, McDonough B, Smoot L, Mullen MP, Woolf PK, Wigle ED, Seidman JG, Seidman CE. Mutations in sarcomere protein genes as a cause of dilated cardiomyopathy. N Engl J Med. 2000;343:1688–1696. doi: 10.1056/NEJM200012073432304. [DOI] [PubMed] [Google Scholar]
- 36.Bienengraeber M, Olson TM, Selivanov VA, Kathmann EC, O'Cochlain F, Gao F, Karger AB, Ballew JD, Hodgson DM, Zingman LV, Pang YP, Alekseev AE, Terzic A. ABCC9 mutations identified in human dilated cardiomyopathy disrupt catalytic KATP channel gating. Nat Genet. 2004;36:382–387. doi: 10.1038/ng1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Knoll R, Hoshijima M, Hoffman HM, Person V, Lorenzen-Schmidt I, Bang ML, Hayashi T, Shiga N, Yasukawa H, Schaper W, McKenna W, Yokoyama M, Schork NJ, Omens JH, McCulloch AD, Kimura A, Gregorio CC, Poller W, Schaper J, Schultheiss HP, Chien KR. The cardiac mechanical stretch sensor machinery involves a Z disc complex that is defective in a subset of human dilated cardiomyopathy. Cell. 2002;111:943–955. doi: 10.1016/s0092-8674(02)01226-6. [DOI] [PubMed] [Google Scholar]
- 38.Daehmlow S, Erdmann J, Knueppel T, Gille C, Froemmel C, Hummel M, Hetzer R, Regitz-Zagrosek V. Novel mutations in sarcomeric protein genes in dilated cardiomyopathy. Biochem Biophys Res Commun. 2002;298:116–120. doi: 10.1016/s0006-291x(02)02374-4. [DOI] [PubMed] [Google Scholar]
- 39.Vatta M, Mohapatra B, Jimenez S, Sanchez X, Faulkner G, Perles Z, Sinagra G, Lin JH, Vu TM, Zhou Q, Bowles KR, Di Lenarda A, Schimmenti L, Fox M, Chrisco MA, Murphy RT, McKenna W, Elliott P, Bowles NE, Chen J, Valle G, Towbin JA. Mutations in Cypher/ZASP in patients with dilated cardiomyopathy and left ventricular non-compaction. J Am Coll Cardiol. 2003;42:2014–2027. doi: 10.1016/j.jacc.2003.10.021. [DOI] [PubMed] [Google Scholar]
- 40.Duboscq-Bidot L, Xu P, Charron P, Neyroud N, Dilanian G, Millaire A, Bors V, Komajda M, Villard E. Mutations in the Z-band protein myopalladin gene and idiopathic dilated cardiomyopathy. Cardiovasc Res. 2008;77:118–125. doi: 10.1093/cvr/cvm015. [DOI] [PubMed] [Google Scholar]
- 41.Olson TM, Illenberger S, Kishimoto NY, Huttelmaier S, Keating MT, Jockusch BM. Metavinculin mutations alter actin interaction in dilated cardiomyopathy. Circulation. 2002;105:431–437. doi: 10.1161/hc0402.102930. [DOI] [PubMed] [Google Scholar]
- 42.Schmitt JP, Kamisago M, Asahi M, Li GH, Ahmad F, Mende U, Kranias EG, MacLennan DH, Seidman JG, Seidman CE. Dilated cardiomyopathy and heart failure caused by a mutation in phospholamban. Science. 2003;299:1410–1413. doi: 10.1126/science.1081578. [DOI] [PubMed] [Google Scholar]
- 43.Li D, Tapscoft T, Gonzalez O, Burch PE, Quinones MA, Zoghbi WA, Hill R, Bachinski LL, Mann DL, Roberts R. Desmin mutation responsible for idiopathic dilated cardiomyopathy. Circulation. 1999;100:461–464. doi: 10.1161/01.cir.100.5.461. [DOI] [PubMed] [Google Scholar]
- 44.Mogensen J, Murphy RT, Shaw T, Bahl A, Redwood C, Watkins H, Burke M, Elliott PM, McKenna WJ. Severe disease expression of cardiac troponin C and T mutations in patients with idiopathic dilated cardiomyopathy. J Am Coll Cardiol. 2004;44:2033–2040. doi: 10.1016/j.jacc.2004.08.027. [DOI] [PubMed] [Google Scholar]
