

Abstract
We present a set of protocols showing how to use the 3DNA suite of programs to analyze, rebuild, and visualize three-dimensional nucleic-acid structures. The software determines a wide range of conformational parameters, including the identities and rigid-body parameters of interacting bases and base-pair steps, the nucleotides comprising helical fragments, the area of overlap of stacked bases, etc. The reconstruction of three-dimensional structure takes advantage of rigorously defined rigid-body parameters, producing rectangular block representations of the nucleic-acid bases and base pairs and all-atom models with approximate sugar-phosphate backbones. The visualization components create vector-based drawings and scenes that can be rendered as raster-graphics images, allowing for easy generation of publication-quality figures. The utility programs use geometric variables to control the view and scale of an object, for comparison of related structures. The commands run in seconds even for large structures. The software and related information are available at http://3dna.rutgers.edu/.
Keywords: base interactions, conformational analysis, DNA bending, DNA/RNA junctions, double helix, fiber models, higher-order base associations, nucleic-acid structure, model building, molecular images, non-canonical base pairs, RNA folding, solvation patterns
INTRODUCTION
In addition to the genetic message, DNA base sequence carries structural information important to its biological packaging and processing. This structural code governs how the double-helical molecule deforms in response to proteins and when and where the genetic information is expressed. For example, the binding of DNA onto the surface of the nucleosome (the protein-DNA assembly that constitutes the basic packaging unit in the nucleus) involves an indirect structural response dependent on the ease of deforming the constituent base-pair steps1. Most regulatory proteins and many enzymes, by contrast, recognize specific, sequence-dependent structural features of individual base pairs, making direct contact with DNA atoms unique to their binding sites2. Other enzymes that perform cutting or sealing operations of the phosphodiester backbone take advantage of the natural susceptibility of particular sequences to switch from the B to the A helical form3, binding to sites on DNA normally hidden within the B-type duplex. Thus DNA is not just a passive substrate of cellular proteins but an active player with physical properties capable of influencing the three-dimensional organization of genetic sequences and the activity of regulatory proteins and processing enzymes.
The diverse functions of RNA, ranging from the transmission of genetic information to chemical catalysis4, similarly reflect the sequences and structures of the heterocyclic base side groups. In contrast to double-stranded DNA, RNA molecules are generally single-stranded, with the ribonucleotide building blocks assembling into intricate, functionally specific, three-dimensional forms. The RNA chain folds back on itself, with double-helical elements interspersed between unpaired bulges, internal loops, and multi-armed junctions and the structure as a whole stabilized by tertiary interactions and/or bound ligands4. Whereas the complementary strands of double-helical DNA associate almost exclusively through canonical, Watson-Crick base pairing, RNA incorporates both Watson-Crick and non-canonical base pairs, including interactions of the bases with the 2′-hydroxyl group unique to the ribose sugar5,6.
Growing interest in understanding the relationship between the global folding of nucleic acids and the sequence-dependent arrangements of the constituent bases and base pairs has stimulated the development of new approaches to analyze and depict DNA and RNA structures7,8. The spatial configurations of both associated bases and neighboring bases or base pairs depend upon the sequence9,10. Clear differences arise between the six rigid-body parameters (three angular variables and three translational variables) that describe the arrangements of different bases and base pairs (Figure 1). The arrangements of paired bases are defined with respect to the orientation and displacement of an ideal, planar Watson-Crick base pair11, where the rigid-body parameters (buckle κ, propeller π, opening σ angles; shear Sx, stretch Sy, stagger Sz displacements7) are zero. The configurations of successive bases or base pairs are specified by an analogous set of variables: two bending angles (roll ρ and tilt τ), a rotation angle (twist ω), two in-plane dislocations (shift Dx and slide Dy), and a vertical displacement (rise Dz)7. The local geometry of each dinucleotide step can also be described by set of six helical parameters (x-displacement dx, y-displacement dy, helical rise h, inclination η, tip θ, and helical twist (Ωh ) that describe the regularity of the helix in which it occurs7.
Figure 1.

Pictorial definitions of rigid-body parameters used to describe the geometry of complementary (or non-complementary) base pairs and sequential base pair steps. The base pair reference frame (lower left) is constructed such that the x-axis points away from the (shaded) minor groove edge of a base or base pair and the y-axis points toward the sequence strand (I). The relative position and orientation of successive base pair planes are described with respect to both a dimer reference frame (upper right) and a local helical frame (lower right). Images illustrate positive values of the designated parameters. Reproduced with permission from Nucleic Acids Research.
In contrast to the analysis of the Watson-Crick base pairs in double-helical DNA structures, the study of ‘globular’ RNA and other complex nucleic-acid structures requires preliminary knowledge of the bases comprising the helices, loops, and folds of the polynucleotide chains. While such information is self-evident in the spatial arrangements of small molecules, the sheer enormity of structures like the ribosome (2876 nucleotides in the large 50S subunit12) hampers direct visual identification of secondary structural motifs, non-Watson-Crick pairs, and long-range interactions. Studying such complicated nucleic-acid structures requires automated procedures to identify the short- and long-range base associations.
The arrangements of bases in nucleic-acid structures are closely tied to the immediate chemical environment in which they occur. That is, the deformations of helical structure occur in the context of the water molecules, proteins, and other species in direct contact with the double helix. Prior to our work, analyses of such interactions were literal and statistical in the sense of counting the number of different contacts in known structures13,14, or protein-centric in the sense of cataloging the various amino acids and folding motifs in close contact with nucleic acid15–18. Many of the tools developed to visualize protein-DNA complexes, e.g., NUCPLOT19, similarly focus on the identities of amino acids or secondary structural elements with respect to DNA primary structure. Understanding the effects of solvation on nucleic-acid sequence and structure requires a DNA- or RNA-centric perspective, which takes account of the precise locations of solvent, including closely associated protein and drug atoms, with respect to the heterocyclic bases or nucleotide steps20–23.
Software overview
3DNA is a versatile, integrated software system for the analysis, rebuilding, and visualization of three-dimensional nucleic-acid-containing structures, including their complexes with proteins and other ligands8. At its core, the software uses a simple matrix-based scheme24–28 for calculating the complete set of rigid-body parameters that characterize the orientation and displacement of the base pairs, base-pair steps, and single-stranded nucleotide steps that make up DNA and RNA structures. The description of structure is geometrically straightforward and the computation of parameters is mathematically rigorous8,27,28, allowing for exact rebuilding of a molecular structure based on the derived parameters. The software evolved from our efforts to resolve the discrepancies among seven commonly used nucleic-acid conformational analysis programs29,30, and makes use of the standard base-centered reference frame11 recommended by the 1999 Tsukuba Workshop and later approved by the Nomenclature Committee of IUBMB (NC-IUBMB)/IUPAC-IUBMB Joint Commission on Biochemical Nomenclature (JCBN).
3DNA is written in strict ANSI C computer language (~25,000 lines of code), with connecting Perl scripts. The software package consists of over 30 executable programs that can be run directly from the command-line in a Unix/Linux environment (including Cygwin on Windows). Running each program with the “-h” option (e.g., find_pair -h) provides the user with detailed usage information, and worked examples. Once the user is familiar with the package, it is straightforward to combine the various pieces and other command-line-driven (or capable) tools in a script to automate commonly repeated tasks, as demonstrated by the examples detailed herein.
The 3DNA software was first made available on the Internet in late 1999 and the currently used version (v1.5) was released in late 2002. A paper published in 20038 describes the major functionality available in the software. The 3DNA website includes unpublished technical details, a user’s manual, and answers to frequently asked questions (FAQ). Over the years, we have answered hundreds of 3DNA-related questions from users all over the world, initially via email and more recently through the 3DNA forum, and have taken each and every query as an opportunity to improve the functionality of the software. We strive to respond to users as quickly and concretely as possible, often with a step-by-step recipe, until an issue is resolved. This process has helped us to refine and improve the software and has prompted us to add new functionality. As a result, 3DNA has found widespread use and the 3DNA homepage is linked from the websites of various bioinformatics databases and computer-graphics tools, such as the Nucleic Acid Database (NDB)31, the RNA World website at Jena32, and the Raster3D graphics toolset33.
The widespread use of 3DNA in the scientific community is evident from the rapidly growing number of citations of the 2003 paper8. While it is not practical to summarize the literature on all of the applications of the suite of programs, we draw attention here to several projects where 3DNA has played a particularly significant role: (1) the NDB31 reports 3DNA base-pair and base-pair-step parameters for the double-helical fragments of each structure in the database; (2) both the NDB31 and the PDB (Protein Data Bank)34 report the simple and informative molecular images generated with the blocview component of 3DNA; (3) the nucleic-acid Solvation Web Service35 (SwS) uses 3DNA to find base pairs, collect conformational patterns, and set the orientation of molecular images; (4) the DNA structural bioinformatics server36 (MDDNA) uses the 3DNA rebuild routine to construct models of arbitrary DNA sequences based on predictions obtained from molecular-dynamics simulations; (5) the ARTS37 (Alignment of RNA Tertiary Structures) software uses 3DNA to identify both canonical and non-canonical base pairs for its seed-match construction; (6) the information-driven protein-DNA docking method HADDOCK38 uses 3DNA to build canonical B-DNA models, generate custom DNA libraries, calculate various base-pair and base-pair-step parameters, determine the torsion angles of the sugar-phosphate backbone, and evaluate the puckering of the sugar ring; (7) the base-pair server (BPS) from this laboratory uses find_pair and other utility programs from 3DNA to build a comprehensive database and to generate illustrative molecular images of the base pairs in RNA structures; (8) our recent identification of a roll-slide-induced DNA folding mechanism in chromatin1 takes advantage of the rigorous matrix-based analysis and rebuilding functions of 3DNA, and (9) our generalization of the 3DNA code to treat the rigid-body parameters of planar molecular fragments39 facilitates the classification of small, flexible drug molecules and identification of representative structures for three-dimensional quantitative structure-activity relationship analyses.
