

Abstract
Monocular lid closure (MC) causes a profound shift in the ocular dominance (OD) of neurons in primary visual cortex (V1). Anatomical studies in both cat and mouse V1 suggest that large-scale structural rearrangements of eye-specific thalamocortical (TC) axons in response to MC occur much more slowly than the shift in OD. Consequently, there has been considerable debate as to whether the plasticity of TC synapses, which transmit competing visual information from each eye to V1, contributes to the early functional consequences of MC or is simply a feature of long-term deprivation. Here, we used quantitative immuno-electron microscopy to examine the possibility that alterations of TC synapses occur rapidly enough to impact OD after brief MC. The effect of short-term deprivation on TC synaptic structure was examined in male C57BL/6 mice that underwent 3 and 7 d of MC or monocular retinal inactivation (MI) with tetrodotoxin. The data show that 3 d of MC is sufficient to induce substantial remodeling of TC synapses. In contrast, 3 d of MI, which alters TC activity but does not shift OD, does not significantly affect the structure of TC synapses. Our results support the hypothesis that the rapid plasticity of TC synapses is a key step in the sequence of events that shift OD in visual cortex.
Introduction
The mouse has emerged as the preferred species to study the mechanisms of ocular dominance (OD) plasticity in visual cortex. As in other animals with binocular vision, monocular lid closure (MC) causes a rapid shift in cortical OD that degrades vision through the deprived eye (Gordon and Stryker, 1996; Prusky and Douglas, 2003; Frenkel and Bear, 2004). The structural and molecular basis for this plasticity is a question of extraordinary interest.
It is well established that extended periods (weeks to years) of MC lead to dramatic structural rearrangements of thalamocortical (TC) axons in layer 4 of primary visual cortex (V1) (Wiesel, 1982; Antonini and Stryker, 1993; Antonini et al., 1999). However, the relationship between these structural rearrangements and physiological plasticity remains unclear. In kittens, the onset of TC arbor modifications can occur rapidly after MC, but they still lag physiological plasticity by several days (Antonini and Stryker, 1993, 1996; Antonini et al., 1998). In mice, the relationship between anatomical and functional plasticity is even less clear. The OD shift reaches an asymptote with as few as 3 d of MC (Frenkel and Bear, 2004; Liu et al., 2008), several weeks before TC axonal changes are demonstrable (Antonini et al., 1999). Furthermore, current evidence suggests that the effect of MC on individual TC axonal arbors in mice is most likely attributable to the arrested growth of arbors serving the deprived eye rather than a disconnection of these inputs from their synaptic targets in layer 4 (Antonini et al., 1999). Thus, the question has remained open as to whether plasticity of TC synapses is an expression mechanism for the early OD shift in visual cortex.
The aim of the current study was to investigate the possibility of fine structural modifications of TC synapses after brief MC (3–7 d). We used quantitative immuno-electron microscopy (EM) to examine the state of TC synapses in layer 4 of mouse V1. We first confirmed that antibodies against the VGluT2 vesicular glutamate transporter selectively label TC synapses in the mouse, as previously described in the ferret (Nahmani and Erisir, 2005). We then compared the structural consequences of MC with those of monocular retinal inactivation (MI), which eliminates visual input but does not cause deprived-eye response depression (Rittenhouse et al., 1999; Frenkel and Bear, 2004). Our data show that a robust anatomical correlate of the rapid OD shift is a modification of the structure and density of TC synapses in layer 4 of mouse V1. We suggest that modification of TC synaptic transmission is a primary expression mechanism for OD plasticity in the mouse.
Materials and Methods
Animals
Male C57BL/6 mice [postnatal day 21 (P21) to P28] were obtained from Charles River and housed under 12 h light/dark conditions at Massachusetts Institute of Technology (MIT). All animal procedures were performed in accordance with National Institutes of Health guidelines for humane handling of animals and were approved by the Institutional Animal Care and Use Committee at MIT.
Eyelid sutures and intraocular injections
Monocular eyelid sutures were performed as previously described (Frenkel and Bear, 2004). Only animals whose eyelids were completely fused were included in the study. Intraocular tetrodotoxin (TTX) and saline injections were performed daily as follows. To access the vitreous chamber for TTX and saline injections, the temporal portion of the globe was reflected anteriorly by gently pulling a 7-0 silk suture threaded through the conjunctiva and securing it to the operating surface. The conjunctiva was then trimmed using fine angled scissors to expose the sclera. A small puncture was made at the corneoscleral junction into the vitreous chamber with a fine needle. A glass micropipette with a tip diameter of 40–50 μm was attached to a manually driven microinjection apparatus (MMP; World Precision Instruments) and inserted into the vitreous chamber at a depth of ∼2 mm. For delivery of TTX, we used a method that allowed for controlled, sustained delivery of the drug. Approximately 0.3 μl of 2.4 mm TTX or saline in a solution of 1% sodium alginate (1:1 mixture of 4.7 mm TTX and 2% sodium alginate; Sigma-Aldrich) was injected into one eye. Injected eyes were randomized. Before removing the injection pipette, a drop of 0.1 mm CaCl2 solution was placed around the injection site to polymerize the exposed alginate solution, creating a soft gel that facilitated the slow release of TTX into the vitreous chamber. The eye was then rinsed with sterile eye drops and coated with an antibiotic ointment to prevent infection. Experimental animals and littermate controls received injections of TTX or saline, respectively, once per day. Before each injection, pupillary reflexes were tested using direct white light stimulation with a direct ophthalmoscope. Any TTX-injected animals with detectable pupil constriction at any time point up to perfusion were excluded from the study. This method for monitoring chronic retinal blockade was confirmed using diagnostic and electrophysiological methods as described below.
Visually evoked potential recordings and pupillometry
We chronically monitored visually evoked potentials (VEPs) to ensure that cortical input activity was abolished by intraocular TTX and that retinal activity block was well correlated with changes in the direct and consensual pupillary light reflex. Field electrodes were surgically implanted into the binocular zone of V1 as previously described (Sawtell et al., 2003) in a group of young mice (∼P25). Baseline VEPs were recorded in awake, head-restrained mice from the hemisphere contralateral to the eye chosen for injection of either TTX or vehicle, as described by Frenkel and Bear (2004). The first recording was made immediately before the intraocular injection, and VEPs were recorded again after the animals were allowed to recover from isoflurane (2%) for 1–2 and 17–20 h after injection. Direct and consensual pupillary reflexes to white light stimulation were documented for both eyes in conjunction with each postinjection VEP recording session using a Nikon digital camera fitted to a surgical microscope under red light illumination. To avoid confounds associated with stimulus-selective response potentiation, which can be induced in mice by brief presentations of sinusoidal gratings, novel stimulus orientations were used in each subsequent recording session as previously described (Frenkel et al., 2006). Each session was classified as containing a VEP if the peak-to-peak response amplitude was greater than noise, defined as twice the SD of the activity recorded during presentation of a gray screen. If the VEP magnitude was less than noise, it was classified as containing no VEP. The pupillary response was quantified by measuring the dilated pupil radius under red light illumination and the minimal pupil radius achieved during white light stimulation and calculating the percentage change in the radius during light stimulation. The pupil radius was estimated by calculating the center of the circle identified by three points selected on a visible arc of the pupil immediately before and during illumination. All scoring was performed blind to recording session.
Light microscopy and colocalization analysis
Tracer injections.
Mice were anesthetized with a mixture of ketamine and xylazine and prepared for stereotaxic injections as previously described (Frenkel et al., 2006). For binocular V1, the injection site was positioned 3.0 mm lateral from lambda and 300–600 μm below the dural surface. For targeting the binocular segment of the dorsal lateral geniculate nucleus (dLGN), injections were made 2.10 mm posterior to bregma, 2.19 mm lateral from the midline, and 2.5–2.6 mm below the dural surface. A Nanoject II injection system (Drummond Scientific) was used to deliver 100–200 nl of tracer (1% micro-ruby in PBS; 3000 molecular weight; Invitrogen). Animals were allowed to recover for 4–6 d before fixation.
Tissue preparation and immunohistochemistry.
Animals were transcardially perfused with saline (1–2 min) followed by 30–40 ml of 4% paraformaldehyde (PFA) (EM grade; Electron Microscopy Sciences) in 0.1 m phosphate buffer (PB), pH 7.4. Brains were removed and postfixed in 4% PFA overnight at room temperature (RT) or 1–3 d at 4°C. There were no discernible differences between postfix procedures with regard to tissue or staining quality. Sections of V1 (60 μm) were cut from the occipital lobe using a vibratome and collected into 0.01 m PBS, pH 7.2. Free-floating sections were blocked at RT for 30–60 min in PBS containing 0.3% Triton X-100, 0.9% NaCl, 1% bovine serum albumin (BSA), and 0.05% NaN3. Sections were then incubated in guinea pig anti-VGluT2 (1/4000; Millipore Bioscience Research Reagents) in blocking buffer with gentle shaking for 48 h at RT. In some cases, an anti-biotin rhodamine red-X-conjugated IgG (1/125; Jackson ImmunoResearch) was added to the primary anti-VGluT2 mixture. Sections were then rinsed and incubated in donkey anti-guinea pig IgG tagged with Alexa 488 (1/400 in PBS with 1% BSA; Invitrogen) for 2 h at RT. Sections were rinsed, mounted onto Superfrost Plus slides (Thermo Fisher Scientific) in an aqueous-based mounting media (Biomeda GelMount), and coverslipped.
Laser-scanning confocal microscopy.
All images were acquired with an Olympus FluoView 300 laser-scanning confocal microscope and FluoView 500 acquisition software using appropriate filter sets and excitation lasers (channel 1, BA510IF/BA530RIF filters, Ar, 488 nm; channel 2, BA585 filter, HeNe green, 543 nm). Each channel was sequentially acquired to avoid spectral bleed-through. A 10× air objective [Olympus UPLanFl; numerical aperture (NA), 0.30] was used to acquire low-magnification images with the confocal aperture (CA) set to 3 to maximize sensitivity. Low-magnification images were acquired by scanning five optical sections with a step size of 2.0 μm, converted to maximum-intensity Z-projections, and adjusted using ImageJ software for presentation purposes. The layer 4/5 border was identified by differences in the packing density and size of neurons, and found to consistently lie at a depth of one-half the distance between pia and white matter. For colocalization analysis, all images were acquired with a 60× water-immersion objective (Olympus UPLanFl; NA, 1.2) at 3× optical zoom and CA set to 2 with a frame size of 1024 × 1024 pixels (pixel scale, 0.09 × 0.09 μm). A step size of 0.2 μm was used to scan from the surface of each section down to the depth at which VGluT2 signal began to dissipate (∼30–40 optical sections or 6–8 μm of focal depth).
Image segmentation and colocalization analysis.