- 45.Durand JB, Bachinski LL, Bieling LC, Czernuszewicz GZ, Abchee AB, Yu QT, Tapscott T, Hill R, Ifegwu J, Marian AJ, Brugada R, Daiger S, Gregoritch JM, Anderson JL, Quiñones M, Towbin JA, Roberts R. Localization of a gene responsible for familial dilated cardiomyopathy to chromosome 1q32. Circulation. 1995;92:3387–3389. doi: 10.1161/01.cir.92.12.3387. [DOI] [PubMed] [Google Scholar]
- 46.Gerull B, Heuser A, Wichter T, Paul M, Basson CT, McDermott DA, Lerman BB, Markowitz SM, Ellinor PT, MacRae CA, Peters S, Grossmann KS, Drenckhahn J, Michely B, Sasse-Klaassen S, Birchmeier W, Dietz R, Breithardt G, Schulze-Bahr E, Thierfelder L. Mutations in the desmosomal protein plakophilin-2 are common in arrhythmogenic right ventricular cardiomyopathy. Nat Genet. 2004;36:1162–1164. doi: 10.1038/ng1461. [DOI] [PubMed] [Google Scholar]
- 47.Bowles NE, Bowles KR, Towbin JA. The "final common pathway" hypothesis and inherited cardiovascular disease. The role of cytoskeletal proteins in dilated cardiomyopathy. Herz. 2000;25:168–175. doi: 10.1007/s000590050003. [DOI] [PubMed] [Google Scholar]
- 48.Schönberger J, Seidman CE. Many roads lead to a broken heart: the genetics of dilated cardiomyopathy. Am J Hum Genet. 2001;69:249–260. doi: 10.1086/321978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nelson SD, Sparks EA, Graber HL, Boudoulas H, Mehdirad AA, Baker P, Wooley C. Clinical characteristics of sudden death victims in heritable (chromosome 1p1-1q1) conduction and myocardial disease. J Am Coll Cardiol. 1998;32:1717–1723. doi: 10.1016/s0735-1097(98)00424-0. [DOI] [PubMed] [Google Scholar]
- 50.Malhotra R, Mason PK. Lamin A/C deficiency as a cause of familial dilated cardiomyopathy. Curr Opin Cardiol. 2009;24:203–208. doi: 10.1097/HCO.0b013e32832a11c6. [DOI] [PubMed] [Google Scholar]
- 51.van Berlo JH, de Voogt WG, van der Kooi AJ, van Tintelen JP, Bonne G, Yaou RB, Duboc D, Rossenbacker T, Heidbuchel H, de Visser M, Crijns HJ, Pinto YM. Meta-analysis of clinical characteristics of 299 carriers of LMNA gene mutations: do lamin A/C mutations portend a high risk of sudden death? J Mol Med. 2005;83(1):79–83. doi: 10.1007/s00109-004-0589-1. [DOI] [PubMed] [Google Scholar]
- 52.Hershberger RE, Lindenfeld J, Mestroni L, Seidman CE, Taylor MR, Towbin JA. Genetic evaluation of cardiomyopathy--a Heart Failure Society of America practice guideline. J Card Fail. 2009;15:83–97. doi: 10.1016/j.cardfail.2009.01.006. [DOI] [PubMed] [Google Scholar]
- 53.Marcus FI, Fontaine GH, Guiraudon G, Frank R, Laurenceau JL, Malergue C, Grosgogeat Y. Right ventricular dysplasia: a report of 24 adult cases. Circulation. 1982;65:384–398. doi: 10.1161/01.cir.65.2.384. [DOI] [PubMed] [Google Scholar]
- 54.Dalal D, Nasir K, Bomma C, Prakasa K, Tandri H, Piccini J, Roguin A, Tichnell C, James C, Russell SD, Judge DP, Abraham T, Spevak PJ, Bluemke DA, Calkins H. Arrhythmogenic right ventricular dysplasia: a United States experience. Circulation. 2005;112:3823–3832. doi: 10.1161/CIRCULATIONAHA.105.542266. [DOI] [PubMed] [Google Scholar]