Relationship to other programs
3DNA originated in the SCHNAaP/ SCHNArP software27,28 written to implement the computation of DNA rigid-body parameters with the mathematical formalism (CEHS) developed by El Hassan and Calladine26 and to rebuild structures from those parameters, including the generation of ‘standardized’ base-stacking diagrams. Our efforts to resolve the discrepancies among nucleic-acid conformational analysis programs29,30 and the definition of a standard base reference frame by the structural biology community11, prompted us to take advantage of various features in the earlier programs and to adopt the standard frame in 3DNA. Specifically, we use the least-squared fitting procedure, developed by Babcock et al. in RNA40 (Running Nucleic Acids), to superimpose a standard base41 on an experimental structure in the determination of local base reference frames, and the procedure, employed in Newhelix42, to fit a global linear axis to a helix that does not deviate substantially from a regular structure.
The computation of DNA dimeric parameters is sensitive to the way in which a reference frame is assigned to each base and base pair29. Since the reference frames used in 3DNA roughly coincide with the frames used in CompDNA43 and Curves44,45, the 3DNA base-pair and base-pair step parameters closely resemble the values determined with CompDNA and the local parameters from Curves (except when DNA is highly kinked and the Curves values tend to be larger29). The discrepancies are more pronounced with programs, such as SCHNAaP/CEHS26–28, Newhelix/Freehelix42, and RNA40, that employ very different reference frames30. The cehs routine in 3DNA, nevertheless, allows the user to calculate authentic SCHNAaP/CEHS parameters, which (owing to the similar reference frames) closely match the values obtained with Newhelix/Freehelix, and with the “-r” option within cehs to find authentic RNA40 parameters. The 3DNA software also lets the user take advantage of the unique capability of Curves to find a global curvilinear helical axis and related global parameters, such as the widths and depths of the major and minor grooves, by providing an option “-c” in the find_pair routine that generates an input file to Curves from a PDB file.
3DNA is unique among the many available nucleic-acid model-building programs in that it is purely geometrically based and that it allows for exact construction of a model from a set of straightforward and well understood, user-supplied parameters. A number of other programs make use of rigid-body methods, although for other purposes: NAB46 (Nucleic Acid Building) provides “a programming environment for geometric and force-field manipulations of nucleic acids”; NAMOT47 (Nucleic Acid MOdeling Tool) builds and manipulates models based on a set of reduced coordinates developed by the authors; and Mc-Sym48, a knowledge-based software, builds RNA structures from molecular subfragments.
The visualization components in 3DNA yield drawings in vector-based EPS and Xfig formats. The latter can be manipulated easily using the free Xfig interactive drawing tool. The software also creates Raster3D scenes that can be fed directly into render or PyMOL for high-quality graphic images. RasMol49 is especially effective for interactive visualization of the rectangular schematic presentations generated in Alchemy format with 3DNA. Thus, 3DNA complements many commonly used visualization tools (Raster3D, RasMol, MolScript, and PyMOL).
3DNA is tailored for automatic visualization of structural variations in nucleic-acid base-pair geometry, a feature not found in other interactive and schematic drawing programs, such as DRAWNA50 and UCSF Chimera51. 3DNA has built-in capabilities to generate and modify the sizes and shapes of Calladine-Drew-style base and base-pair ‘blocks’, and to highlight the minor-groove edges of such base or base-pair representations. As emphasized here and seen from the images of nucleic-acid containing structures at the NDB/PDB websites, the blocview-generated images are highly effective in revealing the key features of small to medium-sized nucleic-acid structures, and are used to represent each nucleic-acid containing structure in both the NDB31 and PDB34. The programmatic generation of publication-quality diagrams of the base-stacking and higher-order base associations is also unique to 3DNA. Recently added tools for generating images of the ensembles of nucleic acid structures found with nuclear magnetic resonance (NMR) methods, and for converting “.r3d” files for PyMOL ray-traced images are expected to gain wide use.
Strengths and limitations of 3DNA
3DNA has many unique features that make it a practical tool in various real-world applications. For example, the automatic detection of all possible base pairs, higher-order base associations, and helical fragments using find_pair makes analyzing nucleic-acid structures with 3DNA straightforward. To our knowledge, there are no other tools with such functionality. The real power of 3DNA, however, lies in its integrated approach of combining the analysis, reconstruction, and visualization of three-dimensional nucleic-acid structures in a single software package. The examples given herein exemplify some of the possible applications. Except for some final touches or format conversion for use of commonly used, freely available software (RasMol, Raster3D, MolScript, Xfig, Ghostscript, and ImageMagick), all the images in this paper are generated directly from the program using scripts to automate an otherwise tedious, error-prone process.
As with any software, 3DNA has become more sophisticated and robust over the years as it becomes more widely used, with user-reported bugs fixed, the code internally refined, and new functionality added. The interactions with users from different backgrounds have given us the incentive to adapt the programs for further applications in related fields, e.g., RNA structure-motif identification and alignment, structural analysis of DNA-protein complexes, and modeling RNA folds. The reference-frame-based description of three-dimensional spatial geometry makes the methodology and algorithms in 3DNA directly applicable to these problems, and treats them in a rigorous and consistent fashion. Similar extension and application of 3DNA to the growing number of DNA quadruplex structures should contribute to the analysis and understanding of these novel forms.
On the other hand, the command-line driven style of 3DNA, while flexible and efficient, has a downside in that it is not very user-friendly, especially for educational purposes and to novices (non-Linux/Unix users). Our next goal is to create a web-based interface for some commonly used features of the package, e.g., generation of fiber models of arbitrary sequences, creation of blocview images, analysis of a structure, generation of stacking diagrams, etc. We will continue to support the 3DNA forum, and maintain the on-line documentation to get the community involved in providing better documentation and more working examples.
Sample applications of 3DNA
This procedure describes five protocols for: (A) Preparation of schematic diagrams of representative base-pair parameters; (B) Determination of DNA curvature associated with different roll distributions; (C) Analysis and visualization of a DNA structure with a B-Z junction; (D) Automatic identification of double-helical regions in a DNA-RNA junction; and (E) Identification of higher-order base associations in ribosomal RNA.
(A) Preparation of schematic diagrams of representative base-pair parameters
Although it is possible, albeit tedious, to obtain reasonable quality schematic images (typically rectangular blocks) of the rigid-body parameters describing the nucleic-acid base pairs and base-pair steps with any modern computer-graphics tool, it is not trivial to control the relative position and orientation of the rectangular blocks precisely, to set the exact viewpoint, and to maintain the same viewpoint and scale in different diagrams. While the many representations in the literature of nucleic-acid rigid-body parameters, such as the propeller twist of complementary base pairs, the roll at deformed base-pair steps, etc., may look similar, they differ subtly from one another. 3DNA provides the necessary tools to generate such schematics more professionally by allowing for exact control of the conformational parameters as well as for the viewpoint and scale of the images (see, for example, Figures 1 and 4 in the 2003 paper8). Because many of the recent users of 3DNA have expressed an interest in illustrations such as those described in our earlier work8 (reproduced here in Figure 1), we first present the detailed steps needed to generate block images of each of the three types of rigid-body parameters, using propeller as a representative base-pair parameter, rise as a representative local base-pair-step parameter, and inclination as a representative local helical parameter (Fig. 2).
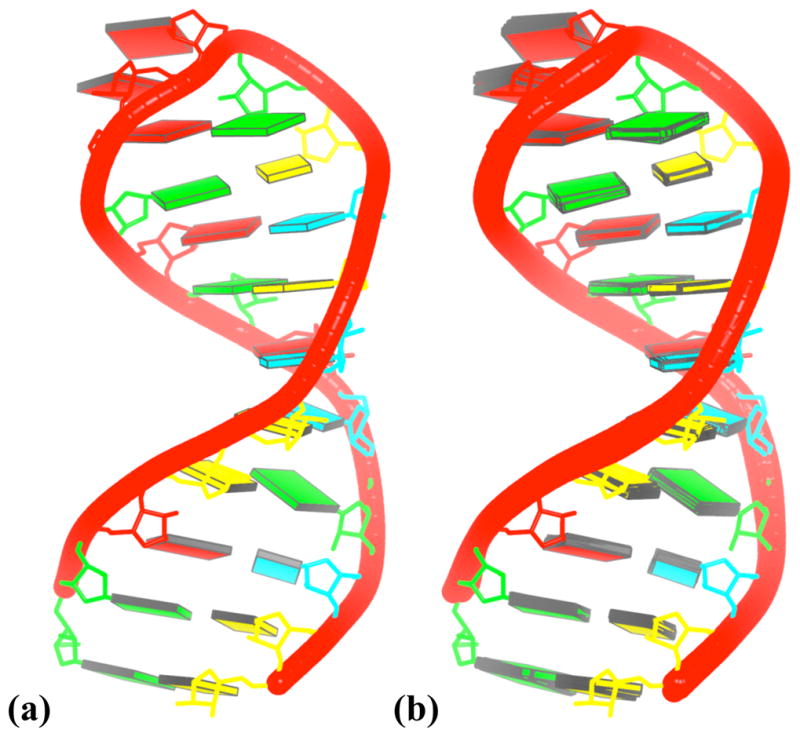
Figure 4. Images of a B-Z-junction structure63.

(a) blocview-generated image highlighting the ‘melted’ and flipped-out A11-T26 pair and the inverted base pairs at the C10T12/A25G27 junction step in the middle of the duplex; (b) standard stacking diagram of the junction obtained directly from 3DNA showing the intuitively negative twist angle, the appreciable shift and slide, and the same-strand base overlap of stacked base pairs. Base color-coding conforms to the NDB31 convention: A (red); C (yellow); G (green); T (blue); U (aqua).
Figure 2. Schematic diagrams of three representative rigid-body parameters.

(a) propeller (45°); (b) rise (3.4 Å); (c) inclination (36°). Images in the left column were created automatically with 3DNA, and those on the right side are final, touched-up versions generated with Xfig. The six images were merged and aligned, and the composite figure shown here was exported into Encapsulated PostScript (EPS) format and subsequently converted to PNG format at 300 DPI using Ghostscript. Note that, with the automatic generation procedure in 3DNA, one can control the magnitude and sign of the parameters precisely and specify the orientation and scale of the image.