Image stacks were acquired from labeled sections obtained from three different animals for each injected region and analyzed using MetaMorph software (version 6.1; Molecular Devices). Regions of interest were drawn to include the area immediately surrounding clearly identifiable axonal boutons in maximum-intensity Z-projections, typically within 0.5 μm in any one direction. The bouton regions were then transferred to single-channel image stacks for analysis. Data in each channel were binarized using an intensity threshold so that only the brightest pixels above background comprising puncta and boutons within each optical section were analyzed. For channel 1 (VGluT2), a threshold of 44% was applied to all images (to include pixels >1800 on a scale of 0–4095). For channel 2 [TC and intracortical (IC) boutons], the threshold was determined using median-filtered images. Boutons were more clearly separable from background than puncta and the threshold was determined by including pixel areas whose average intensity was at least 3.5 times greater than that of the surrounding axon. The colocalization module in MetaMorph was used to determine the area of axon pixels (red, R), VGlut2 pixels (green, G), colocalized pixels (yellow, RG), and total pixels within each region in single optical sections. In some figure panels, pixel values were scaled for presentation purposes, but images were always adjusted equally for comparisons.
In previous studies, various correlation coefficients have been used to quantify colocalization data, but determining statistical significance can be somewhat arbitrary. In some instances, a “correlation threshold” or “colocalization index” is empirically determined by performing parallel experiments with markers that are known not to colocalize (Colonnese and Constantine-Paton, 2006) or by randomly shuffling data (Silver and Stryker, 2000). Such procedures can be cumbersome and prone to systematic errors because of the need for multiple thresholding steps and additional immunostaining and imaging parameters. To circumvent these pitfalls, we adopted the use of the Pearson's correlation coefficient for binary variables (ρ, Eq. 1), which has been used as a robust test for temporally correlated events such as the firing probabilities of connected neurons and can be used to determine whether two events are temporally or spatially correlated beyond that expected by chance (Aertsen et al., 1989; Linden et al., 2009) as follows:
 |
P R(P G) is the probability of bouton pixels (VGluT2 pixels) occurring in a defined space, and P RG is the probability of overlap between bouton and VGluT2 pixels within the defined space (P R = n R/n total, etc., where n is the number of pixels). Positive (or negative) departures of ρ from zero indicate colocalization above (or below) that expected by chance (for more details, see supplemental Fig. S1, available at www.jneurosci.org as supplemental material).
Statistical analysis.
As illustrated in supplemental Figure S1 (available at www.jneurosci.org as supplemental material), correlation coefficients were calculated separately for each bouton region (ρ-bouton) or by combining pixels from all bouton regions analyzed in one animal to calculate a single ρ value for each animal (ρ-total). This allowed us to (1) test for differences in colocalization between TC and IC bouton groups and (2) determine whether colocalization within each population of boutons was greater than expected by chance. A Kolmogorov–Smirnov (K-S) test was used to test for differences between the distributions of ρ-bouton values for each group, where p < 0.05 was the criterion for statistical significance. To test whether colocalization was statistically significant, the lower (a) and upper (b) bounds of the 95% confidence interval (CI) for ρ were calculated as previously described by Aertsen et al. (1989) and as shown below. In addition, a p value was calculated for each ρ value. The correlation coefficient can be either positive (correlation) or negative (anticorrelation), and the corresponding p value indicates whether the correlation or anticorrelation is significantly different from zero. Because the biological relevance of anticorrelation in the context of our analysis is uncertain, the following criterion had to be met for colocalization to be considered statistically significant: ρ and a had to be >0, and the p value had to be <0.05.
Correlation coefficients are not normally distributed, so Fisher's transformation (Eq. 2.1) was used to transform them into variables (z 1), which are normally distributed with SD as given in Equation 2.2 (n = total pixel number in region of interest). Next, 95% CIs for z 1 were calculated as shown in Equation 2.3 (z α /2 = 1.96 for a 95% interval). The 95% CIs for each correlation coefficient (ρ-bouton and ρ-total) were calculated as follows:
 |
 |
 |
To obtain the 95% CIs for each ρ value, the inverse Fisher's transformation (Eq. 3.1) was applied to the 95% confidence bounds (a and b) of z 1. The inverse Fisher's transformation is monotonically increasing, so if a < z 1 < b then f(a) < f(z1) = ρ < f(b).
 |
 |
The p values for each correlation coefficient were obtained using Equation 4, where Φ is the cumulative distribution function of the standard normal distribution, n is the total number pixels within the region, and z 1 is from Equation 2.1 above:
 |
Methodological considerations and experimental design.
One potential source of experimental bias lies in the masking of non-neuropil areas because axon labeling is sparse after tracer injections. For example, if few axons are present in the image, colocalization will be artificially low. In contrast, the density of VGluT2 label should be constant across samples so as not to influence the probability of colocalization within any given region. We directly confirmed this by calculating the percentage of green pixels in a 145 × 145 μm region that was centered on “bouton regions” from all depths and animals used for analysis and found no significant difference between groups (p = 0.41, K-S test). Therefore, variations in VGluT2 immunostaining efficiency or image acquisition settings should not influence colocalization. In addition, antibody penetration issues were readily controlled for by restricting analysis to optical sections located <7 μm of the section surface where labeling was the strongest and most consistent. Furthermore, restricting the pixel counts to “bouton regions” eliminates much of the noise attributable to factors that are not of biological interest (for extensive discussion of these and other factors to consider in colocalization analyses with traced axons, see Silver and Stryker, 2000). As an additional precaution against biasing the selection of regions for analysis, all boutons were identified and marked in maximum intensity Z-projections blind to VGluT2 label.
Electron microscopy
Immunohistochemistry, embedding, imaging, and image analysis were performed blind to the history of the tissue.
Perfusions.
Animals were killed as described above and transcardially perfused with artificial CSF (ACSF) containing heparin (10 U/ml; Sigma-Aldrich) until vasculature was flushed clear (<2 min). Perfusion with ACSF was then immediately followed by 100–150 ml of 4% PFA/0.5% glutaraldehyde in PB (EM grade; Electron Microscopy Sciences). Experimental animals were always perfused on the same day as littermate controls. Tissue was postfixed for 1–2 d at RT.
Immunohistochemistry.
Occipital lobe blocks were placed in a vibratome and cut at 60 μm. For each group of animals, yoked, littermate control tissue was always processed with experimental tissue. Sections were subsequently immersed in a 1% sodium borohydride/PB solution, followed by a series of PB rinses until borohydride solution was cleared. Free-floating sections were stored in PBS containing 0.05% NaN3 at 4°C. VGluT2 was visualized using guinea pig anti-VGluT2 (Millipore Bioscience Research Reagents) diluted 1/5000 in PBS, pH 7.4, containing 0.9% NaCl, 1% BSA, and 0.05% NaN3 at RT for 36–48 h. Next, sections of V1 were immersed in biotinylated anti-guinea pig IgG (Vector Laboratories) secondary antibody at 1/100 for 2 h. Sections were then incubated in ABC solution for 2 h, followed by three rinses in PBS for 3 min. Sections were then exposed to a 1% diaminobenzidine (DAB) solution in the presence of 0.003% H2O2 with gentle agitation for 5–8 min.
Embedding.
The embedding procedure was performed blind to the history of the tissue using previously published protocols, thereby eliminating any biases in selecting regions for analysis (Erisir et al., 2001; Nahmani and Erisir, 2005). DAB-treated sections were rinsed in PB and treated with 1% osmium tetroxide in PB for 30–45 min. Osmicated sections were then treated with 4% uranyl acetate in 70% ethanol for 2 h, and they were dehydrated in a series of ethanols. Sections were then dehydrated in acetone and placed in a 1:1 acetone to resin (Epon 812; Electron Microscopy Sciences) mixture for 2 h. Next, sections were placed in full resin for 2 h, flat-embedded between two acetate sheets (Aclar; Ted Pella), and cured in a 60°C oven. Binocular V1 was delineated using a brain atlas based on C57BL/6J mice, and the region of V1 that represents the upper binocular hemifield, which served as the site for previous electrophysiological recordings (Frenkel and Bear, 2004), was identified. Tissue containing all six cortical layers was excised from the flat embed and placed in a plastic capsule, which was then filled with resin. Capsules were cured in a 60°C oven overnight or until polymerized. Embedded sections were traced using a camera lucida, capsule blocks were then trimmed down to a trapezoid that contained only layer 4, and ultrathin sections (∼70 nm) were collected on 400 mesh copper grids (Ted Pella) using an ultramicrotome (Ultracut UCT; Leica). As antibody penetration can be limited to several micrometers from the surface of individual thick vibratome sections, ultrathin sections were collected at a plane that is nearly parallel to the surface, thereby maximizing the width of the tissue that contained labeled regions. Using this cutting approach, we obtained strips of labeled tissue that were at least 50 (and up to 150) μm wide. Data collection was performed within the first 10–100 μm from the tissue–resin transition zone on ultrathin sections. Particular care was paid during ultrathin sectioning to obtain tissue “strips” that contain the tissue–resin transition zone and that span all of layer 4 dorsoventrally. Collecting sections in this manner allowed for ample sample area (∼25 photographs; 20–30 μm2 per electron micrograph) within each ultrathin section containing cortical layer 4. Moreover, the width of the labeled strip enabled us to choose optimal areas for both VGluT2 labeling and ultrastructural preservation. Although the specific dimensions of this labeled strip necessarily varied from section to section, in the rare instance wherein ultrathin sections did not adhere to the above criteria they were omitted from all of our analyses. These strips were then systematically photographed (blind to tissue history) to obtain minimally overlapping images of layer 4, regardless of label or profiles present in the sections.
Ultrathin sections were examined on a JEOL 1010 EM. Digital pictures were captured using a 16 megapixel SIA-12C (sia-cam.com) camera coupled with MaxIm DL CCD software (Diffraction Limited). Individual ultrathin sections were photographed at the tissue–resin interface, in which label is present within a 50- to 150-μm-wide strip along the dorsoventral expanse of layer 4 as described above. Images captured at 12,000× magnification yielded 20–30 μm2 per micrograph, excluding blood vessels, myelinated axons, and somata areas.
EM image analysis and quantification.
Each EM micrograph was examined using Image-Pro Plus, version 5.0, software (Media Cybernetics), at a final screen magnification of 60,000–80,000×. Profiles were identified as synaptic terminals if they met the following criteria: (1) the presence of at least three vesicles, (2) parallel alignment of presynaptic terminal membrane in apposition with that of the postsynaptic membrane (Colonnier, 1968; Peters et al., 1991), and (3) a pronounced asymmetric postsynaptic density (PSD). In each digital picture, vesicle-filled profiles that displayed a synapse at the level of cross section (i.e., synaptic terminals) were identified and classified as containing VGluT2-DAB chromogen (labeled) or unlabeled. For each labeled synaptic profile, the postsynaptic target was classified as a dendritic spine or shaft. Dendritic shafts were defined principally by the presence of an organized arrangement of microtubules or at least one mitochondrion. When there were no microtubules or mitochondria present inside a profile, it was classified as a dendritic spine. Presence of a spine apparatus also aided in identifying dendritic spines.