- 55.Marcus FI, Nava A, Thiene G. Arrhythmogenic right ventricular cardiomyopathy/dysplasia: recent advances. Milan: Springer Verlag; 2007. [Google Scholar]
- 56.Basso C, Corrado D, Marcus FI, Nava A, Thiene G. Arrhythmogenic right ventricular cardiomyopathy. Lancet. 2009;373:1289–1300. doi: 10.1016/S0140-6736(09)60256-7. [DOI] [PubMed] [Google Scholar]
- 57.McKenna WJ, Thiene G, Nava A, Fontaliran F, Blomstrom-Lundqvist C, Fontaine G, Camerini F. Diagnosis of arrhythmogenic right ventricular dysplasia/cardiomyopathy. Task Force of the Working Group Myocardial and Pericardial Disease of the European Society of Cardiology and of the Scientific Council on Cardiomyopathies of the International Society and Federation of Cardiology. Br Heart J. 1994;71:215–218. doi: 10.1136/hrt.71.3.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Marcus FI, McKenna WJ, Sherrill D, Basso C, Bauce B, Bluemke DA, Calkins H, Corrado D, Cox MG, Daubert JP, Fontaine G, Gear K, Hauer R, Nava A, Picard MH, Protonotarios N, Saffitz JE, Sanborn DM, Steinberg JS, Tandri H, Thiene G, Towbin JA, Tsatsopoulou A, Wichter T, Zareba W. Diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia: proposed modification of the task force criteria. Circulation. 2010;121:1533–1541. doi: 10.1161/CIRCULATIONAHA.108.840827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nava A, Bauce B, Basso C, Muriago M, Rampazzo A, Villanova C, Daliento L, Buja G, Corrado D, Danieli GA, Thiene G. Clinical profile and long-term follow-up of 37 families with arrhythmogenic right ventricular cardiomyopathy. J Am Coll Cardiol. 2000;36:2226–2233. doi: 10.1016/s0735-1097(00)00997-9. [DOI] [PubMed] [Google Scholar]
- 60.Nava A, Thiene G, Canciani B, Scognamiglio R, Daliento L, Buja G, Martini B, Stritoni P, Fasoli G. Familial occurrence of right ventricular dysplasia: a study involving nine families. J Am Coll Cardiol. 1988;12:1222–1228. doi: 10.1016/0735-1097(88)92603-4. [DOI] [PubMed] [Google Scholar]
- 61.Prakasa KR, Dalal D, Wang J, Bomma C, Tandri H, Dong J, James C, Tichnell C, Russell SD, Spevak P, Corretti M, Bluemke DA, Calkins H, Abraham TP. Feasibility and variability of three dimensional echocardiography in arrhythmogenic right ventricular dysplasia/cardiomyopathy. Am J Cardiol. 2006;97:703–709. doi: 10.1016/j.amjcard.2005.11.020. [DOI] [PubMed] [Google Scholar]
- 62.Marcus FI, McKenna WJ, Sherrill D, Basso C, Bauce B, Bluemke DA, Calkins H, Corrado D, Cox MG, Daubert JP, Fontaine G, Gear K, Hauer R, Nava A, Picard MH, Protonotarios N, Saffitz JE, Sanborn DM, Steinberg JS, Tandri H, Thiene G, Towbin JA, Tsatsopoulou A, Wichter T, Zareba W. Diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia: proposed modification of the task force criteria. Circulation. 2010;121:1533–1541. doi: 10.1161/CIRCULATIONAHA.108.840827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bomma C, Dalal D, Tandri H, Prakasa K, Nasir K, Roguin A, Piccini J, Dong J, Mahadevappa M, Tichnell C, James C, Lima JA, Fishman E, Calkins H, Bluemke DA. Evolving role of multidetector computed tomography in evaluation of arrhythmogenic right ventricular dysplasia/cardiomyopathy. Am J Cardiol. 2007;100:99–105. doi: 10.1016/j.amjcard.2007.02.064. [DOI] [PubMed] [Google Scholar]