(B) Determination of DNA curvature associated with different roll distributions
DNA is not a monotonous, regular straight helix, but rather deforms in response to external stresses in a sequence-dependent manner9,52–56. For example, the catabolic activator protein (CAP) introduces two sharp kinks at pyrimidine-purine (YR) steps in its binding site, bending the DNA at these steps into the major groove via large positive roll57,58. By contrast, the DNA wrapped tightly around the assembly of histone proteins on the surface of the nucleosome core particle deforms via both positive and negative roll, with pronounced bending into the minor groove occurring at regularly spaced YR steps59. In between these two extremes of bending are other possible modes of deformation, such as the patterns of roll in the idealized curved DNA pathways presented in the Calladine et al. textbook on DNA structural mechanics60. The complicated three-dimensional nature of local bending makes it difficult, even for a seasoned scientist, to visualize how the variation in roll at different dinucleotide steps leads to global curvature. With 3DNA, one simply prepares a data file with any prescribed set of parameters and builds the structure to see what it looks like (Fig. 3). The matrix-based scheme adopted in the 3DNA analysis/rebuilding programs makes this a completely reversible and rigorous process. The second protocol shows how one can mimic the idealized global fold of a 22 base-pair fragment of DNA illustrated in Figure 4.8(a) of the Calladine et al textbook60.
Figure 3. 3DNA-generated images of 22-bp DNA duplexes with the same overall 45° curvature per helical turn.

(a-d) four different theoretical distributions of roll angle achieving such curvature, according to models in the Calladine et al. textbook60, namely two steps with 45° roll separated by a complete (10 bp) helical turn, five bends with 22.5° roll spaced at increments of a half helical turn and with alternating sign, two tracts of five 14°-rolled steps separated by a half helical turn, and a smoothly curved structure with a repeating sinusoidal-like variation in roll (−9.00°, −7.28°, −2.78°, 2.78°, 7.28°, 9.00°, 7.28°, 2.78°, −2.78°,−7.28°); (e) ideal B-DNA reference structure with uniform (zero) roll.
(C) Analysis and visualization of a DNA structure with a B-Z junction
From a purely structural point of view, right-handed B-DNA has a positive twist (+36°, on average, per base-pair step) and left-handed Z-DNA has a negative twist angle (−60° per CGC/GCG repeating unit). Importantly, compared to B-DNA, the base pairs in Z-DNA are inverted along the long axis27 (Fig. 4). While various B-Z junction models have been offered for decades61,62, the first crystal structure of such a junction, in the presence of the human editing enzyme, double-stranded RNA-specific adenosine deaminase, was only recently solved by the Rich group at MIT63.
The 3DNA analysis routine ( analyze) checks for possible base-pair inversion within a double helix and classifies such structures as “unusual” in the main output file. Prior to the publication of the B-Z junction paper, we worked with the lead author on the analysis of a partial structure containing only the two base pairs at the B-Z junction, using the opportunity to update the 3DNA code to handle such structures automatically (Sung Chul Ha, personal email communication). In order for meaningful parameters to be calculated, the program reverses the x- and z-axes of the Z-DNA base pair at the junction (in the same way that the program must reverses the y- and z-axes of bases on complementary strands to compute sensible values of the base-pair parameters describing a Watson-Crick pair). Following publication of the B-Z junction structure, we were able to check the complete structure and verify that the program modification works properly. The third protocol shows how one can analyze and illustrate this interesting structural feature.
(D) Automatic identification of double-helical regions in a DNA-RNA junction
By taking advantage of the standard base reference frame and selected geometric features, the find_pair program within 3DNA can identify all possible nucleic-acid base pairs, whether they are canonical Watson-Crick or non-canonical pairs and are made up of normal or modified bases, in any tautomeric form or protonation state. The program identifies helical regions from a purely base-stacked perspective by tracing each base pair against its neighborhood vertically, i.e., in a direction perpendicular to the base-pair plane, regardless of whether there is a direct backbone connection between stacked base pairs. This program is the key application that makes the analysis of arbitrary nucleic-acid structures an easy job with 3DNA. Previously one had to identify each base pair and each helical region manually or semi-automatically, a tedious and error-prone task, especially for RNA structures and DNA-protein or RNA-protein complexes.
The crystal structure of the RNA-DNA four-way junction (PDB entry 1EGK) solved by Nowakowski et al64 serves as an excellent example of the capabilities of 3DNA to identify base pairs and helical regions automatically, and to draw a cylinder to visualize each relatively straight helix within the structure. The fourth protocol outlines the steps for finding the helical regions and generating a color-coded image of the overall structure (Fig. 5).
Figure 5. 3DNA-generated view of a four-way DNA-RNA junction64.
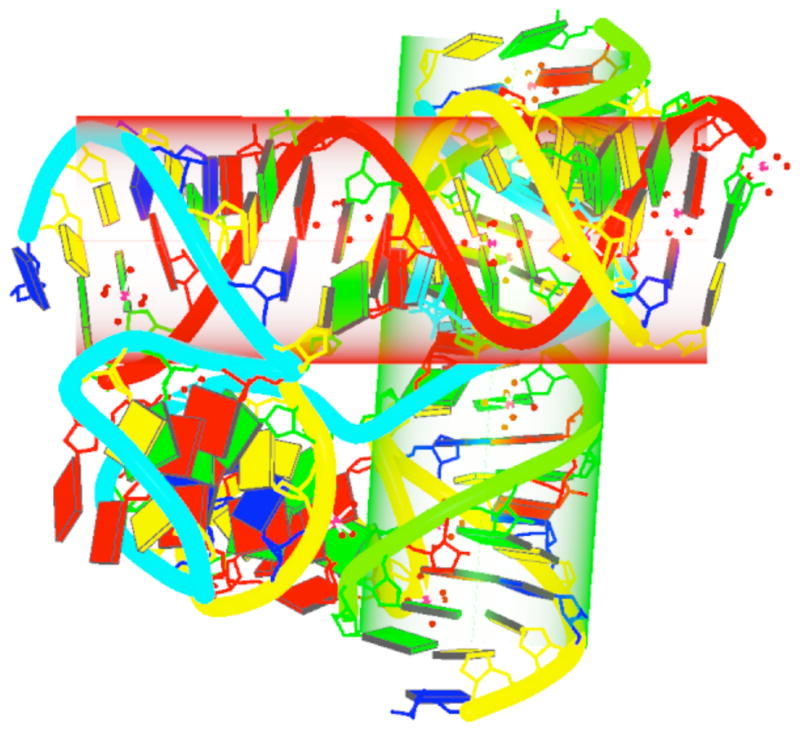
(PDB entry 1EGK), highlighting, with cylinders, the two straight helical regions detected by the program and orienting the structure into a view perpendicular to the long axes of the cylinders. Backbones are color-coded by chain identifiers, cylinders by helical identity, and nucleotides by chemical type. Magnesium ions and the waters to which they are coordinated in the grooves of the structure are represented by red dots. See text for chain and helix color-coding and the legend to Figure 4 for base color-coding.
(E) Identification of higher-order base associations in ribosomal RNA
The same find_pair program used for locating base pairs and helical regions (Option (D)) also identifies triplets, quadruplets, and higher-order base associations. Starting from the identified base pairs, the program searches horizontally in the planes of the associated bases for further connections/interactions, and checks for pairwise ‘compatibility’ to eliminate unreasonable associations. The program then collects the hydrogen bonds (H-bonds) formed from the nitrogen and oxygen atoms on the base and sugar-phosphate backbones of the contacted residues, including bifurcated H-bonds.
The last protocol demonstrates the capability of 3DNA to identify and generate publication-quality images of the two pentaplets, i.e., associations of five paired bases, found in the fully refined crystal structure of the H. marismortui large ribosomal subunit65 (PDB entry 1JJ2; Fig. 6).
Figure 6. Pentaplets from the refined crystal structure of the H. marismortui large ribosomal subunit65 (PDB entry 1JJ2) automatically detected and illustrated with 3DNA.

Note the direct involvement of the O2′ sugar and O2P backbone atoms in H-bonding and the bridging role of the adenine. See legend to Figure 4 for base color-coding.
MATERIALS
Equipment
A computer running a Linux/Unix based operating system (such as one of those listed below).
Equipment setup
3DNA is a software product that is distributed in binary form for common Linux/Unix operating systems, including various flavors of Linux distribution (e.g., Ubuntu, SuSE, RedHat, etc.), SunOS, SGI, Mac OS X, and Cygwin on Windows. The package distribution includes a subdirectory named NP_Recipes/, which contains all the scripts, associated data files, and graphics images used in this manuscript. The new 3DNA homepage, located at http://3dna.rutgers.edu/, has links to the 3DNA forum, the online documentation, answers to FAQs, technical details, and other related information. See Box 1 for detailed instructions on the installation of 3DNA.
Box 1. 3DNA installation instructions.
Setting up 3DNA is a straightforward process, facilitated by a Perl script. Essentially, two things need to be done: (1) set up an environment variable X3DNA to point to the location where 3DNA is installed on your local system so that the various parameter files can be found; (2) put the 3DNA binary directory into your command line search path so that you can run the 3DNA commands conveniently. Here, we use the binary distribution compiled on Ubuntu Linux with the tarball file named “ Linux_X3DNA_v2.0.tar.gz” as an example to illustrate the process.
To install 3DNA, first download the tarball from http://3dna.rutgers.edu/ to a directory of your choice on your local computer, presumably your home directory, and then follow the instructions below:
-
tar zxvf Linux_X3DNA_v2.0.tar.gz
The command creates a directory named X3DNA that contains a complete 3DNA distribution. The directory has several sub-directories, including bin/ with the entire C binaries and Perl scripts, and NP_Recipes/ that contains all the data files, scripts, the images used in this work.
-
cd X3DNA/bin
The command changes the current working directory to the 3DNA binary directory.
-
x3dna_setup
The command will produce an output message on the screen, like the following (on an Ubuntu Linux box with a default “bash” shell):
To run X3DNA, you need to set up the following: ○ the environment variable X3DNA ○ add $X3DNA/bin to your command-line search path for your “bash” shell, please add the following into ~/.bashrc: -------------------------------------------------------------- export X3DNA=/home/xiangjun/X3DNA export PATH=/home/xiangjun/X3DNA/bin:$PATH --------------------------------------------------------------
To complete setting up 3DNA, simply follow the output from x3dna_setup as shown above, i.e., set the environment variable X3DNA and add $X3DNA/bin into your command-line search path by copying and pasting the two lines between the dashed lines into your current command window for the current session, or into your startup script (e.g., ~/.bashrc) for a permanent effect. For verification, type find_pair –h. Detailed command-line help related to the find_pair routine will be displayed on the screen (the same “-h” option can be used to obtain help with all of the 3DNA programs and Perl scripts stored in the bin/ directory).