In each micrograph, the numbers of synaptic labeled and unlabeled terminals were counted, terminal profile areas and synaptic lengths (PSD lengths) were quantified, and the effective sampling area was measured. Effective sampling area was calculated as the total area of an individual micrograph minus the area occupied by cell bodies, blood vessels, and myelinated axons, as these elements may otherwise inflate the amount of neuropil available for synaptic interaction nonuniformly across photographs. Synapses were traced by following the entire length of the postsynaptic density along the parallel membranes of the synapse. In the event that a postsynaptic density was perforated, the synapse was assumed to continue along the length of the perforation. In the analysis of overall layer 4 terminal morphology (i.e., terminals located >200 μm below the tissue–resin interface), additional images were captured from the same material that was prepared for labeled terminal analysis, but from the unstained portions of these identical sections. All terminals that had a synapse at the cross section were traced to obtain terminal area size distributions in layer 4.
Because experimental manipulations can potentially introduce variations in synaptic elements that could bias the sampling procedure, the size–frequency method was used to calculate volumetric densities of labeled and unlabeled synaptic terminal profiles as previously described (Nv = NA/d, where NA is the number of terminals per square micrometer and d is the average synapse length in micrometers (Colonnier and Beaulieu, 1985; DeFelipe et al., 1999; Erisir and Harris, 2003). This quantification method allows for estimations of synaptic terminal densities and has been shown to yield a reliable measure of profile density for asymmetric synapses in cerebral cortex (DeFelipe et al., 1999; Erisir and Harris, 2003). The size–frequency method is sometimes preferred over the optical disector method because it accounts for sampling biases introduced by the fact that the sampled unit (synapse) is several times larger than the tissue section thickness (Merchán-Pérez et al., 2009). Although the volumetric density method yields nearly identical results to the optical disector method applied in the same tissue samples, the former is advantageous because it allows analysis of considerably larger areas in cortex, reducing the variation across samples (DeFelipe et al., 1999; Benes and Lange, 2001). Furthermore, in immunostained ultrathin sections, as used in this study, the sampling is performed at the border of tissue and resin, which progresses in consecutive sections; this makes volumetric density a more feasible method to obtain enough data for meaningful statistical comparisons between experimental and control groups after multiple manipulations (Erisir and Harris, 2003; May et al., 2008). The percentage of VGluT2-labeled synaptic terminals in layer 4 was determined as follows: %Nv labeled = 100 × (Nv labeled/Nv all). To control for variations in signal intensity, tissue obtained from experimental and control animals (yoked controls) were always processed together and all values were normalized to the means of yoked controls. Unpaired t tests (two-tailed) were performed on means of VGluT2-labeled synaptic terminal densities (normalized to yoked controls) to test for statistically significant differences between the means of control and MC groups. K-S tests were used to test for statistically significant differences in the distributions of terminal and PSD sizes between groups. Differences were considered statistically significant if values of p < 0.05.
Results
VGluT2 colocalization
The assumption that VGluT2 is selectively expressed by TC synapses in visual cortex, as shown previously for ferret (Nahmani and Erisir, 2005), was validated in P28 mouse visual cortex. Previous immunofluorescence and EM studies in ferret have shown that VGluT2 puncta colocalize with TC axon terminals, but not IC axon terminals, in layer 4 of V1 (Nahmani and Erisir, 2005). Consistent with these observations, images from the Allen Brain Atlas (http://mouse.brain-map.org) (Lein et al., 2007) show negligible VGluT2 mRNA expression in mouse visual cortex at either P14 or P56, although robust expression is observed in the dLGN. However, there is evidence from both rat and mouse that VGluT2 mRNA is weakly expressed in a population of neurons in some cortical regions at some stages of early postnatal development (De Gois et al., 2005; Nakamura et al., 2007). Although VGluT2 protein has never been shown to be expressed in intracortical synapses in mature visual cortex (Nakamura et al., 2007), it was important to critically evaluate whether VGluT2 could be used as a selective marker for TC axon terminals in layer 4 of mouse V1 as was described in ferret. Our strategy was to label axons of thalamic or cortical origin with anterograde tracer injections, and assess the degree to which labeled boutons also contained immunoreactive VGluT2.
Anterogradely labeled TC axons in binocular V1 (Fig. 1 A1) showed the characteristic arborization pattern previously described for mouse where deep layer 2/3 and layer 4 receive the heaviest input (Antonini et al., 1999). Individual TC axonal segments were marked by boutons resulting in the expected “beads on a string” appearance (Fig. 1 B1–C1) (Nahmani and Erisir, 2005). Direct injection of micro-ruby into layers 2/3 through 6 of binocular V1 (supplemental Fig. S2, available at www.jneurosci.org as supplemental material) also labeled axons in layer 4 that were marked by varicosities resembling boutons (Fig. 1 D1–E1; supplemental Fig. S2, available at www.jneurosci.org as supplemental material). For colocalization analysis, immunofluorescence staining for VGluT2 was performed on sections with labeled axons. Consistent with previous reports that VGluT2 is specific for TC terminals in V1, the pattern of VGluT2 staining closely matched the pattern of TC axonal input (Fig. 1 A2,A3). The density and span of VGluT2 puncta was highest in layer 4 and fell off sharply in layer 5. In contrast, there was a gradient in the intensity of VGluT2 staining going toward the pial surface with an increase again in layer 1, which also receives significant TC input in mouse (Antonini et al., 1999). In addition, VGluT2 puncta were present in layer 6, which receives weak TC input (Sherman and Guillery, 2006).
Figure 1.
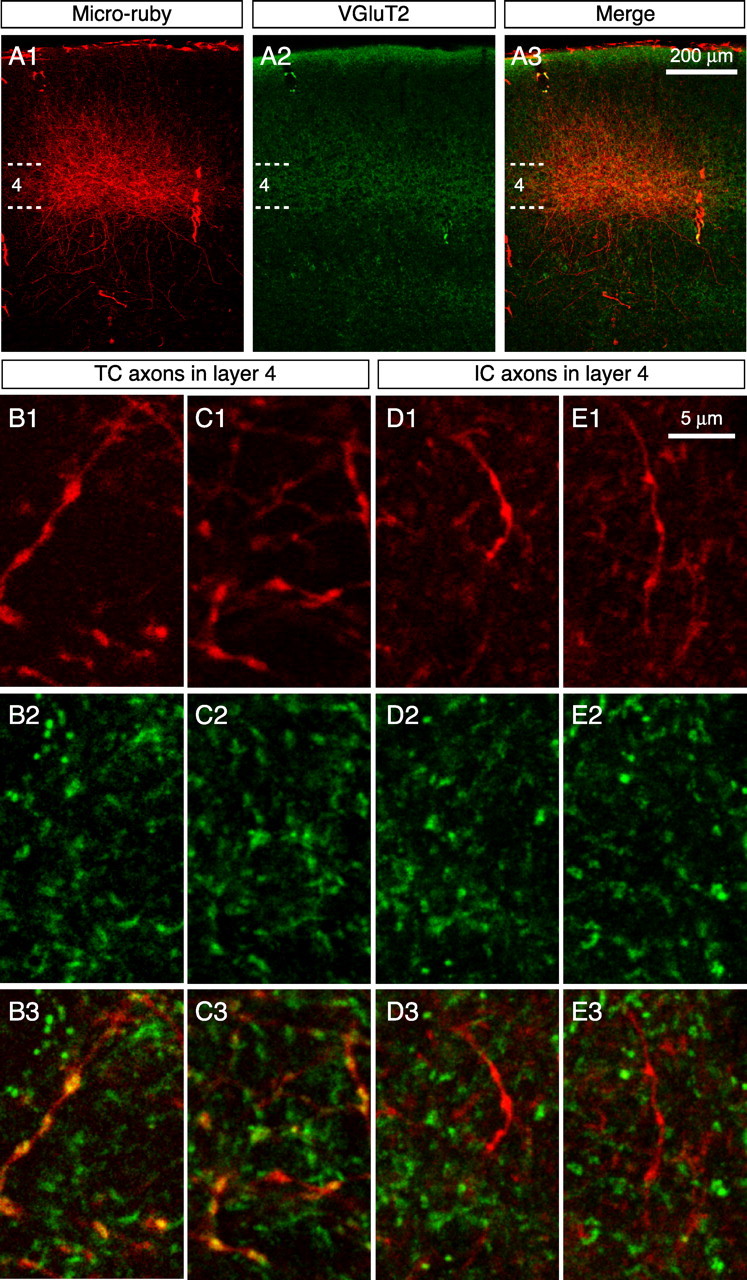
VGluT2 labeling is specific for TC boutons in layer 4 of mouse V1. A1–A3, Confocal images of TC axons labeled with micro-ruby. The dashed lines mark the upper and lower bounds of layer 4. Note the correspondence of TC axonal elaborations with the pattern of VGluT2 staining. Dense labeling of both axons and puncta is evident in layers 2/3 and 4. B–E, Representative high-magnification confocal images of dual labeling with TC boutons (B1–B3, C1–C3) and IC boutons (D1–D3, E1–E3) in layer 4. Note the obvious colocalization (yellow) between TC boutons and VGluT2 puncta (B3, C3), but not IC boutons (D3, E3).
On close inspection of confocal image stacks, there was obvious colocalization of VGluT2 puncta and TC boutons (Figs. 1 B,C, 2 A), but not with IC boutons (Figs. 1 D,E, 2 A). To quantify these observations, data were analyzed using two approaches (Fig. 2 A,B; supplemental Fig. S1, available at www.jneurosci.org as supplemental material). First, we compared the distributions of correlation coefficients calculated for all TC and IC boutons (ρ-bouton) and found a significant difference between groups (Fig. 2 C) (p < 10−13, K-S test; n = 47 TC boutons from 3 animals; n = 50 IC boutons from 3 animals). In addition, 64% of TC boutons exhibited overlap greater than expected by chance (30 of 47 with ρ-bouton > 0, 95% CI lower bounds > 0 and values of p < 0.05). In contrast, 0% of IC boutons exhibited overlap greater than expected by chance (only 3 of 50 with ρ-bouton > 0, and these values had 95% CI lower bounds that were <0 and values of p > 0.05). Second, we calculated a ρ value for each group (ρ-total) by combining pixel data obtained for each bouton region in all images acquired from all animals (supplemental Fig. S1D, available at www.jneurosci.org as supplemental material). Colocalization was greater than expected by chance for TC boutons (ρ-total = 0.226; 95% CI bounds: a = 0.211, b = 0.241; p < 10−179) but not for IC boutons (ρ-total = −0.036; 95% CI bounds: a = −0.053, b = −0.019). Together, these results demonstrate a strong correlation between TC boutons and VGluT2 immunoreactivity, but not IC boutons, and suggest that the VGluT2 antibody can be used to discriminate between these two populations in layer 4 of mouse V1.
Figure 2.