- 64.Tandri H, Saranathan M, Rodriguez ER, Martinez C, Bomma C, Nasir K, Rosen B, Lima JA, Calkins H, Bluemke DA. Noninvasive detection of myocardial fibrosis in arrhythmogenic right ventricular cardiomyopathy using delayed-enhancement magnetic resonance imaging. J Am Coll Cardiol. 2005;45:98–103. doi: 10.1016/j.jacc.2004.09.053. [DOI] [PubMed] [Google Scholar]
- 65.Tandri H, Calkins H, Nasir K, Bomma C, Castillo E, Rutberg J, Tichnell C, Lima JA, Bluemke DA. Magnetic resonance imaging findings in patients meeting task force criteria for arrhythmogenic right ventricular dysplasia. J Cardiovasc Electrophysiol. 2003;14:476–482. doi: 10.1046/j.1540-8167.2003.02560.x. [DOI] [PubMed] [Google Scholar]
- 66.Tandri H, Bomma C, Calkins H, Bluemke DA. Magnetic resonance and computed tomography imaging of arrhythmogenic right ventricular dysplasia. J Magn Reson Imaging. 2004;19:848–858. doi: 10.1002/jmri.20078. [DOI] [PubMed] [Google Scholar]
- 67.Bomma C, Rutberg J, Tandri H, Nasir K, Roguin A, Tichnell C, Rodriguez R, James C, Kasper E, Spevak P, Bluemke DA, Calkins H. Misdiagnosis of arrhythmogenic right ventricular dysplasia/cardiomyopathy. J Cardiovasc Electrophysiol. 2004;15:300–306. doi: 10.1046/j.1540-8167.2004.03429.x. [DOI] [PubMed] [Google Scholar]
- 68.Roffe C. Ageing of the heart. Br J Biomed Sci. 1998;55:136–148. [PubMed] [Google Scholar]
- 69.Tung K, Raman SV, King MA, Dephilip RM. Correlation of magnetic resonance imaging with histopathology in arrhythmogenic right ventricular dysplasia/cardiomyopathy (ARVD/C) Clin Anat. 2006;19:44–50. doi: 10.1002/ca.20186. [DOI] [PubMed] [Google Scholar]
- 70.Tansey DK, Aly Z, Sheppard MN. Fat in the right ventricle of the normal heart. Histopathology. 2005;46:98–104. doi: 10.1111/j.1365-2559.2005.02054.x. [DOI] [PubMed] [Google Scholar]
- 71.Basso C, Ronco F, Marcus F, Abudureheman A, Rizzo S, Frigo AC, Bauce B, Maddalena F, Nava A, Corrado D, Grigoletto F, Thiene G. Quantitative assessment of endomyocardial biopsy in arrhythmogenic right ventricular cardiomyopathy/dysplasia: an in vitro validation of diagnostic criteria. Eur Heart J. 2008;29:2760–2771. doi: 10.1093/eurheartj/ehn415. [DOI] [PubMed] [Google Scholar]
- 72.Basso C, Thiene G. Adipositas cordis, fatty infiltration of the right ventricle, and arrhythmogenic right ventricular cardiomyopathy. Just a matter of fat? Cardiovasc Pathol. 2005;14:37–41. doi: 10.1016/j.carpath.2004.12.001. [DOI] [PubMed] [Google Scholar]
- 73.Grothues F, Moon JC, Bellenger NG, Smith GS, Klein HU, Pennell DJ. Interstudy reproducibility of right ventricular volumes, function, and mass with cardiovascular magnetic resonance. Am Heart J. 2004;147:218–223. doi: 10.1016/j.ahj.2003.10.005. [DOI] [PubMed] [Google Scholar]