Data format
Input for the analysis of a nucleic-acid structure is a PDB-formatted file listing the Cartesian coordinates and chemical features (atom type, residue name, and chain identifier) of all atoms in the structure of interest (see the RCSB PDB website for a detailed description of the PDB format). The PDB files can be those determined experimentally (e.g., X-ray or NMR from the PDB or NDB), or from modeling studies (e.g., molecular dynamics simulations).
For rectangular block schematic representations of bases and base pairs, 3DNA makes use of the Alchemy file format commonly used to depict the three-dimensional structures of small molecules. This format allows for explicit assignment of the types of atoms and chemical bond linkages (single, double, or aromatic) that make up a given structure. We adopt the Alchemy format in 3DNA in order to take advantage of the popular molecular graphics program RasMol49, which reads and writes information in this format. While no-detailed, authoritative description is found on the Internet, as for the PDB format, it is easy to understand Alchemy format from a few examples (see the examples/calladine_drew/ directory, or load a PDB structure into RasMol and then write back its coordinates in Alchemy format).
Input for the generation of a three-dimensional structure uses a simple 3DNA-specific text file format: the first line contains the number of base pairs or nucleotides in the desired (double- or single-stranded) structure; the second contains either a 0 or a 1 to denote whether the set of rigid-body variables will be in the form of base-pair/nucleotide step parameters (slide, roll, etc.) or helical parameters (x-displacement, inclination, etc.), respectively; the third line allows the user to insert a comment for the header of the parameter list. The lines that follow, equal to the number of base pairs or nucleotides specified in line 1, contain (i) the identities of the base pairs or nucleotides (e.g., G-C or G) and (ii) either the six rigid-body variables that relate each base pair or nucleotide to its predecessor or the 12 parameters that detail the positioning of the bases in the given pairs and the spatial relationship of each pair to its predecessor, i.e., both the six base-pair parameters and the six dimeric step parameters. The lines are arranged so that the base sequence can be read down the first column and rigid-body parameters occur in order of the axis of operation, with displacement variables listed before angular parameters, i.e., (shear, stretch, stagger, buckle, propeller, opening) in the case of base-pair parameters, (shift, slide, rise, tilt, roll, twist) in the case of base-pair-step parameters, (x-displacement, y-displacement, helical rise, inclination, tip, helical twist) in the case of helical parameters. The step parameters of the first base pair or nucleotide are naturally zero (given the lack of a preceding residue). While the omission of pair parameters has no effect on unpaired nucleotides, missing (i.e., null) values of the base-pair parameters automatically fix the associated bases in ideal, planar Watson-Crick arrangements. Data in this form provide a short-hand way to generate structures in which each base pair is represented as a single rectangular block (see Step 1 Option (B) of the procedure).
The accompanying supplemental materials at the Nature Protocols website contain a sample Alchemy file of a base-pair rectangular block (“Block_BP.alc”), and two sample input files for rebuild: one for the generation of a base pair with a 45° propeller angle (“propeller.dat”, Recipe#1 of Box 3), and another one for the construction of a 22 base-pair DNA with 45° curvature per helical turn (“roll_a.dat”, Recipe#2 of Box 3). Other worked examples can be found in the NP_recipes/ directory that is distributed with 3DNA. Checking the examples is the most effective way to understand the manner in which nucleic-acid structure is specified and to use the software effectively.
Box 2. External programs that complement the visualization components of 3DNA.
RasMol (http://www.umass.edu/microbio/rasmol/), although not directly used in generating the images made for this paper, is a popular, easy-to-use program for molecular structure visualization and a useful product for gaining experience with Alchemy files (with options -alchemy and -noconnect).
MolScript (http://www.avatar.se/molscript/) is a program for displaying molecular three-dimensional structures in both schematic and detailed representations. It is used in 3DNA for generating a Raster3D input file for rending protein and nucleic-acid backbone ribbons.
Raster3D (http://skuld.bmsc.washington.edu/raster3d/) is a set of tools for generating high-quality raster images. 3DNA makes use of render to generate raster-graphics images in Raster3D “.r3d” format.
PyMOL (http://pymol.sourceforge.net/) is a user-sponsored molecular visualization system with scripting capabilities (“.pml” script) used by 3DNA for integration with other command-line driven tools. A user can also load the “.r3d” files created by 3DNA into PyMOL to manipulate them interactively, in the same way that one would use the normal Raster3D/MolScript to PyMOL interface.
Ghostscript (http://sourceforge.net/projects/ghostscript/) is an interpreter for the Adobe PostScript language and can serve as a file-format converter. It is used by 3DNA for converting an EPS file to a PNG image at a specific resolution, 300 dots per inch (DPI) for the figures in the paper. Commercial products like Adobe Illustrator® can substitute for Ghostscript for interactive uses.
ImageMagick (http://www.imagemagick.org/script/index.php) is a useful, general-purpose, image-processing toolset for creating, editing, and composing bitmap images. 3DNA uses the display and convert components of the software. Adobe Photoshop® can also substitute for ImageMagick for interactive uses.
Xfig (http://www.xfig.org/) is an interactive drawing tool chosen by 3DNA for its simplicity, quality, and availability. The software fulfills our needs for line drawing and composing composite figures and also allows for direct editing of the Xfig files generated by 3DNA.
Third-party software
3DNA is designed to be as self-contained as possible with minimal (clearly delineated) connections to third-party software. The analysis and rebuilding parts of 3DNA are totally independent of any third-party tools. The seven external programs listed in Box 2 complement the visualization components of 3DNA, helping to create all the figures reported in this work. All of these programs are freely available and highly valuable to research problems of the sort that we describe.
Timing
With the above software tools properly installed, re-running each of the scripts given below (on an ordinary Ubuntu Linux box) takes less than a minute, even for the large 50S ribosomal subunit. Most tasks take only a few seconds or less.
PROCEDURE
In this protocol, we show five real-world applications of 3DNA, inspired largely by our interactions with users over the years. Combined together, they illustrate the versatile capabilities of 3DNA, made possible by its unique integrated approaches on the analysis, rebuilding and visualization of three-dimensional nucleic acid structures.
To prepare schematic diagrams of representative base-pair parameters, in particular, to generate an image of a base pair with a propeller of 45°, follow the steps in Option (A). For determining the curvature of DNA associated with different roll distributions, follow the steps in Option (B). To analyze and visualize a DNA structure with a B-Z junction, follow the steps in Option (C). For automatic identification of double-helical regions in a DNA-RNA junction, go to Option (D). To identify higher-order base associations in ribosomal RNA go to Option (E).
(A) Preparation of schematic diagrams of representative base-pair parameters, using propeller of 45° as an example (Recipe #1, Box 3)
Box 3. Five step-by-step recipes illustrating various capabilities of 3DNA.
Recipe #1: command-line script to create a schematic image for propeller of 45°
01 \cp $X3DNA/config/block/Block_M.alc Block_R.alc 02 \cp $X3DNA/config/block/Block_M.alc Block_Y.alc 03 rebuild -block2 propeller.dat propeller.alc 04 frame_mol -1 -m -g -L=8 ref_frames.dat propeller.alc propeller_mg.alc 05 rotate_mol -r=side_view.dat propeller_mg.alc propeller_sv.alc 06 alc2img -f -g -s=166 propeller_sv.alc propeller_sv.fig
Recipe #2: command-line script to create a 22 base-pair long schematic duplex structure with a 45° curvature per helical turn
01 rebuild roll_a.dat roll_a_raw.alc 02 frame_mol -m -11,12 ref_frames.dat roll_a_raw.alc roll_a_mm.alc 03 rotate_mol -r=roty90.dat roll_a_mm.alc roll_a_ok.alc 04 alc2img -f -l -s=128 roll_a_ok.alc roll_a.fig 05 alc2img -l -s=7 roll_a_ok.alc roll_a.eps
Recipe #3: command-line script to analyze and visualize a B-Z junction structure
01 get_part 2acj.pdb 2acj_dna.pdb 02 blocview -t=300 -i=2acj_dna.png 2acj_dna.pdb 03 find_pair 2acj.pdb 2acj.inp 04 analyze 2acj.inp 05 ex_str -8 stacking.pdb bz_step.pdb 06 stack2img -c -d -o -l -t bz_step.pdb bz_step.eps
Recipe #4: command-line script to illustrate the three helices in a four-way RNA-DNA junction
01 find_pair 1egk.pdb stdout 02 ex_str -1 hel_regions.pdb h1.pdb 03 ex_str -2 hel_regions.pdb h2.pdb 04 ex_str -3 hel_regions.pdb h3.pdb 05 find_pair h1.pdb stdout | analyze 06 find_pair h2.pdb stdout | analyze 07 find_pair h3.pdb stdout | analyze 08 rotate_mol -t=h1x_h3y.rot 1egk.pdb 1egk_h1x_h3y.pdb 09 find_pair 1egk_h1x_h3y.pdb stdout 10 ex_str -1 hel_regions.pdb h1x.pdb 11 ex_str -3 hel_regions.pdb h3y.pdb 12 find_pair h1x.pdb stdout | analyze 13 \mv poc_haxis.r3d h1x.r3d 14 find_pair h3y.pdb stdout | analyze 15 \mv poc_haxis.r3d h3y.r3d 16 blocview -o 1egk_h1x_h3y.pdb 17 cat t.r3d transparent.r3d h1x_ok.r3d h3y_ok.r3d > 1egk_ok.r3d 18 x3dna_r3d2png 1egk_ok.r3d 1egk_ok.png
Recipe #5: command-line script to identify pentaplets in the large ribosomal subunit (1JJ2)
01 find_pair -p 1jj2.pdb 1jj2.bps 02 ex_str -117 multiplets.pdb p1.pdb 03 ex_str -135 multiplets.pdb p2.pdb 04 stack2img -s=300 -f -cdol p1.pdb p1.fig 05 stack2img -s=300 -f -cdol p2.pdb p2.fig
Prepare a data file entitled “propeller.dat” with all base-pair parameters other than propeller (45°) set to zero in the simple 3DNA-specific text format described above (see data format in the Materials section). This file can be created and edited with any text editor (e.g., vi and emacs), or more easily done using the 3DNA utility program regular_dna with the number of repeating units set to unity (only one base pair). It should be noted that sequence identity does not matter in the construction of a general base-pair schematic.