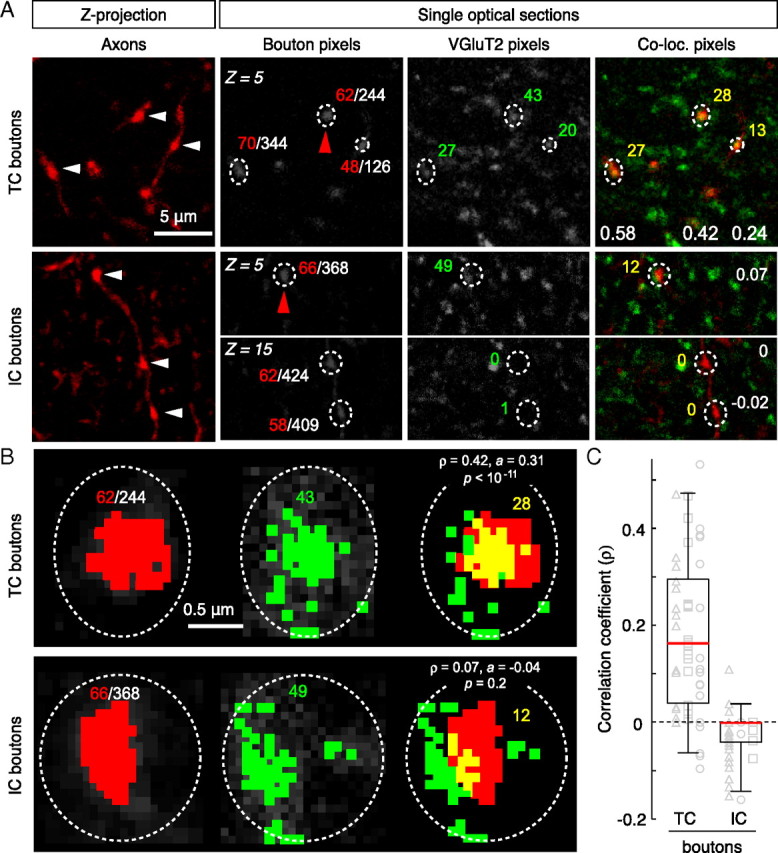
Colocalization analysis in layer 4. A, Examples of micro-ruby-filled TC axonal boutons (arrowheads) in maximum-intensity Z-projections of 15 optical sections (Z = plane number). The grayscale images show single optical sections for each channel and regions of interest with corresponding pixel areas (white denominator: total pixels within region; colored numbers: red, axon; green, VGluT2; yellow, overlap). The white numbers in the colocalized panels are the respective ρ-bouton values. B, Higher magnification views of single boutons from the above panels (indicated by red arrowheads in A). The colored pixels have gray values above the threshold (see Materials and Methods). Note that there is a significant spatial correlation between red and green pixels for the TC bouton, but not the IC bouton. Although ρ is positive for both examples, colocalization is only significant for the TC bouton and is an example of how spurious overlap can be discerned with a relatively straightforward pixel-based analysis. C, Summary of colocalization analysis for each bouton population. The box plots show the distribution of correlation coefficients calculated for each bouton class. The horizontal lines mark the 25th, 50th (red), and 75th percentiles, and the ends of the whiskers mark the 2.5th and 97.5th percentiles (n = 47 TC, n = 50 IC; n = 3 animals per group). The two distributions were statistically different from each other (p < 10−13, K-S test). Correlation coefficients for individual boutons are plotted as gray symbols; data from each animal are represented by different shapes.
Experimental design of quantitative immuno-EM studies
In previous work, chronic VEP recordings from layer 4 were used to monitor input-specific visual responses in the hemisphere contralateral to the deprived eye in monocularly deprived mice (Frenkel and Bear, 2004). Four findings from this and related work contributed to the design of the current study. (1) The contralateral eye provides two to three times more input to binocular V1 than the ipsilateral eye (Coleman et al., 2009). (2) After 3 d of MC, the OD shift occurs solely by depression of deprived-eye responses. Because closure of the contralateral eyelid has no effect on the ipsilateral eye responses, there is a loss of total TC synaptic drive in V1 contralateral to the deprived eye after 3 d of MC. (3) After 7 d of MC, the compensatory potentiation of ipsilateral eye responses leads to full recovery of total TC synaptic drive. (4) Intraocular retinal activity blockade fails to induce synaptic depression (Frenkel and Bear, 2004) and prevents the depression of deprived-eye responses after 1–3 d of MC (Rittenhouse et al., 1999; Heynen et al., 2003).
Although VGluT2 immunostaining allows us to resolve TC synapses at the EM level (Fig. 3 A), it does not enable us to determine whether a given synapse serves the deprived (contralateral) or nondeprived (ipsilateral) eye. Thus, our experiments were designed to detect changes in total TC input after MC. Based on the considerations listed above, we hypothesized that there would be a detectable structural change in TC synapses after 3, but not 7 d of MC, and that simply abolishing retinal activity in the contralateral eye with TTX would not alter TC input. The design of experiments to test these hypotheses is shown in Figure 3, B and C.
Figure 3.
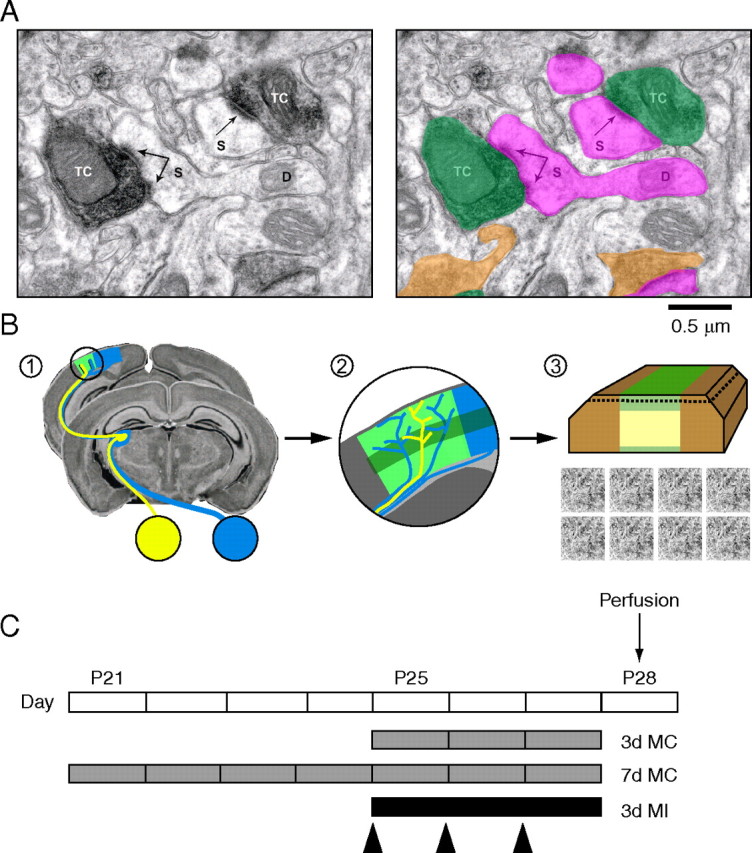
VGluT2 immuno-EM and experimental design summary. A, High-magnification micrograph showing VGluT2-labeled synaptic terminals (TC/green highlight, VGluT2-positive terminal; orange highlight, unlabeled terminal; S/pink highlight, dendritic spine head; D, dendritic shaft; arrows are pointing to PSDs). VGluT2 label was confined to presynaptic terminals, the majority of which made synaptic contact with dendritic spines. For labeled synaptic terminals (n = 634), 77.4 ± 2.6% contacted spines and 22.5 ± 2.6% contacted dendritic regions. For unlabeled synaptic terminals (n = 1341), 71.1 ± 4.1% contacted spines and 28.7 ± 4.1% targeted dendritic shafts. VGluT2-labeled synaptic terminals represented a distinct subclass in layer 4 (supplemental Fig. S3A, available at www.jneurosci.org as supplemental material). B, Diagram summary of the sampling methods used for quantifying the effects of monocular deprivation on TC synaptic input to layer 4. Step 1, Coronal sections through binocular V1 (circled area) were cut from occipital lobes. The schematic of the mouse primary visual pathway shows how information from the two eyes is transmitted to segregated regions within dorsal lateral geniculate nucleus and is then relayed to the binocular segment of V1 (green patch) via TC afferents. Yellow indicates the ipsilateral (nondeprived eye) pathway and blue indicates the contralateral (deprived eye) pathway. Step 2, Before embedding for EM, immunohistochemistry (IHC) for VGluT2 (darker band indicates higher density staining in layer 4) was performed on vibratome sections prepared from MC animals and yoked, littermate controls. Step 3, Layer 4 was then blocked and processed for EM. The portion of tissue that remained unlabeled because of limited antibody penetration is indicated in light yellow. Ultrathin sections cut near-parallel to the surface contain the labeled portion at the tissue–resin interface (indicated by the dashed line). Several nonoverlapping images (total area, >1000 μm2) were then acquired from ultrathin tissue close to the tissue–resin interface and analyzed blind to condition. C, Experimental timeline showing ages at which MC, eyeball injections (arrowheads), and perfusions were performed.
TC synapse density and size are reduced after 3 d of MC
We first examined the effects of 3 d MC on TC input. We observed a qualitative difference in the prevalence of VGluT2-labeled synapses between 3 d MC and control hemispheres (Fig. 4). Quantitative analysis revealed a significant decrease (∼30%) in the prevalence of VGluT2-labeled synapses after 3 d of MC relative to control hemispheres (Fig. 5 A) (control, n = 7; 3 d MC, n = 5; p = 0.0009, unpaired t test). The distribution of PSD lengths of remaining TC synaptic terminals in 3 d MC hemispheres was significantly different from that of control hemispheres (Fig. 5 B) (p = 0.02, K-S test; n = 306 control synapses; n = 283 MC synapses). Furthermore, the distributions of cross-sectional areas of VGluT2-labeled synaptic terminals were significantly different in control and 3 d MC hemispheres (Fig. 5 C) (p = 0.0002, K-S test; n = 300 control terminals; n = 278 MC terminals). In MC hemispheres, only 7% of the TC synaptic terminal cross-sectional areas were >0.6 μm2, as opposed to 16% in control hemispheres. Together, these results show that a significant fraction of TC synapses are lost in layer 4 after 3 d of MC and that the remaining TC terminals and synapses are smaller in size relative to controls.
Figure 4.

VGluT2-labeled synaptic terminal density after 3 d MC. Example low-magnification EM micrographs from a control (normal) and 3 d MC case. The diagram below each image highlights identified synaptic terminals and PSDs in each case. Note the reduction in both the number and size of VGluT2-labeled terminals in the 3 d MC case.
Figure 5.