- 74.Dalal D, Tandri H, Judge DP, Amat N, Macedo R, Jain R, Tichnell C, Daly A, James C, Russell SD, Abraham T, Bluemke DA, Calkins H. Morphologic variants of familial arrhythmogenic right ventricular dysplasia/cardiomyopathy a genetics-magnetic resonance imaging correlation study. J Am Coll Cardiol. 2009;53:1289–1299. doi: 10.1016/j.jacc.2008.12.045. [DOI] [PubMed] [Google Scholar]
- 75.Corrado D, Thiene G. Cardiac sarcoidosis mimicking arrhythmogenic right ventricular cardiomyopathy/dysplasia: the renaissance of endomyocardial biopsy? J Cardiovasc Electrophysiol. 2009;20:477–479. doi: 10.1111/j.1540-8167.2008.01387.x. [DOI] [PubMed] [Google Scholar]
- 76.Patel MR, Cawley PJ, Heitner JF, Klem I, Parker MA, Jaroudi WA, Meine TJ, White JB, Elliott MD, Kim HW, Judd RM, Kim RJ. Detection of myocardial damage in patients with sarcoidosis. Circulation. 2009;120:1969–1977. doi: 10.1161/CIRCULATIONAHA.109.851352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Sen-Chowdhry S, Syrris P, Prasad SK, Hughes SE, Merrifield R, Ward D, Pennell DJ, McKenna WJ. Left-dominant arrhythmogenic cardiomyopathy: an under-recognized clinical entity. J Am Coll Cardiol. 2008;52:2175–2187. doi: 10.1016/j.jacc.2008.09.019. [DOI] [PubMed] [Google Scholar]
- 78.Jain A, Shehata ML, Stuber M, Berkowitz SJ, Calkins H, Lima JA, Bluemke DA, Tandri H. Prevalence of Left Ventricular Regional Dysfunction in Arrhythmogenic Right Ventricular Dysplasia: A Tagged MRI Study. Circ Cardiovasc Imaging. 2010;3:290–297. doi: 10.1161/CIRCIMAGING.109.911313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Norman M, Simpson M, Mogensen J, Shaw A, Hughes S, Syrris P, Sen-Chowdhry S, Rowland E, Crosby A, McKenna WJ. Novel mutation in desmoplakin causes arrhythmogenic left ventricular cardiomyopathy. Circulation. 2005;112:636–642. doi: 10.1161/CIRCULATIONAHA.104.532234. [DOI] [PubMed] [Google Scholar]
- 80.Rampazzo A, Nava A, Danieli GA, Buja G, Daliento L, Fasoli G, Scognamiglio R, Corrado D, Thiene G. The gene for arrhythmogenic right ventricular cardiomyopathy maps to chromosome 14q23-q24. Hum Mol Genet. 1994;3:959–962. doi: 10.1093/hmg/3.6.959. [DOI] [PubMed] [Google Scholar]
- 81.Asimaki A, Syrris P, Wichter T, Matthias P, Saffitz JE, McKenna WJ. A novel dominant mutation in plakoglobin causes arrhythmogenic right ventricular cardiomyopathy. Am J Hum Genet. 2007;81:964–973. doi: 10.1086/521633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Beffagna G, Occhi G, Nava A, Vitiello L, Ditadi A, Basso C, Bauce B, Carraro G, Thiene G, Towbin JA, Danieli GA, Rampazzo A. Regulatory mutations in transforming growth factor-beta3 gene cause arrhythmogenic right ventricular cardiomyopathy type 1. Cardiovasc Res. 2005;65:366–373. doi: 10.1016/j.cardiores.2004.10.005. [DOI] [PubMed] [Google Scholar]
- 83.Merner ND, Hodgkinson KA, Haywood AF, Connors S, French VM, Drenckhahn JD, Kupprion C, Ramadanova K, Thierfelder L, McKenna W, Gallagher B, Morris-Larkin L, Bassett AS, Parfrey PS, Young TL. Arrhythmogenic right ventricular cardiomyopathy type 5 is a fully penetrant, lethal arrhythmic disorder caused by a missense mutation in the TMEM43 gene. Am J Hum Genet. 2008;82:809–821. doi: 10.1016/j.ajhg.2008.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Norgett EE, Hatsell SJ, Carvajal-Huerta L, Cabezas JC, Common J, Purkis PE, Whittock N, Leigh IM, Stevens HP, Kelsell DP. Recessive mutation in desmoplakin disrupts desmoplakin-intermediate filament interactions and causes dilated cardiomyopathy, woolly hair and keratoderma. Hum Mol Genet. 2000;9:2761–2766. doi: 10.1093/hmg/9.18.2761. [DOI] [PubMed] [Google Scholar]