Assign blocks of the same size (4.5 × 5.0 × 0.5 Å) to the complementary purine (R) and pyrimidine (Y) residues of a Watson-Crick base pair using a “Block_M.alc” file for both bases, i.e., “Block_R.alc” and “Block_Y.alc” (lines 01 and 02). The edges of the blocks run parallel to the axes of the bases, i.e., 4.5 Å across the short axis (joining the minor-and major-groove edges) of each base, 5.0 Å along the long axis (pointing toward the sugar-phosphate backbone), and 0.5 Å along the normal. Note that the “\” character in front of “cp”, i.e., “\cp”, forces the copy command to run non-interactively.
-
Build the schematic structure of the base pair in Alchemy format, by running rebuild with the two-block option “-block2”, i.e., using one rectangular block for each base, and naming the resultant set of coordinates “propeller.alc” (line 03). The Alchemy file can be conveniently viewed with RasMol49 (using the “-alchemy” and “-noconnect” options).
CAUTION: The option “-noconnect” makes sure that RasMol uses only the linkage information specified in the Alchemy file (by setting the CalcBondsFlag to false). It is not required for the “propeller.alc” file, but is critical for the “propeller_mg.alc” (step 4) and “propeller_sv.alc” files (step 6). The latter two Alchemy files contain explicitly specified coordinate axes, which would interfere with the default bond-calculation algorithm in RasMol.
Reorient the structure with respect to the base-pair reference frame (“-1”) using frame_mol and naming the transformed coordinate file “propeller_mg.alc” (line 04). The option “-m” specifies that the minor groove faces the viewer, and the options “-g - L=8” that the global reference axes are 8.0 Å in length. Note that the viewing options, i.e., “-1 -m -g -L=8”, can be listed in any order in 3DNA, in either lower or upper case (although “-L” for length is used here to avoid any confusion between a lower case “l” and the number one “1” introduced to specify the reference frame), and that single-letter options can be bundled (e.g., “-m” and “-g” can be combined as either “-mg” or “-gm”). Strictly speaking, the “-1” option is redundant since rebuild places the global reference frame by default on the first base-pair frame of a generated structure.
Create the data file “side_view.dat” containing the following two lines to transform the image so that the imposed propeller angle can be seen more clearly. The first line rotates the structure about the (vertical) y-axis by −30° and the second about the (horizontal) x-axis by +20°.
by rotation y -30 by rotation x 20
Rotate the base-pair block (“propeller_mg.alc”) using the preceding rotation file (“-r=side_view.dat”) to create the new output in side view (“propeller_sv.alc”, line 05).
Create the final schematic image (“propeller_sv.fig”) in Xfig (“-f”) format by applying alc2img to the above set of rotated coordinates (“propeller_sv.alc”), with the global axes represented by arrows (“-g”) and the scale factor set to a specified number (“-s=166”) (line 06). The latter value is expressed in Xfig-defined units, where a value of 8333 units corresponds, at a resolution of 1200 units per inch, to a maximum figure dimension of 6.94 inches or 17.6 cm. The 3DNA image-generating programs ( alc2img, stack2img, r3d_atom, blocview, etc.) automatically output the currently used scale factor in the diagnostic messages for drawing images of different styles (PostScript, Xfig, Raster3D). Setting the scale factor to a fixed value with the “-s” option ensures that a series of related structures are drawn, as illustrated in Figure 2 (propeller, rise, and inclination), in exactly the same scale.
(B) Determination of DNA curvature associated with different roll distributions, using two 45° kinked steps separated by 10 base pairs as an example (Recipe #2, Box 3)
Prepare a data file entitled “roll_a.dat” specifying the base-pair step parameters that relate each base pair to its predecessor in a 22 base-pair fragment. The base pairs in the structure span 2.1 double-helical turns of DNA, i.e., 21 base-pair steps, each twisted by 36°. The overall curvature of 45° per helical turn is produced by two properly phased kinks at steps 6 and 16, each with a 45° roll angle. Thus, in the data file, the rise is set to 3.34 Å and the twist to 36° throughout the structure and all remaining step parameters, other than the 45° roll at steps 6 and 16, are fixed at zero. The omission of base-pair parameters (buckle, propeller, etc.) in each line ensures that these values are all zero (see discussion of file format in the Materials section). Here each base pair is represented by a single (4.5 × 10.0 × 0.5 Å) rectangular block.
Build the structure (using rebuild) from the base-pair step parameters specified in the above input file “roll_a.dat” and store the coordinates of the base-pair blocks in the output file named “roll_a_raw.alc” (line 01). By default, rebuild constructs a structure using base-pair blocks (“-block1”) in Alchemy format, as we want here.
Place the molecular coordinate system in the middle frame of the central 11–12 dinucleotide step (“-11,12”) by employing frame_mol, set the view so that the minor groove faces the viewer (“-m”), and name the resultant file “roll_a_mm.alc” (line 02). It should be noted that the “ref_frames.dat” file, which contains the reference-frame information for each base pair, can be generated by a number of programs in 3DNA, including find_pair, analyze, and rebuild. Here this information comes from the previously executed rebuild step.
Rotate the DNA (using rotate_mol) by 90° around the (vertical) y-axis of the global coordinate frame (“–r=roty90.dat”, where “roty90.dat” is a file with the instruction “by rotation y 90”) so that the structure adopts the desired view (“roll_a_ok.alc”) (line 03). As pointed out in Option (A) above, the various Alchemy files generated with 3DNA can be conventionally viewed and manipulated (e.g., rotated) with RasMol49.
Create the final illustrated image “roll_a.fig” in Xfig format (“-f”) using alc2img, with consecutive base-pair centers connected by dotted lines (“-l”) (line 04). The fixed scale factor (“-s=128”) makes sure that all five images (Fig. 3) are in exactly the same scale. Finally, for illustration purposes, the last line (05) generates the same image in EPS, the default image format from alc2img. The “-l” and “-s=7” options in this instruction have the same meaning as the “l” and “s” symbols used above to generate an image in Xfig format.
(C) Analysis and visualization of a DNA structure with a B-Z junction (Recipe #3, Box 3)
Extract (with get_part) the nucleic-acid part (“2acj_dna.pdb”) of the protein-Z-DNA complex63 (PDB entry 2ACJ) (line 01).
Use blocview to obtain a simplified, yet informative color-coded image (“2acj_dna.png”) of the DNA (Fig. 4a) (line 02). The recipe includes instructions for the creation of a PyMOL ray-traced image (“-i=2acj_dna.png”) at a resolution of 300 DPI (“-t=300”).
Determine the base-pair identities and content directly from the original protein-bound DNA file (“2acj.pdb”) using find_pair (line 03), generating the file (“2acj.inp”) to be used as input for the analysis routine. Note that find_pair also outputs a diagnostic message “^^vv opposite bp direction: 1(14) 8(8)-9(9)”, indicating that the z-axes of base pairs 8 and 9 in this 14 base-pair double helix (helix 1, the only helical fragment in the structure) run in opposite directions, as characteristic of a B-Z junction.
Calculate structural parameters using analyze from the base-pairing information in file “2acj.inp” (line 04), storing the output in the file “2acj.out”. The “Structure classification” component of analyze correctly identifies the structure as containing a junction located between a right-handed helix and a left-handed one. The analysis program also creates a fixed-named file called “stacking.pdb” that stores the coordinates of all dinucleotide steps in terms of their middle frame.
Extract (using ex_str) the B-Z junction at step 8 (“-8”) from the file “stacking.pdb” and save the coordinates in the PDB-formatted file “bz_step.pdb” (line 05).
Generate a standardized stacking diagram of the B-Z junction with stack2img (line 06). The completed image (Fig. 4b) is color-coded by residue (“-c”), with hydrogen bonds drawn (“-d”), base rings filled (“-o”), bases labeled (“-l”) at the centers of the six-membered rings, and the base pair oriented in a top-down view (“-t”) such that the (middle-frame) long axis of the step is horizontal and the leading strand lies on the left. As noted above, these five single-letter options can be combined as “-cdolt”.
(D) Automatic identification of double-helical regions in a DNA-RNA junction (Recipe #4, Box 3)
Using RasMol or any other molecular-graphics program (e.g., PyMOL), examine the structure of the DNA-RNA junction64 (PDB entry 1EGK) and confirm that it forms three helices.
Locate the three helical regions (here termed h1, h2, and h3) in the 1EGK coordinate file (“1egk.pdb”) using find_pair and store the helical content information in the fixed-named file “hel_regions.pdb” (line 01). Note that “stdout” in the script stands for the standard output stream, normally connected to the screen, and that a fixed-named file “hel_regions.pdb” is generated by default and thus not given in the command line.
Extract the atomic coordinates of the three helices into separate files (“h1.pdb”, “h2.pdb”, “h3.pdb”) using ex_str (lines 02–04) and calculate the structural parameters of each with analyze (lines 05–07). The “|” is the pipe used to connect the standard output from find_pair (“stdout”) directly into analyze without resorting to an intermediate data file (as in line 03 of Recipe #3 above). The 3DNA default settings, defined in the file “misc_3dna.par” (see Recipe #5 below), show that helices h1 and h3 are relatively straight, and thus a linear least-squared global helical axis is fitted to each. The analysis program also provides a normalized helical-axis vector for h1 and h3, and the two end points that each axis passes through, expressed in the original coordinate frame of the PDB file. Such information is useful for calculating the global bending angle between the two helices (86° here) and the shortest distance between the centers of the helices (20.2 Å), the latter value indicating that the two helices are nearly touching (given the characteristic 20 Å diameter of double-helical DNA/RNA).
-
Reorient the structure as a whole (“1egk.pdb”) with rotate_mol such that the two helical-axis vectors (associated with h1 and h3) lie in the x-y plane (line 08). Here h1 is set horizontally (along the x-axis), and h3 is set vertically, with its component orthogonal to the (cylindrical) axis of h1 along the y-axis. The z-axis is chosen to complete a right-handed coordinate system. The above transformation is expressed in a transformation matrix file “h1x_h3y.rot”, which is used with the “-t” option of rotate_mol (“-t=h1x_h3y.rot”) to transform the PDB data file “1egk.pdb” into the desired orientation in the file “1egk_h1x_h3y.pdb”.