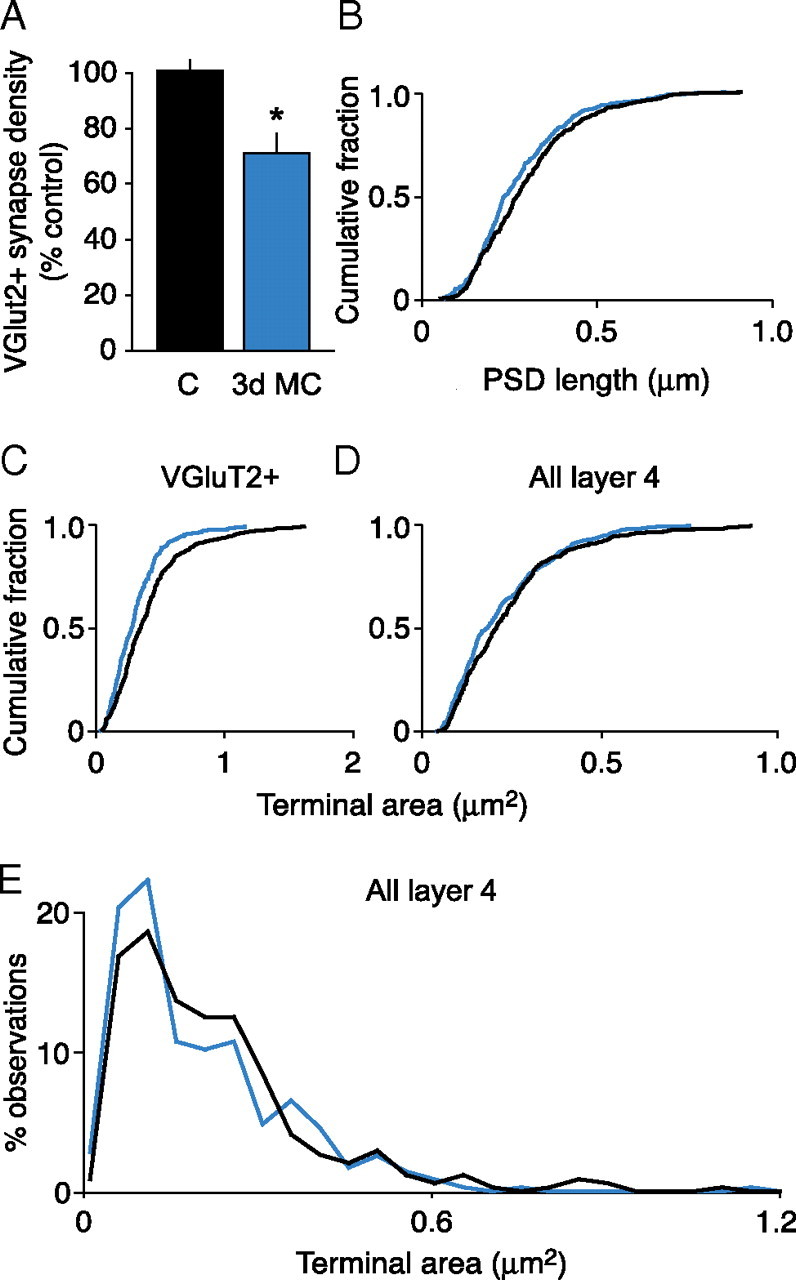
The effect of 3 d MC on the density and terminal morphology of VGluT2-labeled synapses. A, Summary of labeled (VGluT2+) synaptic density (mean ± SEM). The asterisk in A indicates statistically significant difference from control (p = 0.0002, unpaired t test) (C, control). B–D, Cumulative probability histograms of PSD lengths, VGluT2-positive terminal size, and terminal size of all layer 4 terminals (black, control; blue, MC). The distributions of PSD lengths (B) and synaptic terminal sizes (C) were significantly different between 3 d MC and control animals (p = 0.02, K-S test). The distributions of synaptic terminal sizes in layer 4 (D) were significantly different between 3 d MC (n = 355 terminals) and control (n = 345 terminals) groups, which was attributable to a loss of larger-sized terminals (p = 0.03, K-S test). The terminals in D were sampled in layer 4, >200 μm from the surface of vibratome sections cut from control (non-MC) and 3 d MC brains. E, Frequency distribution histogram of data shown in D.
To ensure that the decrease in labeled terminals shown in Figures 4 and 5 was not attributable to immunostaining artifacts (e.g., poor or differential antibody penetration in control vs MC tissues), we examined the terminal size distributions of layer 4 terminals >200 μm below the tissue–resin interface. In deeper portions of tissue, immunostaining is greatly diminished or absent, but the ultrastructure preservation is overall better than the surface portions of the vibratome sections. We reasoned that, since TC synaptic terminals are among the largest in layer 4 (Ahmed et al., 1997; Erisir and Dreusicke, 2005), we should be able to detect a shift in the distribution of the cross-section areas of layer 4 terminals if TC synapses were indeed affected by MC.
First, we confirmed that the distributions of VGluT2-labeled terminals in layer 4 of mouse V1 were different from unlabeled (VGluT2-negative) terminals in the same sections. Similar to what has been previously reported for ferret, we found that VGluT2-labeled synaptic terminals represent a subpopulation of larger-sized terminals in layer 4 (supplemental Fig. S3A, available at www.jneurosci.org as supplemental material). Second, we compared the distribution of all terminals (regardless of origin) within layer 4 in deeper sections with that of VGluT2-labeled terminals. The results of this analysis showed again that VGluT2 antibody recognizes a specific subpopulation of larger terminals in layer 4 in control and MC tissues (supplemental Fig. S3B,C, available at www.jneurosci.org as supplemental material). Furthermore, the population of larger VGluT2-labeled terminals (supplemental Fig. S3B,C, dashed lines, available at www.jneurosci.org as supplemental material) was clearly shifted after 3 d of MC, whereas the effect on the entire population of synaptic terminals was less pronounced (supplemental Fig. S3B,C, solid lines, available at www.jneurosci.org as supplemental material). Finally, we compared the distributions of all layer 4 terminals from two control and two 3 d MC brains and found that the distribution of all terminals in layer 4 was shifted toward smaller terminal areas after MC (Fig. 5 D,E) (p = 0.03; n = 345 control terminals; n = 355 MC terminals). Together, these results confirm that there is an effect of MC on some fraction of larger presynaptic terminals in layer 4 and strongly suggest that the affected population is of TC origin.
Retinal activity is required for the effects of MC on VGluT2-labeled synaptic input
To determine whether the anatomical effects of 3 d MC share a common cellular mechanism with deprived-eye response depression, we performed monocular injections of TTX or saline in a second group of animals. In contrast to the 3 d MC results, 3 d MI had no measurable effect on the prevalence of VGluT2-labeled synapses (Fig. 6 A) (p = 0.67, unpaired t test; n = 3 saline hemispheres; n = 5 TTX hemispheres). Furthermore, there was no measurable effect of MI on VGluT2-labeled synaptic terminal morphology. PSD length was unaffected by MI with TTX as the distributions of lengths were not significantly different between the two groups (Fig. 6 B) (p = 0.14, K-S test; n = 149 saline synapses; n = 145 TTX synapses). In addition, the distributions of synaptic terminal cross-section areas were not different between TTX and saline-control animals (Fig. 6 C) (p = 0.18, K-S test; n = 137 saline terminals; n = 199 TTX terminals). Thus, there was no measurable TC synapse loss or change in morphology to TC synaptic terminals after 3 d MI.
Figure 6.
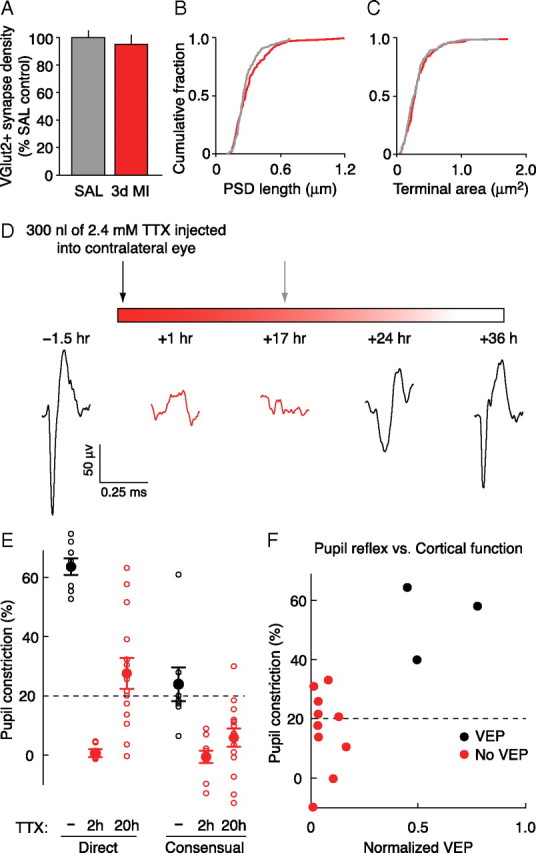
The effect of 3 d MI on VGluT2-labeled synapses and the relationship between the direct pupillary reflex and VEPs. A, Summary of VGluT2+ synaptic density (mean ± SEM) (C, control; SAL, saline). B, C, Cumulative probability histograms of PSD lengths (B) and VGluT2-positive terminal size (C) (gray, saline; red, TTX). The distributions of PSD lengths and synaptic terminal sizes were not significantly different between 3 d MI and control animals (p > 0.05, K-S test). D, Experimental timeline and representative traces of the average field potentials evoked by a patterned visual stimulus viewed through the TTX-injected eye. Recordings were made in the binocular region of V1 contralateral to the injected eye. In this example, evoked cortical activity returned after 24 h and was fully restored by 36 h. For the MI experiments used in the EM analysis, a second TTX injection (gray arrow) would have been administered before the appearance of visually evoked activity in V1, maintaining full retinal blockade. E, Summary of direct and consensual pupillary reflexes with no TTX (black) and 2 or 20 h after TTX (red). The magnitude of pupillary constriction is represented by the percentage change in the pupil radius on direct light stimulation of a dark-adapted eye. Thus, 0% indicates a nonreflexive pupil (tonically dilated). Pupillary responses >20% would have been excluded from the MI experiments (E, F, dashed line). Each open circle represents an individual animal, and the filled circles show group means ± SEM [ipsilateral (direct) pupil constriction values: uninjected, 63 ± 3%, n = 8; 2 h TTX, 0 ± 1%, n = 10; 20 h, 27 ± 5%, n = 14; contralateral (consensual) pupil constriction values: uninjected, 24 ± 6%, n = 8; 2 h TTX, −1 ± 2%, n = 10; 20 h, 6 ± 3%, n = 15]. F, Constriction of the TTX-injected eye evoked by direct stimulation plotted as a function of cortical VEPs (17–22 h after injection, normalized to pre-TTX baseline values). Note that the absence of a direct pupillary reflex in the TTX-injected eye, as described in Materials and Methods, is sufficient to confirm retinal blockade as assessed by the presence of a categorical VEP. Recorded field potentials were classified as containing “no VEP” if the scored amplitude was <2 SDs of the noise (field potentials recorded in response to gray screen). Responses were recorded ∼17–22 h after TTX.