- 85.Pilichou K, Nava A, Basso C, Beffagna G, Bauce B, Lorenzon A, Frigo G, Vettori A, Valente M, Towbin J, Thiene G, Danieli GA, Rampazzo A. Mutations in desmoglein-2 gene are associated with arrhythmogenic right ventricular cardiomyopathy. Circulation. 2006;113:1171–1179. doi: 10.1161/CIRCULATIONAHA.105.583674. [DOI] [PubMed] [Google Scholar]
- 86.Rampazzo A, Nava A, Malacrida S, Beffagna G, Bauce B, Rossi V, Zimbello R, Simionati B, Basso C, Thiene G, Towbin JA, Danieli GA. Mutation in human desmoplakin domain binding to plakoglobin causes a dominant form of arrhythmogenic right ventricular cardiomyopathy. Am J Hum Genet. 2002;71:1200–1206. doi: 10.1086/344208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Syrris P, Ward D, Evans A, Asimaki A, Gandjbakhch E, Sen-Chowdhry S, McKenna WJ. Arrhythmogenic right ventricular dysplasia/cardiomyopathy associated with mutations in the desmosomal gene desmocollin-2. Am J Hum Genet. 2006;79:978–984. doi: 10.1086/509122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Tiso N, Stephan DA, Nava A, Bagattin A, Devaney JM, Stanchi F, Larderet G, Brahmbhatt B, Brown K, Bauce B, Muriago M, Basso C, Thiene G, Danieli GA, Rampazzo A. Identification of mutations in the cardiac ryanodine receptor gene in families affected with arrhythmogenic right ventricular cardiomyopathy type 2 (ARVD2) Hum Mol Genet. 2001;10:189–194. doi: 10.1093/hmg/10.3.189. [DOI] [PubMed] [Google Scholar]
- 89.McKoy G, Protonotarios N, Crosby A, Tsatsopoulou A, Anastasakis A, Coonar A, Norman M, Baboonian C, Jeffery S, McKenna WJ. Identification of a deletion in plakoglobin in arrhythmogenic right ventricular cardiomyopathy with palmoplantar keratoderma and woolly hair (Naxos disease) Lancet. 2000;355:2119–2124. doi: 10.1016/S0140-6736(00)02379-5. [DOI] [PubMed] [Google Scholar]
- 90.Corrado D, Thiene G. Arrhythmogenic right ventricular cardiomyopathy/dysplasia: clinical impact of molecular genetic studies. Circulation. 2006;113:1634–1637. doi: 10.1161/CIRCULATIONAHA.105.616490. [DOI] [PubMed] [Google Scholar]
- 91.Sen-Chowdhry S, Syrris P, McKenna WJ. Role of genetic analysis in the management of patients with arrhythmogenic right ventricular dysplasia/cardiomyopathy. J Am Coll Cardiol. 2007;50:1813–1821. doi: 10.1016/j.jacc.2007.08.008. [DOI] [PubMed] [Google Scholar]
- 92.van Tintelen JP, Entius MM, Bhuiyan ZA, Jongbloed R, Wiesfeld AC, Wilde AA, van der Smagt J, Boven LG, Mannens MM, van Langen IM, Hofstra RM, Otterspoor LC, Doevendans PA, Rodriguez LM, van Gelder IC, Hauer RN. Plakophilin-2 mutations are the major determinant of familial arrhythmogenic right ventricular dysplasia/cardiomyopathy. Circulation. 2006;113:1650–1658. doi: 10.1161/CIRCULATIONAHA.105.609719. [DOI] [PubMed] [Google Scholar]