CAUTION: Two warning messages, “Warning: x- and y-axes are not orthogonal” and “Warning: z-axis is not perpendicular to xy-plane”, are not a matter of concern. The program ( rotate_mol) checks if the three input axes are orthogonal, and performs vector normalization/decomposition as necessary. Here the h1 and h3 axes are not orthogonal, and the z-axis is randomly chosen and not necessarily perpendicular to the plane defined by the h1 and h3 axes. The content of the “h1x_h3y.rot” file is as follows:
1 # x-, y-, z-axes row-rise 0.000 0.000 0.000 # translation −0.348 0.922 −0.167 # x: h1 axis, from h1.out 0.597 0.161 −0.785 # y: h3 axis, from h3.out 0.000 0.000 1.000 # z: can be anything Repeat the process of identifying helical regions in the newly reoriented structure, i.e., apply find_pair to the transformed PDB file “1egk_h1x_h3y.pdb”, and extract (using ex_str) the two straight helices h1 and h3 into the files “h1x.pdb” and “h3y.pdb”, respectively (lines 09–11).
Re-compute the structural parameters of the residues in the reoriented helix h1, i.e., apply find_pair followed by analyze to “h1x.pdb” (line 12). The output includes an auxiliary fixed-named file, “poc_haxis.r3d”, which contains the Raster3D scenes for the cylinders with radii based on the mean displacement of the P, O4′, and C1′ atoms.
Rename (“ \mv”) the Raster3D file “poc_haxis.r3d” into file “h1x.r3d” (line 13) and manually modify “h1x.r3d”, by uncommenting (i.e., selecting) the cylinder scenes based on the phosphorus atoms, changing the color to red (by modifying its RGB color components). The updated Raster3D file is now called “h1x_ok.r3d”. Compare the two files (“h1x.r3d” and “h1x_ok.r3d”) to see the difference, e.g., use the Unix command diff.
Repeat steps 6 and 7 for the reoriented helix h3 to obtain the file “h3y_ok.r3d”, with the color of the cylinder set to green (lines 14–15).
Run blocview (line 16) on the previously transformed PDB file “1egk_h1x_h3y.pdb” to get the color-coded-block Raster3D scenes in the fixed-named file “t.r3d” using the original (“-o”) coordinates (i.e., without automatically setting the ‘best-view’ based on the longest principal axes of the structure).
Employ cat (a simple Unix utility program) to combine all the “.r3d” files (“t.r3d” from step 9, “h1x_ok.r3d” and “h3y_ok.r3d” from steps 6–8) into a single file (by file redirection “>”) called “1egk_ok.r3d”, including the “transparent.r3d” scenes from Raster3D distribution that make the two cylinders transparent. The file “t.r3d” must be in the first position in the list for its header information, and “transparent.r3d” should be second followed by “h1x_ok.r3d” and “h3y_ok.r3d” (in either order) to ensure transparency in the two cylinders (line 17).
Transform the scenes into a PNG image (“1egk_ok.png”) with the utility Perl script x3dna_r3d2png (line 18) that employs the render program of Raster3D (Fig. 5).
(E) Identification of higher-order base associations in ribosomal RNA (Recipe #5, Box 3)
Find all pairwise interactions and higher-order associations (“1jj2.bps”) in the H. marismortui large ribosomal subunit65 (“1jj2.pdb”) using find_pair with the “-p” option (line 01). The program generates a PDB coordinate file named “multiplets.pdb”, which contains all triplets and higher base associations (187 cases), each defined with respect to its middle reference frame. Specifically, the z-axis of the assembly lies along the average of all the base normals, giving an objective, top-down view of the multi-base association. Here, a primary H-bond distance cut-off of 3.3 Å, a bifurcated H-bond distance cut-off of 3.1 Å, and a vertical base-separation (stagger) cut-off of 2.0 Å are used by modifying the parameters file “misc_3dna.par”.
-
Extract (with ex_str) the two pentaplets, numbered 117 and 135 in the file “multiplets.pdb”, into two separate PDB files (“p1.pdb” and “p2.pdb”, lines 02–03)
CAUTION: The serial numbers given here for the two pentaplets, 117 and 135, depend on the parameter settings in “misc_3dna.par”. They will differ if, for example, the H-bonding distance cut-off is changed.
Generate images of the pentaplets in Xfig (“-f”) with options similar to those used in Option (C) (Recipe #3 of Box 3), and a fixed scale factor (“-s=300”) that ensures that the two molecular representations are scaled exactly the same (lines 04–05).
ANTICIPATED RESULTS
(A) Preparing schematic diagrams of representative base-pair parameters
The detailed procedure given in PROCEDURE Option (A) (see Recipe #1 of Box 3) can be put into a shell script, and run automatically to yield the desired image (Fig. 2a, left). With Xfig, it is then straightforward to touch-up the 3DNA-generated diagram (“propeller_sv.fig”) directly, e.g., changing the font, extending the axes, etc. (Fig. 2a, right). The advantage of the procedure is the precise control of the value, viewpoint, and scale of the illustrated parameter. The script makes it easy to set propeller to another value, e.g., the −15° value typical of the base pairs found in A- and B-DNA crystal structures11, and repeat the procedure to generate another block diagram.
The procedure needed to make a schematic diagram of rise (Fig. 2b) or inclination (Fig. 2c) is similar to that described for propeller, except for the replacement of the complementary bases by two successive base pairs. The two base blocks in each pair will touch each other if they are of the same size and if all base-pair parameters are zero, generating an image (4.5 × 10 × 0.5 Å) in size with a line running along the short x-axis through the middle of the base-pair block. (The central line is actually the superimposed edges of the two base blocks that make up each base pair (Fig. 2b, c)). The images of nucleic-acid parameters in Figure 1 of the 2003 paper8, reproduced here, also as Figure 1, were generated in this way with an early version of 3DNA, where each of the illustrated displacement parameters in the figure, except for rise (3.4 Å), is 2 Å, and each of the angular parameters, except for inclination and tip (each 36°), is 45°.
(B) DNA curvature associated with different roll distributions
Application of PROCEDURE Option (B) yields an image that mimics the global DNA fold illustrated in Figure 4.8(a) of the Calladine et al. textbook60 (Fig. 3a). The overall curvature of 45° per helical turn — produced by two properly phased kinks at steps 6 and 16, each with a 45° roll angle — is striking compared to a regular (unkinked) duplex of the same length (Fig. 3e).
This built-as-desired strategy for nucleic-acid structure generation, made possible with 3DNA, has broader implications in real-world applications. Firstly, one can easily modify the parameter file and rebuild a structure to see how selected local parameters affect global structure. For example, the images (Figs. 3b–d), created with the same procedure as Option (B) (see Recipe #2 of Box 3) but incorporating different parameter files, show how different patterns of roll yield the same 45° global bend of DNA. On the other hand, while conventional models attribute the global fold of DNA to the bending and twisting of the constituent base-pair steps, a recent paper from our group1 shows that simply by setting the slide, the rigid-body parameter that specifies the displacement of successive base pairs along their long axis (Fig. 1), to zero (typical of B-DNA11), the superhelical fold of nucleosomal DNA collapses to a circle. Indeed, we uncovered this novel roll-slide mechanism of DNA folding in chromatin by using the 3DNA rebuild routine to model nucleosomes. Secondly, rebuild allows for the construction of atomic-level nucleic-acid structures (“-atomic” option) with sugar-phosphate backbones in pre-assigned, fixed conformations (specified by standard residue files; see the FAQ section at the 3DNA website). Such models, which have precise base-pair geometry but approximate (sometimes distorted) backbone connections, provide a useful starting point66 and basis for analysis36 of all-atom simulations. Thirdly, as demonstrated in SCHNArP28, various DNA bending ‘rules’, with different sets of tilt/roll/twist values at the constituent dinucleotide or trinucleotide steps, can be easily incorporated within a script that transforms a sequence with an assigned ‘bending model’ into an input file that feeds into rebuild.
(C) Analysis and visualization of a DNA structure with a B-Z junction
In the molecular image (Fig. 4a) created with PROCEDURE Option (C), the unpaired bases at the chain ends and the broken and flipped-out A-T base pair in the middle of the B-Z structure63 immediately stand out. Here A, C, G, and T are colored by default in red, yellow, green, and blue, respectively, following the NDB base color-coding convention. The stacked C10T12/A25G27 B-Z junction abuts the flipped-out A-T pair in the center of the duplex. Whereas the (long, unshaded) major-groove edge of the C10-G27 base pair in the lower Z-DNA half of the structure faces the viewer, the (long, shaded) minor-groove edge of the T12-A25 base pair in the upper B-DNA half faces outward. The obvious buckling of the two base pairs in the opposite sense leads to the small rise (2.6 Å) at the junction step29.
The stacking diagram that is also produced (Fig. 4b) clearly shows the inversion of the two base pairs, with their opposing connections to the sugar rings and differently exposed base-pair edges. The B-Z junction step has a twist angle of around −17°, as well as noticeable shift (1.26 Å) and slide (0.80 Å) (given in the file “2acj.out” from step 4 of Option (C)). The stacking diagram also reveals the significant overlap of bases on the same-strand (4.88 Å2 for C10-T12 and 7.56 Å2 for A25-G27, including exocyclic atoms) and the total absence of overlap of bases on complementary strands (i.e., cross-strand overlap) at the junction.
(D) Automatic identification of double-helical regions in a DNA-RNA junction
The procedure in Option (D) illustrates how 3DNA can automatically identity the three helical regions in the DNA-RNA junction64 (Fig. 5). Helix 1, highlighted by the red cylinder, is made up of one continuous RNA chain along one strand (chain A in red) and two DNA chains along the other strand (chains B and D in order of their 5′-3′directions). Helix 3, highlighted by the green cylinder, also contains one RNA chain along one strand (chain C in green) and two DNA chains along the other strand (chains D and B in order of their 5′-3′directions). Helix 2, consists primarily of DNA (chains B and D) with a single residue (A101) from the RNA chain inserted in one strand and another residue (A1) of the RNA along the other strand. Unlike helices 1 and 3, helix 2 is irregular and not represented by a cylinder. Thus, the backbones are not continuous along each helix. The chosen view allows for easy visualization and protractor measurement of the overall bending angle between the two relatively straight helices (the red horizontal helix h1 and the green vertical helix h3). This intuitive approach, initially developed to quantify the bending angles of a series of CAP-DNA complexes67, yields sensible results when other methods fail.