In several previous studies (Frenkel and Bear, 2004; Linden et al., 2009), TTX injections into the vitreous humor were used to inactivate retinal ganglion cells (confirmed by the absence of light-evoked activity in the dorsal lateral geniculate nucleus) (Linden et al., 2009). However, unlike previous studies, we did not have a functional measure of retinal blockade in our experimental animals. A convenient way to assess the duration of retinal inactivation is to monitor the direct pupillary light reflex—the constriction of the pupil in response to illumination of the same eye. When the retina is inactivated, the pupil dilates and no longer responds to direct illumination. However, if TTX were to spread from the vitreous humor to the anterior chamber of the eye, this measure could be corrupted by paralysis of the sphincter pupillae muscle. This potential confound is avoided by using the consensual pupillary light reflex—the constriction of the pupil in the uninjected (contralateral) eye when the injected (ipsilateral) eye is illuminated. In practice, however, this measure is cumbersome for routine monitoring because the response is smaller and more difficult to assess qualitatively in the diminutive, laterally positioned mouse eye. Therefore, we felt it was worthwhile to validate use of the direct reflex by quantitatively comparing this measure of retinal inactivation with the consensual reflex and light-evoked activity in visual cortex.
Another group of mice were prepared at P25 for chronic VEP recordings as described by Frenkel and Bear (2004). VEPs evoked in visual cortex by contralateral eye stimulation were collected before and after TTX injections of the same eye (Fig. 6 D). A single injection of TTX was sufficient to completely eliminate VEPs for 17–22 h (n = 9 animals). Full recovery was usually observed within 36 h (Fig. 6 D). The TTX treatment also completely eliminated both the direct and consensual pupillary light reflexes (Fig. 6 E). By 20 h after injection, there was a partial recovery, on average, of both pupillary light reflexes, which was more pronounced for the direct reflex. This observation is consistent with that of Stryker and Harris (1986), who also reported that the direct reflex returns earlier than the consensual.
Next, we asked whether the absence of a direct pupillary reflex in TTX-injected eyes correlated with an absence of visually driven activity in cortex. As shown in Figure 6 F, all injected eyes lacking a robust direct pupillary reflex 17–22 h after TTX injection failed to elicit a VEP (defined as a response >2 SD of the noise) in V1. Although some animals did recover both a VEP and a direct pupillary reflex within this time window (Fig. 6 F), it is important to note that they would have been excluded from analysis in this and previous studies.
These results demonstrate two important points. First, a single TTX injection using our method is sufficient to completely block ascending retinal activity for at least 17–22 h, the time at which subsequent injections have been made to achieve chronic blockade over a period of 3 d (Fig. 3 C). Second, the direct pupillary reflex is a conservative measure of the duration of block of retino-geniculo-cortical activity. These experiments confirm that chronic retinal activity blockade was achieved in the current and previous studies that have used intraocular TTX injections in mice (Frenkel and Bear, 2004; Linden et al., 2009).
TC synaptic density and size appear normal after 7 d of MC
We next examined the effects of 7 d MC on TC synaptic input. In contrast to 3 d of MC, there were no qualitative or quantitative changes in the prevalence of VGluT2-labeled synapses after 7 d of MC (Fig. 7 A) (p = 0.50, unpaired t test; n = 3 control hemispheres; n = 4 MC hemispheres). Furthermore, the overall morphology of VGluT2-labeled synaptic terminals after 7 d MC was indistinguishable from controls as the distributions of both PSD lengths and terminal cross-section areas were not significantly different (Fig. 7 B,C) (PSD length: p = 0.74, K-S test; n = 134 control synapses; n = 134 MC synapses; terminal area: p = 0.15, K-S test; n = 143 control terminals; n = 276 MC terminals). Hence, after 7 d of MC, TC synapse density and synaptic terminal morphology were indistinguishable from those of control animals.
Figure 7.
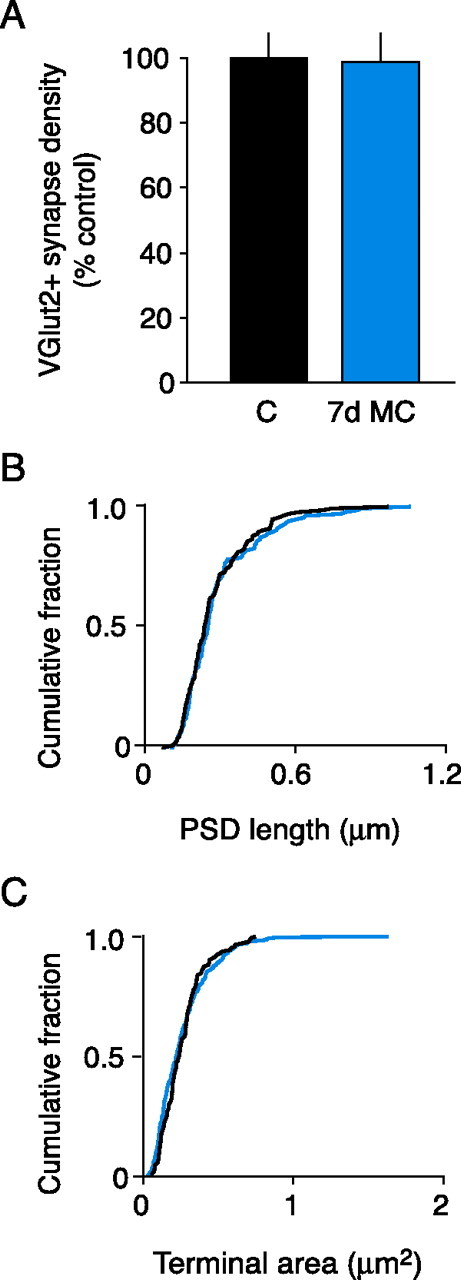
The effect of 7 d MC on VGluT2-labeled synapse density and morphology. A, Summary of VGluT2+ synaptic density (mean ± SEM). B, C, Cumulative probability histograms of PSD lengths (B) and VGluT2-positive terminal size (C) (black, control; blue, MC). The distributions of PSD lengths and synaptic terminal sizes were not significantly different between 7 d MC and control animals (p > 0.05, K-S test).
Discussion
Two important findings emerged from this study: (1) VGluT2 protein is a reliable marker for TC terminals in layer 4 of mouse V1 at ∼P30, and (2) the structural modification of TC synapses in response to MC is much faster than has been previously thought. Although the earliest changes to gross TC axonal structure reportedly occur days (kitten) or weeks (mouse) after the maximal OD shift, by examining ultrastructure we have demonstrated that TC synaptic remodeling is actually coincident with the maximal OD shift.
The current findings provide direct anatomical evidence that TC input is altered by brief periods of MC in vivo and that the remodeling of this input closely mirrors physiological OD shifts. The decrease in total TC synapse density we observed after 3 d MC coincides with maximal depression of responses to deprived-eye and binocular stimulation (Frenkel and Bear, 2008), whereas the apparently normal density after 7 d of MC coincides with the compensatory recovery of visual responsiveness resulting from potentiation of nondeprived-eye input. Thus, our findings show that modification of TC synaptic transmission is a likely substrate for rapid OD plasticity.
Recent reports have shown that VGluT1 and VGluT2 expression patterns can be highly variable through development and that the complementary expression pattern of their mRNAs in cortex and thalamus, respectively, may be less clear-cut than previously thought (De Gois et al., 2005; Barroso-Chinea et al., 2007; Nakamura et al., 2007). De Gois et al. (2005) reported coexpression of VGluT1 and VGluT2 in cultured rat cortical neurons, a preparation that is tenuously related to the situation in situ because expression levels depend on culture conditions, cellular composition, and time in vitro. However, they did find weak (orders of magnitude less than VGluT1) expression of VGluT2 mRNA in rat auditory and parietal cortex, raising the possibility that some small fraction of cortical neurons express VGluT2 (in addition to VGluT1) in situ. In a more recent study, Nakamura et al. (2007) examined VGluT1 and VGluT2 expression in mouse neocortex. One important finding of this study was significant variation in the degree of colocalization across cortical areas during early postnatal development (<P14). However, neither study provided evidence that these two proteins are coexpressed at the same synapses nor that any VGluT2-positive synapses are of intracortical origin in visual cortex older than P21.
We performed experimenter-blind colocalization studies to address the substantive issue of whether the extremely weak and sparse expression of VGluT2 mRNA reported in cortex is enough to cause VGluT2 protein immunoreactivity at synapses other than those of thalamic origin. We found that, in mouse visual cortex at 4–5 weeks of age, TC synapses contained immunoreactive VGluT2, whereas intracortical synapses did not. This result is consistent with the original observation that thalamic lesions in rats deplete VGluT2 immunoreactivity in cortex without affecting VGluT1 (Fujiyama et al., 2001), as well as the fact that VGluT2 staining pattern in mouse barrel cortex precisely matches the distinctive pattern of TC axonal input to this area (She et al., 2009).
Interpretation of our results would also be complicated if VGluT2 protein expression at TC terminals is itself regulated by experience. However, since VGluT2+ terminals were not affected by either 3 d MI or 7 d MC, this possibility seems remote. Moreover, direct analysis of colocalization did not reveal any significant effect of 3 d of MC on the expression of VGluT2 protein in TC boutons (supplemental Fig. S4, available at www.jneurosci.org as supplemental material). Together, our results considered with available data in the literature support the conclusion that VGluT2 is selective for TC synapses in layer 4 of mouse V1 and that the effects of MC on this population of synapses is indicative of experience-dependent modification of the structure of TC input.
Axon-labeling methods, suitable only for detecting gross changes in axonal arbors, have thus far failed to reveal rapid TC changes in the mouse (Antonini et al., 1999). In considering the reasons for this apparent discrepancy, it is interesting to note that the territory occupied by an individual mouse TC axon is relatively large (Antonini et al., 1999) and is likely to provide input to neurons with varying degrees of binocularity, including monocular neurons. Depression of deprived-eye responses fails to occur in neurons that lack converging input from the nondeprived eye (Mrsic-Flogel et al., 2007; Smith et al., 2009). Thus, synapses along branches of an eye-specific axon may display a mosaic of changes depending on the postsynaptic target. Accordingly, the fate of all branches (and synapses) of a single axon after MC could be more varied, thereby rendering changes to overall axonal complexity more discrete and difficult to detect in the mouse than in the cat.