- 93.Sen-Chowdhry S, Syrris P, Ward D, Asimaki A, Sevdalis E, McKenna WJ. Clinical and genetic characterization of families with arrhythmogenic right ventricular dysplasia/cardiomyopathy provides novel insights into patterns of disease expression. Circulation. 2007;115:1710–1720. doi: 10.1161/CIRCULATIONAHA.106.660241. [DOI] [PubMed] [Google Scholar]
- 94.Dalal D, James C, Devanagondi R, Tichnell C, Tucker A, Prakasa K, Spevak PJ, Bluemke DA, Abraham T, Russell SD, Calkins H, Judge DP. Penetrance of mutations in plakophilin-2 among families with arrhythmogenic right ventricular dysplasia/cardiomyopathy. J Am Coll Cardiol. 2006;48:1416–1424. doi: 10.1016/j.jacc.2006.06.045. [DOI] [PubMed] [Google Scholar]
- 95.Bauce B, Basso C, Rampazzo A, Beffagna G, Daliento L, Frigo G, Malacrida S, Settimo L, Danieli G, Thiene G, Nava A. Clinical profile of four families with arrhythmogenic right ventricular cardiomyopathy caused by dominant desmoplakin mutations. Eur Heart J. 2005;26:1666–1675. doi: 10.1093/eurheartj/ehi341. [DOI] [PubMed] [Google Scholar]
- 96.Antoniades L, Tsatsopoulou A, Anastasakis A, Syrris P, Asimaki A, Panagiotakos D, Zambartas C, Stefanadis C, McKenna WJ, Protonotarios N. Arrhythmogenic right ventricular cardiomyopathy caused by deletions in plakophilin-2 and plakoglobin (Naxos disease) in families from Greece and Cyprus: genotype-phenotype relations, diagnostic features and prognosis. Eur Heart J. 2006;27:2208–2216. doi: 10.1093/eurheartj/ehl184. [DOI] [PubMed] [Google Scholar]
- 97.Sen-Chowdhry S, Prasad SK, Syrris P, Wage R, Ward D, Merrifield R, Smith GC, Firmin DN, Pennell DJ, McKenna WJ. Cardiovascular magnetic resonance in arrhythmogenic right ventricular cardiomyopathy revisited: comparison with task force criteria and genotype. J Am Coll Cardiol. 2006;48:2132–2140. doi: 10.1016/j.jacc.2006.07.045. [DOI] [PubMed] [Google Scholar]
- 98.Xu T, Yang Z, Vatta M, Rampazzo A, Beffagna G, Pilichou K, Scherer SE, Saffitz J, Kravitz J, Zareba W, Danieli GA, Lorenzon A, Nava A, Bauce B, Thiene G, Basso C, Calkins H, Gear K, Marcus F, Towbin JA. Compound and digenic heterozygosity contributes to arrhythmogenic right ventricular cardiomyopathy. J Am Coll Cardiol. 2010;55:587–597. doi: 10.1016/j.jacc.2009.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Bauce B, Nava A, Beffagna G, Basso C, Lorenzon A, Smaniotto G, De Bortoli M, Rigato I, Mazzotti E, Steriotis A, Marra MP, Towbin JA, Thiene G, Danieli GA, Rampazzo A. Multiple mutations in desmosomal proteins encoding genes in arrhythmogenic right ventricular cardiomyopathy/dysplasia. Heart Rhythm. 2010;7:22–29. doi: 10.1016/j.hrthm.2009.09.070. [DOI] [PubMed] [Google Scholar]
- 100.Hershberger RE, Cowan J, Morales A, Siegfried JD. Progress with genetic cardiomyopathies: screening, counseling, and testing in dilated, hypertrophic, and arrhythmogenic right ventricular dysplasia/cardiomyopathy. Circ Heart Fail. 2009;2:253–261. doi: 10.1161/CIRCHEARTFAILURE.108.817346. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.