(E) Identification of higher-order base associations in ribosomal RNA
The images of the two base pentaplets in Fig. 6, extracted from the large 50S ribosomal subunit65 and visualized with PROCEDURE Option (E), reveal both the canonical hydrogen-bonding of the Watson-Crick base pairs within the complexes and the non-canonical hydrogen bonding of the RNA base atoms with the O2′ atom of ribose and the exocyclic O2P phosphate oxygen, e.g., O2′ in the A2010-G2013 and G1971-U1972 (Fig. 6a) and the U2278-G2471 (Fig. 6b) pairs and O2P in the G1971-U1972 (Fig. 6a) and the U2278-G2471 and G2630-A2633 (Fig. 6b) pairs. The selected viewpoints highlight the very different types of interactions of adenine with the minor-groove edges of the G·C pairs in the two pentaplets, i.e., distinct types of A-minor motifs68. Whereas A2010 approaches G2013 (Fig. 6a) with a different face exposed to the viewer (i.e., the positive z-axes of the two purines point in opposite directions8), A2633 and G2471 (Fig. 6b) reveal the same face, with the two z-axes pointing away from the viewer into the plane of the paper. Moreover, each of the adenines serves as a bridge that connects the two components in each pentaplet, concomitantly interacting with the canonical G·C pair through its minor-groove side and another base via its major-groove edge: with U1972 in a G·U dinucleotide platform69 (Fig. 6a), or G2630 (Fig. 6b). It is also worth noting that the G1971·U1972 platform is stabilized not only by the well-characterized G(N2)···U(O4) H-bond interaction, but also by a little-noticed G(O2′)···U(O2P) sugar-phosphate backbone interaction (Fig. 6a). Examination of the 50S large ribosomal unit (1JJ2) alone reveals ten such double H-bonded G·U platforms, far more occurrences than those registered by any other dinucleotide platform (including A·A) in this structure. Apparently, the G·U platform is more stable than other platforms with only a single base-base H-bond interaction. We are currently investigating this over-represented G·U dinucleotide platform in other RNA structures.
OTHER APPLICATIONS
3DNA contains more programs and features than can be fully documented here. We conclude with a brief mention of three other components of the software package that should be of general interest. Box 4 presents an example showing the automatic generation of schematic images of the ensemble of stem-hairpin structures found by Zoll et al. from NMR studies of the 3′-UTR Y stem from the poliovirus RNA genome70. Box 5 illustrates the procedure within 3DNA used to identify and visualize the structured water molecules found in high-resolution crystal structures, here the waters in contact with the base pairs and backbone of the DNA in the currently best-resolved nucleosome core-particle structure71. Finally, Box 6 describes the large collection of fiber-diffraction models that can be conveniently constructed with 3DNA.
Box 4. Automatic generation of schematic images of an NMR ensemble of an RNA stem-hairpin.
The newly added Perl script nmr_ensemble generates an NMR-ensemble image (Fig. 7b) that combines the unique features of blocview, i.e., the rectangular block representation of bases, color-coding of chains, residues, and shading of base minor-groove edges, with each member of the structural ensemble automatically set in the principal-axis frame of the best representative structure (Fig. 7a). Here we illustrate the set of solution structures, reported by Zoll et al., of variants of the Y domain of the poliovirus single-stranded RNA genome70. We orient the best representative conformer in its most extended view (Fig. 7a) and record the conversion matrix for subsequent transformation of the remaining NMR conformers to the same frame.
The completed set of ensemble images (Fig. 7b) clearly shows the identities of the base residues within the double-helical stem and the unpaired adenines and non-canonical G·A pair in the single-stranded RNA hairpin at the top of the image. The structural variations of nucleotide residues (e.g., the larger, more floppy variation of the unpaired adenines vs. the movement of the G·A pair) are also evident.
Box 5. Automatic generation of structured waters around base pairs and dinucleotide steps in nucleosomal DNA.
When performed with the “-w” option, find_pair identifies the waters in contact with each base pair, and analyze the waters in contact with a dinucleotide step. Although the two programs select, by default, the waters, i.e., the oxygen atoms of the waters, within 3.2 Å of any nitrogen or oxygen atom on a nucleotide pair or dimer fragment, the criteria are easily modified through a parameter file (“misc_3dna.par”). The images shown here — the pattern of waters (denoted by red dots) associated with all the A-T (Fig. 8a) and all the G-C (Fig. 8b) nucleotide pairs, and the waters in contact with the first AT/AT dinucleotide step (Fig. 8c) in the currently best-resolved (1.9 Å) nucleosome core-particle structure71 — are automatically generated using a Perl script that combines various 3DNA programs with PyMOL to generate the ray-traced images. Note that the A-T and G-C base pairs in the composite images are naturally overlapped on the middle base-pair reference frames, generated directly from find_pair. This method forms the basis of the SwS nucleic-acid solvation server35. Here note the distinctive build-up of water near specific base, sugar, and phosphate atoms.
Box 6. Fiber-diffraction models.
The fiber program within 3DNA provides handy access to 55 fiber models of DNA and RNA helices in various polymorphic forms, e.g., A-, B-, C-DNA, etc., and stoichiometries, e.g., single-stranded, double-stranded, triplexes, quadruplexes, and DNA-RNA hybrids. The program generates structure files in standard PDB format, with the “-xml” option allowing for structural output in Protein Data Bank Markup Language (PDBML)72, a format especially useful for very large structures that exceed the 5-digit atom serial number limit and/or the fixed (f8.3) Cartesian-coordinate formatting limit of PDB files. To the best of our knowledge, the collection of fiber models in 3DNA is the most comprehensive of its kind. In preparing this set of fiber models, we have taken great care to ensure the accuracy and consistency of the models. For completeness and user verification, 3DNA includes, in addition to 3DNA-processed files, the original coordinates collected from the literature. Whereas the 3DNA-processed repeating unit is necessary to construct full, atomic-level representations of the models, the structurally equivalent PDB files of the repeating units, expressed in standard base reference frame, prove useful as building blocks when a user wishes to generate a perturbed base-only fiber model, such as an overstretched duplex with variable rise at successive steps.
Figure 7. Schematic nmr_ensemble-generated images of the bases and backbones of.
(a) the best-representative NMR structure; and (b) the complete ensemble of 12 NMR structures of the 3′-UTR Y-stem from the poliovirus RNA genome70. See legend to Figure 4 for base color-coding.
Figure 8. DNA hydration patterns automatically extracted and illustrated with 3DNA from the currently best-resolved nucleosome core-particle structure71.

The depicted waters (red dots) lie within 3.2 Å of the nucleotide oxygen and nitrogen atoms (a) in 84 of the 87 A-T pairs (three pairs are distorted) and (b) in all 60 G-C pairs, and (c) in the first AT/AT dinucleotide step. Composite images (a, b) are generated by superposition of the middle frames of each base pair. See legend to Figure 4 for base color-coding.
Acknowledgments
This work has been generously supported by the U.S. Public Health Service under research grant GM20861. We are grateful to Mauricio Esguerra and Guohui Zheng for their careful reading of the manuscript and independent validation of the protocols presented in this manuscript and to the users of 3DNA for using the software to address real-world problems, and communicating the difficulties that they encounter. Positive interactions with the users have been the driving force behind the development and improvement of 3DNA. We also thank the editor and the anonymous reviewers whose comments helped to clarify the presentation of the protocols.
Footnotes
Competing interests statement: The authors declare no competing financial interests.