Interestingly, the decrease and apparent recovery in total TC synapse density after 3 and 7 d of MC, respectively, correlate remarkably well with the time course of deprived-eye depression and nondeprived-eye potentiation in mouse (Frenkel and Bear, 2004; Cho et al., 2009). Although we were unable to distinguish between deprived and nondeprived TC synapses, our data are consistent with the model proposed in Figure 8. Based on our current observations and previous reports that the retraction of deprived TC arbors precedes the expansion of nondeprived arbors (Antonini and Stryker, 1993, 1996), we speculate that the net loss of TC synapses after 3 d of MC reflects the elimination of a fraction of deprived-eye synapses, which contributes to functional depression. The morphological alterations to the remaining terminals in deprived cortex after 3 d of MC are reminiscent of changes to deprived TC terminals that occur after long-term MC in the cat (Tieman, 1984), thereby suggesting that both synaptic elimination and weakening of remaining TC synapses contribute to the rapid response depression in mouse V1. The apparent recovery of net TC synapse density after 7 d could be attributable to the addition of synaptic terminals serving the nondeprived eye, thereby contributing to the compensatory response potentiation. Furthermore, the finding that the MC-induced loss of TC synapses is abrogated by retinal inactivation strongly suggests that, like the functional consequences of deprivation, structural rearrangements of TC input are driven by decorrelated activity in the deprived-eye channel (Rittenhouse et al., 1999; Frenkel and Bear, 2004; Smith et al., 2009). Together with recent electrophysiological evidence that the weakening of TC transmission can account for deprived-eye depression (Khibnik et al., 2010), these findings point to common cellular mechanisms for functional and structural plasticity of TC input.
Figure 8.

Proposed model for how the experience-dependent structural remodeling of TC synapses maps onto physiological OD plasticity in mouse V1. A, Schematic of the primary visual pathway in mouse. B, Previous studies used chronic VEP recordings to monitor contralateral-eye (blue) and ipsilateral-eye (yellow) responses in V1 contralateral to the deprived eye (DE), which is dominated by contralateral input. Changes in responsiveness to binocular stimulation (B, dashed lines) are detected contralateral to the DE after 3 and 7 d of MC. Total TC synaptic drive in V1 is reduced by one-third after 3 d of MC and is caused by DE depression. After 7 d of MC, total TC synaptic drive is restored to pre-MC levels because of the subsequent potentiation of non-DE responses (yellow bars) [schematized VEP data adapted from Frenkel and Bear (2004)]. C, These changes are mirrored by the structural remodeling of net TC synaptic input detected in layer 4 (Figs. 5 A, 7 A). The diagram illustrates how MC-induced TC synaptic remodeling could occur in layer 4. Because the majority of neurons in mouse V1 are binocularly responsive and the relative magnitude of the input of each eye is reflected in the relative density of afferent input (Coleman et al., 2009), eye-specific input to a dendritic subregion of a binocular neuron is depicted by the relative number of TC synapses (ipsilateral TC input, yellow; contralateral TC input, blue). We propose that the depression of DE responses after 3 d of MC results from the weakening of layer 4 TC synapses, a subset of which is then targeted for elimination. Presynaptic terminals that provide driving input from DE axons (blue) have retracted by the time maximal depression has occurred. After 7 d of MC, two forms of presynaptic change that mirror physiological plasticity could account for deprivation-enabled potentiation: (1) The seemingly normal density of TC synapses could be explained by the addition of new synapses formed along TC axons serving the non-DE, and (2) the growth of existing non-DE synapses may also occur to restore the normal distribution of terminal size.
Our findings fit with the growing body of evidence implicating structural remodeling as a substrate for cortical circuit plasticity (Holtmaat and Svoboda, 2009). Rapid morphological plasticity of postsynaptic spines has already been implicated in the early functional consequences of MC in mice, and axon (and bouton) dynamics closely match those of dendritic spines in both developing and adult sensory cortices (Gogolla et al., 2007). Axons in monkey V1 undergo dynamic changes within 24 h of focal retinal lesions, suggesting that experience-dependent structural plasticity of axons can contribute to the rapid functional reorganization of cortical circuitry (Yamahachi et al., 2009). The observation that as little as 2 d of MC is accompanied by an elevation of spine motility in layer 1 of binocular V1 (Mataga et al., 2004; Oray et al., 2004), which is heavily innervated by TC axons, may reflect a turnover in some fraction of TC synapses. As reported for kitten after >1 year of MC (Tieman, 1984), our finding that PSD length is altered by MC is consistent with a coordinated change in postsynaptic spines associated with deprived TC synapses, as there is a strong correlation between PSD size and spine volume in cortical tissue (Harris and Stevens, 1989; Arellano et al., 2007).
Our data are consistent with the view that the structural remodeling of TC synapses is the culmination of the sequence of molecular and cellular events that underlie the earliest phases of MC-induced response depression (Bear and Rittenhouse, 1999; Heynen et al., 2003). The loss of TC synapses is preceded by input-specific decreases in AMPAR-mediated synaptic transmission, which have been shown to underlie synaptic depression after MC (Heynen et al., 2003; Crozier et al., 2007; Yoon et al., 2009). A relationship between synaptic depression via receptor loss and presynaptic structural plasticity has been elegantly demonstrated at peripheral synapses (Colman et al., 1997; McCann et al., 2008) and recent studies suggest similar relationships exist at central synapses. For example, functional and morphological correlates of synapse elimination can be detected within hours of long-term depression (LTD) induction in hippocampal slices (Bastrikova et al., 2008; Becker et al., 2008) and the rapid (within hours) structural rewiring of presynaptic inputs to form new functional synapses has been demonstrated in vitro (Nagerl et al., 2007). Recent in vivo evidence also shows that rapid morphological plasticity is a substrate for increasing functional synapses within hours (Zito et al., 2009). A challenge for future studies will be to determine whether the mechanisms of LTD are required for structural plasticity in V1 in vivo and whether the induction of LTD in the visual pathway is accompanied by the structural remodeling of TC input.
Footnotes
This research was supported by National Institutes of Health Grants F31NS059189 (M.N.), K25NS052422 (R.H.), RO1EY12138 (A.E.), and partly by RO1EYO12309 (M.F.B.). We thank Sam Cooke, Mikhail Frenkel, Lena Khibnik, Monica Linden, and Marshall Hussain Shuler for helpful discussions and comments. We also thank Suzanne Meagher, Kathleen Oram, Bonnie Shepard, Erik Sklar, and Vivian Tang for administrative and technical support.
References
- Aertsen AM, Gerstein GL, Habib MK, Palm G. Dynamics of neuronal firing correlation: modulation of “effective connectivity”. J Neurophysiol. 1989;61:900–917. doi: 10.1152/jn.1989.61.5.900. [DOI] [PubMed] [Google Scholar]
- Ahmed B, Anderson JC, Martin KA, Nelson JC. Map of the synapses onto layer 4 basket cells of the primary visual cortex of the cat. J Comp Neurol. 1997;380:230–242. [PubMed] [Google Scholar]
- Antonini A, Stryker MP. Rapid remodeling of axonal arbors in the visual cortex. Science. 1993;260:1819–1821. doi: 10.1126/science.8511592. [DOI] [PubMed] [Google Scholar]
- Antonini A, Stryker MP. Plasticity of geniculocortical afferents following brief or prolonged monocular occlusion in the cat. J Comp Neurol. 1996;369:64–82. doi: 10.1002/(SICI)1096-9861(19960520)369:1<64::AID-CNE5>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- Antonini A, Gillespie DC, Crair MC, Stryker MP. Morphology of single geniculocortical afferents and functional recovery of the visual cortex after reverse monocular deprivation in the kitten. J Neurosci. 1998;18:9896–9909. doi: 10.1523/JNEUROSCI.18-23-09896.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonini A, Fagiolini M, Stryker MP. Anatomical correlates of functional plasticity in mouse visual cortex. J Neurosci. 1999;19:4388–4406. doi: 10.1523/JNEUROSCI.19-11-04388.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arellano JI, Benavides-Piccione R, Defelipe J, Yuste R. Ultrastructure of dendritic spines: correlation between synaptic and spine morphologies. Front Neurosci. 2007;1:131–143. doi: 10.3389/neuro.01.1.1.010.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barroso-Chinea P, Castle M, Aymerich MS, Pérez-Manso M, Erro E, Tuñon T, Lanciego JL. Expression of the mRNAs encoding for the vesicular glutamate transporters 1 and 2 in the rat thalamus. J Comp Neurol. 2007;501:703–715. doi: 10.1002/cne.21265. [DOI] [PubMed] [Google Scholar]
- Bastrikova N, Gardner GA, Reece JM, Jeromin A, Dudek SM. Synapse elimination accompanies functional plasticity in hippocampal neurons. Proc Natl Acad Sci U S A. 2008;105:3123–3127. doi: 10.1073/pnas.0800027105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bear MF, Rittenhouse CD. Molecular basis for induction of ocular dominance plasticity. J Neurobiol. 1999;41:83–91. doi: 10.1002/(sici)1097-4695(199910)41:1<83::aid-neu11>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- Becker N, Wierenga CJ, Fonseca R, Bonhoeffer T, Nägerl UV. LTD induction causes morphological changes of presynaptic boutons and reduces their contacts with spines. Neuron. 2008;60:590–597. doi: 10.1016/j.neuron.2008.09.018. [DOI] [PubMed] [Google Scholar]
- Benes FM, Lange N. Two-dimensional versus three-dimensional cell counting: a practical perspective. Trends Neurosci. 2001;24:11–17. doi: 10.1016/s0166-2236(00)01660-x. [DOI] [PubMed] [Google Scholar]
- Cho KK, Khibnik L, Philpot BD, Bear MF. The ratio of NR2A/B NMDA receptor subunits determines the qualities of ocular dominance plasticity in visual cortex. Proc Natl Acad Sci U S A. 2009;106:5377–5382. doi: 10.1073/pnas.0808104106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman JE, Law K, Bear MF. Anatomical origins of ocular dominance in mouse primary visual cortex. Neuroscience. 2009;161:561–571. doi: 10.1016/j.neuroscience.2009.03.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colman H, Nabekura J, Lichtman JW. Alterations in synaptic strength preceding axon withdrawal. Science. 1997;275:356–361. doi: 10.1126/science.275.5298.356. [DOI] [PubMed] [Google Scholar]
- Colonnese MT, Constantine-Paton M. Developmental period for N-methyl-d-aspartate (NMDA) receptor-dependent synapse elimination correlated with visuotopic map refinement. J Comp Neurol. 2006;494:738–751. doi: 10.1002/cne.20841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colonnier M. Synaptic patterns on different cell types in the different laminae of the cat visual cortex. An electron microscope study. Brain Res. 1968;9:268–287. doi: 10.1016/0006-8993(68)90234-5. [DOI] [PubMed] [Google Scholar]
- Colonnier M, Beaulieu C. An empirical assessment of stereological formulae applied to the counting of synaptic disks in the cerebral cortex. J Comp Neurol. 1985;231:175–179. doi: 10.1002/cne.902310205. [DOI] [PubMed] [Google Scholar]
- Crozier RA, Wang Y, Liu CH, Bear MF. Deprivation-induced synaptic depression by distinct mechanisms in different layers of mouse visual cortex. Proc Natl Acad Sci U S A. 2007;104:1383–1388. doi: 10.1073/pnas.0609596104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeFelipe J, Marco P, Busturia I, Merchán-Pérez A. Estimation of the number of synapses in the cerebral cortex: methodological considerations. Cereb Cortex. 1999;9:722–732. doi: 10.1093/cercor/9.7.722. [DOI] [PubMed] [Google Scholar]
- De Gois S, Schäfer MK, Defamie N, Chen C, Ricci A, Weihe E, Varoqui H, Erickson JD. Homeostatic scaling of vesicular glutamate and GABA transporter expression in rat neocortical circuits. J Neurosci. 2005;25:7121–7133. doi: 10.1523/JNEUROSCI.5221-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erisir A, Dreusicke M. Quantitative morphology and postsynaptic targets of thalamocortical axons in critical period and adult ferret visual cortex. J Comp Neurol. 2005;485:11–31. doi: 10.1002/cne.20507. [DOI] [PubMed] [Google Scholar]
- Erisir A, Harris JL. Decline of the critical period of visual plasticity is concurrent with the reduction of NR2B subunit of the synaptic NMDA receptor in layer 4. J Neurosci. 2003;23:5208–5218. doi: 10.1523/JNEUROSCI.23-12-05208.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erisir A, Levey AI, Aoki C. Muscarinic receptor M2 in cat visual cortex: laminar distribution, relationship to gamma-aminobutyric acidergic neurons, and effect of cingulate lesions. J Comp Neurol. 2001;441:168–185. doi: 10.1002/cne.1405. [DOI] [PubMed] [Google Scholar]
- Frenkel MY, Bear MF. How monocular deprivation shifts ocular dominance in visual cortex of young mice. Neuron. 2004;44:917–923. doi: 10.1016/j.neuron.2004.12.003. [DOI] [PubMed] [Google Scholar]
- Frenkel MY, Bear MF. Bidirectional experience-dependent plasticity in primary visual cortex. In: Chalupa LM, Williams RW, editors. Eye, retina, and visual system of the mouse. Cambridge, MA: MIT; 2008. pp. 465–476. [Google Scholar]
- Frenkel MY, Sawtell NB, Diogo AC, Yoon B, Neve RL, Bear MF. Instructive effect of visual experience in mouse visual cortex. Neuron. 2006;51:339–349. doi: 10.1016/j.neuron.2006.06.026. [DOI] [PubMed] [Google Scholar]
- Fujiyama F, Furuta T, Kaneko T. Immunocytochemical localization of candidates for vesicular glutamate transporters in the rat cerebral cortex. J Comp Neurol. 2001;435:379–387. doi: 10.1002/cne.1037. [DOI] [PubMed] [Google Scholar]
- Gogolla N, Galimberti I, Caroni P. Structural plasticity of axon terminals in the adult. Curr Opin Neurobiol. 2007;17:516–524. doi: 10.1016/j.conb.2007.09.002. [DOI] [PubMed] [Google Scholar]
- Gordon JA, Stryker MP. Experience-dependent plasticity of binocular responses in the primary visual cortex of the mouse. J Neurosci. 1996;16:3274–3286. doi: 10.1523/JNEUROSCI.16-10-03274.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris KM, Stevens JK. Dendritic spines of CA 1 pyramidal cells in the rat hippocampus: serial electron microscopy with reference to their biophysical characteristics. J Neurosci. 1989;9:2982–2997. doi: 10.1523/JNEUROSCI.09-08-02982.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heynen AJ, Yoon BJ, Liu CH, Chung HJ, Huganir RL, Bear MF. Molecular mechanism for loss of visual cortical responsiveness following brief monocular deprivation. Nat Neurosci. 2003;6:854–862. doi: 10.1038/nn1100. [DOI] [PubMed] [Google Scholar]
- Holtmaat A, Svoboda K. Experience-dependent structural synaptic plasticity in the mammalian brain. Nat Rev Neurosci. 2009;10:647–658. doi: 10.1038/nrn2699. [DOI] [PubMed] [Google Scholar]
- Khibnik LA, Cho KK, Bear MF. Relative contribution of feedforward excitatory connections to expression of ocular dominance plasticity in layer 4 of visual cortex. Neuron. 2010;66:493–500. doi: 10.1016/j.neuron.2010.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lein ES, Hawrylycz MJ, Ao N, Ayres M, Bensinger A, Bernard A, Boe AF, Boguski MS, Brockway KS, Byrnes EJ, Chen L, Chen TM, Chin MC, Chong J, Crook BE, Czaplinska A, Dang CN, Datta S, Dee NR, Desaki AL, et al. Genome-wide atlas of gene expression in the adult mouse brain. Nature. 2007;445:168–176. doi: 10.1038/nature05453. [DOI] [PubMed] [Google Scholar]
- Linden ML, Heynen AJ, Haslinger RH, Bear MF. Thalamic activity that drives visual cortical plasticity. Nat Neurosci. 2009;12:390–392. doi: 10.1038/nn.2284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu CH, Heynen AJ, Shuler MG, Bear MF. Cannabinoid receptor blockade reveals parallel plasticity mechanisms in different layers of mouse visual cortex. Neuron. 2008;58:340–345. doi: 10.1016/j.neuron.2008.02.020. [DOI] [PubMed] [Google Scholar]
- Mataga N, Mizuguchi Y, Hensch TK. Experience-dependent pruning of dendritic spines in visual cortex by tissue plasminogen activator. Neuron. 2004;44:1031–1041. doi: 10.1016/j.neuron.2004.11.028. [DOI] [PubMed] [Google Scholar]
- May OL, Erisir A, Hill DL. Modifications of gustatory nerve synapses onto nucleus of the solitary tract neurons induced by dietary sodium-restriction during development. J Comp Neurol. 2008;508:529–541. doi: 10.1002/cne.21708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCann CM, Tapia JC, Kim H, Coggan JS, Lichtman JW. Rapid and modifiable neurotransmitter receptor dynamics at a neuronal synapse in vivo. Nat Neurosci. 2008;11:807–815. doi: 10.1038/nn.2145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merchán-Pérez A, Rodriguez JR, Alonso-Nanclares L, Schertel A, Defelipe J. Counting synapses using FIB/SEM microscopy: a true revolution for ultrastructural volume reconstruction. Front Neuroanat. 2009;3:18. doi: 10.3389/neuro.05.018.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mrsic-Flogel TD, Hofer SB, Ohki K, Reid RC, Bonhoeffer T, Hübener M. Homeostatic regulation of eye-specific responses in visual cortex during ocular dominance plasticity. Neuron. 2007;54:961–972. doi: 10.1016/j.neuron.2007.05.028. [DOI] [PubMed] [Google Scholar]
- Nägerl UV, Köstinger G, Anderson JC, Martin KA, Bonhoeffer T. Protracted synaptogenesis after activity-dependent spinogenesis in hippocampal neurons. J Neurosci. 2007;27:8149–8156. doi: 10.1523/JNEUROSCI.0511-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nahmani M, Erisir A. VGluT2 immunochemistry identifies thalamocortical terminals in layer 4 of adult and developing visual cortex. J Comp Neurol. 2005;484:458–473. doi: 10.1002/cne.20505. [DOI] [PubMed] [Google Scholar]
- Nakamura K, Watakabe A, Hioki H, Fujiyama F, Tanaka Y, Yamamori T, Kaneko T. Transiently increased colocalization of vesicular glutamate transporters 1 and 2 at single axon terminals during postnatal development of mouse neocortex: a quantitative analysis with correlation coefficient. Eur J Neurosci. 2007;26:3054–3067. doi: 10.1111/j.1460-9568.2007.05868.x. [DOI] [PubMed] [Google Scholar]
- Oray S, Majewska A, Sur M. Dendritic spine dynamics are regulated by monocular deprivation and extracellular matrix degradation. Neuron. 2004;44:1021–1030. doi: 10.1016/j.neuron.2004.12.001. [DOI] [PubMed] [Google Scholar]
- Peters A, Palay SL, Webster Hd. New York: Oxford UP; 1991. The fine structure of the nervous system: the neurons and supporting cells. [Google Scholar]
- Prusky GT, Douglas RM. Developmental plasticity of mouse visual acuity. Eur J Neurosci. 2003;17:167–173. doi: 10.1046/j.1460-9568.2003.02420.x. [DOI] [PubMed] [Google Scholar]
- Rittenhouse CD, Shouval HZ, Paradiso MA, Bear MF. Monocular deprivation induces homosynaptic long-term depression in visual cortex. Nature. 1999;397:347–350. doi: 10.1038/16922. [DOI] [PubMed] [Google Scholar]
- Sawtell NB, Frenkel MY, Philpot BD, Nakazawa K, Tonegawa S, Bear MF. NMDA receptor-dependent ocular dominance plasticity in adult visual cortex. Neuron. 2003;38:977–985. doi: 10.1016/s0896-6273(03)00323-4. [DOI] [PubMed] [Google Scholar]
- She WC, Quairiaux C, Albright MJ, Wang YC, Sanchez DE, Chang PS, Welker E, Lu HC. Roles of mGluR5 in synaptic function and plasticity of the mouse thalamocortical pathway. Eur J Neurosci. 2009;29:1379–1396. doi: 10.1111/j.1460-9568.2009.06696.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherman SM, Guillery RW. Exploring the thalamus and its role in cortical function. Cambridge, MA: MIT; 2006. [Google Scholar]
- Silver MA, Stryker MP. A method for measuring colocalization of presynaptic markers with anatomically labeled axons using double label immunofluorescence and confocal microscopy. J Neurosci Methods. 2000;94:205–215. doi: 10.1016/s0165-0270(99)00145-4. [DOI] [PubMed] [Google Scholar]
- Smith GB, Heynen AJ, Bear MF. Bidirectional synaptic mechanisms of ocular dominance plasticity in visual cortex. Philos Trans R Soc Lond B Biol Sci. 2009;364:357–367. doi: 10.1098/rstb.2008.0198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stryker MP, Harris WA. Binocular impulse blockade prevents the formation of ocular dominance columns in cat visual cortex. J Neurosci. 1986;6:2117–2133. doi: 10.1523/JNEUROSCI.06-08-02117.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tieman SB. Effects of monocular deprivation on geniculocortical synapses in the cat. J Comp Neurol. 1984;222:166–176. doi: 10.1002/cne.902220203. [DOI] [PubMed] [Google Scholar]
- Wiesel TN. Postnatal development of the visual cortex and the influence of environment. Nature. 1982;299:583–591. doi: 10.1038/299583a0. [DOI] [PubMed] [Google Scholar]
- Yamahachi H, Marik SA, McManus JN, Denk W, Gilbert CD. Rapid axonal sprouting and pruning accompany functional reorganization in primary visual cortex. Neuron. 2009;64:719–729. doi: 10.1016/j.neuron.2009.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon BJ, Smith GB, Heynen AJ, Neve RL, Bear MF. Essential role for a long-term depression mechanism in ocular dominance plasticity. Proc Natl Acad Sci U S A. 2009;106:9860–9865. doi: 10.1073/pnas.0901305106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zito K, Scheuss V, Knott G, Hill T, Svoboda K. Rapid functional maturation of nascent dendritic spines. Neuron. 2009;61:247–258. doi: 10.1016/j.neuron.2008.10.054. [DOI] [PMC free article] [PubMed] [Google Scholar]