References
- 1.Tolstorukov MY, Colasanti AV, McCandlish D, Olson WK, Zhurkin VB. A novel ‘roll-and-slide’ mechanism of DNA folding in chromatin. Implications for nucleosome positioning. J Mol Biol. 2007;371:725–738. doi: 10.1016/j.jmb.2007.05.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Luscombe NM, Laskowski RA, Thornton JM. Amino acid-base interactions: a three-dimensional analysis of protein-DNA interactions at an atomic level. Nucleic Acids Res. 2001;29:2860–2874. doi: 10.1093/nar/29.13.2860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lu XJ, Shakked Z, Olson WK. A-form conformational motifs in ligand-bound DNA structures. J Mol Biol. 2000;300:819–840. doi: 10.1006/jmbi.2000.3690. [DOI] [PubMed] [Google Scholar]
- 4.Tinoco I, Jr, Bustamante C. How RNA folds. J Mol Biol. 1999;293:271–281. doi: 10.1006/jmbi.1999.3001. [DOI] [PubMed] [Google Scholar]
- 5.Auffinger P, Westhof E. Rules governing the orientation of the 2′-hydroxyl group in RNA. J Mol Biol. 1997;274:54–63. doi: 10.1006/jmbi.1997.1370. [DOI] [PubMed] [Google Scholar]
- 6.Leontis NB, Westhof E. Geometric nomenclature and classification of RNA base pairs. RNA. 2001;7:499–512. doi: 10.1017/s1355838201002515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dickerson RE, Bansal M, Calladine CR, Diekmann S, Hunter WN, Kennard O, von Kitzing E, Lavery R, Nelson HCM, Olson WK, Saenger W, Shakked Z, Sklenar H, Soumpasis DM, Tung CS, Wang AHJ, Zhurkin VB. Definitions and nomenclature of nucleic acid structure parameters. J Mol Biol. 1989;208:787–791. doi: 10.1016/0022-2836(89)90324-0. [DOI] [PubMed] [Google Scholar]
- 8.Lu XJ, Olson WK. 3DNA: a software package for the analysis, rebuilding and visualization of three-dimensional nucleic acid structures. Nucleic Acids Res. 2003;31:5108–5121. doi: 10.1093/nar/gkg680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Olson WK, Gorin AA, Lu XJ, Hock LM, Zhurkin VB. DNA sequence-dependent deformability deduced from protein-DNA crystal complexes. Proc Natl Acad Sci, USA. 1998;95:11163–11168. doi: 10.1073/pnas.95.19.11163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Olson WK, Colasanti AV, Lu X-J, Zhurkin VB. Wiley Encyclopedia of Chemical Biology. John Wiley & Sons; New York: 2008. Physico-chemical properties of nucleic acids: character and recognition of Watson-Crick base pairs. in press. [Google Scholar]
- 11.Olson WK, Bansal M, Burley SK, Dickerson RE, Gerstein M, Harvey SC, Heinemann U, Lu XJ, Neidle S, Shakked Z, Sklenar H, Suzuki M, Tung CS, Westhof E, Wolberger C, Berman HM. A standard reference frame for the description of nucleic acid base-pair geometry. J Mol Biol. 2001;313:229–237. doi: 10.1006/jmbi.2001.4987. [DOI] [PubMed] [Google Scholar]
- 12.Ban N, Nissen P, Hansen J, Moore PB, Steitz TA. The complete atomic structure of the large ribosomal subunit at 2.4 Ångstrom resolution. Science. 2000;289:905–920. doi: 10.1126/science.289.5481.905. [DOI] [PubMed] [Google Scholar]
- 13.Mandel-Gutfreund Y, Margalit H, Jernigan RL, Zhurkin VB. A role for C···O interactions in protein-DNA recognition. J Mol Biol. 1998;277:1129–1140. doi: 10.1006/jmbi.1998.1660. [DOI] [PubMed] [Google Scholar]
- 14.Treger M, Westhof E. Statistical analysis of atomic contacts at RNA-protein interfaces. J Mol Recognit. 2001;14:199–214. doi: 10.1002/jmr.534. [DOI] [PubMed] [Google Scholar]
- 15.Suzuki M. DNA recognition code of transcription factors in the helix-turn-helix, probe helix, hormone receptor, and zinc finger families. Proc Natl Acad Sci, USA. 1994;91:12357–12363. doi: 10.1073/pnas.91.26.12357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tatano M, Tamasaki K, Amano N, Kakinuma J, Koike H, Allen MD, Suzuki M. DNA recognition by β-sheets. Biopolymers. 1997;44:335–359. doi: 10.1002/(SICI)1097-0282(1997)44:4<335::AID-BIP3>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 17.Jones S, van Heyningen P, Berman HM, Thornton JM. Protein-DNA interactions: a structural analysis. J Mol Biol. 1999;287:877–896. doi: 10.1006/jmbi.1999.2659. [DOI] [PubMed] [Google Scholar]
- 18.Nadassy K, Wodak SJ, Janin J. Structural features of protein-nucleic acid recognition sites. Biochemistry. 1999;38:1999–2017. doi: 10.1021/bi982362d. [DOI] [PubMed] [Google Scholar]
- 19.Luscombe NM, Laskowski RA, Thornton JM. NUCPLOT: a program to generate schematic diagrams of protein-nucleic acid interactions. Nucleic Acids Res. 1997;25:4940–4945. doi: 10.1093/nar/25.24.4940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Woda J, Schneider B, Patel K, Mistry K, Berman HM. An analysis of the relationship between hydration and protein-DNA interactions. Biophys J. 1998;75:2170–2177. doi: 10.1016/S0006-3495(98)77660-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kono H, Sarai A. Structure-based prediction of DNA target sites by regulatory proteins. Protein-Struct Func Genet. 1999;35:114–131. [PubMed] [Google Scholar]
- 22.Pichierri F, Aida M, Gromiha MM, Sarai A. Free-energy maps of base-amino acid interactions for DNA-protein recognition. J Am Chem Soc. 1999;121:6152–6157. [Google Scholar]
- 23.Ge W, Schneider B, Olson WK. Knowledge-based elastic potentials for docking drugs or proteins with nucleic acids. Biophys J. 2005;88:1166–1190. doi: 10.1529/biophysj.104.043612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhurkin VB, Lysov YP, Ivanov VI. Anisotropic flexibility of DNA and the nucleosomal structure. Nucleic Acids Res. 1979;6:1081–1096. doi: 10.1093/nar/6.3.1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bolshoy A, McNamara P, Harrington RE, Trifonov EN. Curved DNA without A-A: experimental estimation of all 16 DNA wedge angles. Proc Natl Acad Sci, USA. 1991;88:2312–2316. doi: 10.1073/pnas.88.6.2312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.El Hassan MA, Calladine CR. The assessment of the geometry of dinucleotide steps in double-helical DNA: a new local calculation scheme. J Mol Biol. 1995;251:648–664. doi: 10.1006/jmbi.1995.0462. [DOI] [PubMed] [Google Scholar]
- 27.Lu XJ, El Hassan MA, Hunter CA. Structure and conformation of helical nucleic acids: analysis program (SCHNAaP) J Mol Biol. 1997;273:668–680. doi: 10.1006/jmbi.1997.1346. [DOI] [PubMed] [Google Scholar]
- 28.Lu XJ, El Hassan MA, Hunter CA. Structure and conformation of helical nucleic acids: rebuilding program (SCHNArP) J Mol Biol. 1997;273:681–691. doi: 10.1006/jmbi.1997.1345. [DOI] [PubMed] [Google Scholar]
- 29.Lu XJ, Olson WK. Resolving the discrepancies among nucleic acid conformational analyses. J Mol Biol. 1999;285:1563–1575. doi: 10.1006/jmbi.1998.2390. [DOI] [PubMed] [Google Scholar]
- 30.Lu XJ, Babcock MS, Olson WK. Mathematical overview of nucleic acid analysis programs. J Biomol Struct Dynam. 1999;16:833–843. doi: 10.1080/07391102.1999.10508296. [DOI] [PubMed] [Google Scholar]
- 31.Berman HM, Olson WK, Beveridge DL, Westbrook J, Gelbin A, Demeny T, Hsieh SH, Srinivasan AR, Schneider B. The Nucleic Acid Database: a comprehensive relational database of three-dimensional structures of nucleic acids. Biophys J. 1992;63:751–759. doi: 10.1016/S0006-3495(92)81649-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sühnel J. Views of RNA on the World Wide Web. Trends in Genetics. 1997;13:206–207. doi: 10.1016/s0168-9525(97)01134-7. [DOI] [PubMed] [Google Scholar]
- 33.Merritt EA, Bacon DJ. Raster3D: photorealistic molecular graphics. Methods in Enzymology. 1997;277:505–524. doi: 10.1016/s0076-6879(97)77028-9. [DOI] [PubMed] [Google Scholar]
- 34.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The Protein Data Bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Auffinger P, Hashem Y. SwS: a solvation web service for nucleic acids. Bioinformatics. 2007;23:1035–1037. doi: 10.1093/bioinformatics/btm067. [DOI] [PubMed] [Google Scholar]
- 36.Dixit SB, Beveridge DL. Structural bioinformatics of DNA: a web-based tool for the analysis of molecular dynamics results and structure prediction. Bioinformatics. 2006;22:1007–1009. doi: 10.1093/bioinformatics/btl059. [DOI] [PubMed] [Google Scholar]
- 37.Dror O, Nussinov R, Wolfson H. The ARTS web server for aligning RNA tertiary structures. Nucleic Acids Res. 2006;34:W412–W415. doi: 10.1093/nar/gkl312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.van Dijk M, van Dijk ADJ, Hsu V, Boelens R, Bonvin AMJJ. Information-driven protein-DNA docking using HADDOCK: it is a matter of flexibility. Nucleic Acids Res. 2006;34:3317–3325. doi: 10.1093/nar/gkl412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Banerjee A, Misra M, Pai D, Shih LY, Woodley R, Lu XJ, Srinivasan AR, Olson WK, Davé RN, Venanzi CA. Feature extraction using molecular planes for fuzzy relational clustering of a flexible dopamine reuptake inhibitor. J Chem Inf Model. 2007;47:2216–2227. doi: 10.1021/ci7001632. [DOI] [PubMed] [Google Scholar]
- 40.Babcock MS, Pednault EPD, Olson WK. Nucleic acid structure analysis: mathematics for local Cartesian and helical structure parameters that are truly comparable between structures. J Mol Biol. 1994;237:125–156. doi: 10.1006/jmbi.1994.1213. [DOI] [PubMed] [Google Scholar]
- 41.Clowney L, Jain SC, Srinivasan AR, Westbrook J, Olson WK, Berman HM. Geometric parameters in nucleic acids: nitrogenous bases. J Am Chem Soc. 1996;118:509–518. [Google Scholar]
- 42.Dickerson RE. DNA bending: the prevalence of kinkiness and the virtues of normality. Nucleic Acids Res. 1998;26:1906–1926. doi: 10.1093/nar/26.8.1906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gorin AA, Zhurkin VB, Olson WK. B-DNA twisting correlates with base pair morphology. J Mol Biol. 1995;247:34–48. doi: 10.1006/jmbi.1994.0120. [DOI] [PubMed] [Google Scholar]
- 44.Lavery R, Sklenar H. The definition of generalized helicoidal parameters and of axis curvature for irregular nucleic acids. J Biomol Struct Dynam. 1988;6:63–91. doi: 10.1080/07391102.1988.10506483. [DOI] [PubMed] [Google Scholar]
- 45.Lavery R, Sklenar H. Defining the structure of irregular nucleic acids: conventions and principles. J Biomol Struct Dynam. 1989;6:655–667. doi: 10.1080/07391102.1989.10507728. [DOI] [PubMed] [Google Scholar]
- 46.Macke T, Case DA. Modeling unusual nucleic acid structures. In: Leontis NB, SantaLucia J Jr, editors. Molecular Modeling of Nucleic Acids. American Chemical Society; Washington, DC: 1998. pp. 379–393. [Google Scholar]
- 47.Tung CS, Carter ES., II Nucleic acid modeling tool (NAMOT): an interactive graphic tool for modeling nucleic acid structures. Bioinformatics. 1994;10:427–433. doi: 10.1093/bioinformatics/10.4.427. [DOI] [PubMed] [Google Scholar]
- 48.Major F. Building three-dimensional ribonucleic acid structures. IEEE Computing in Science & Engineering. 2003;5:44–53. [Google Scholar]
- 49.Sayle RA, Milnerwhite EJ. RasMol: biomolecular graphics for all. Trends Biochem Sci. 1995;20:374–376. doi: 10.1016/s0968-0004(00)89080-5. [DOI] [PubMed] [Google Scholar]
- 50.Massire C, Gaspin C, Westhof E. DRAWNA: a program for drawing schematic views of nucleic acids. J Mol Graph. 1994;12:201–206. doi: 10.1016/0263-7855(94)80088-x. [DOI] [PubMed] [Google Scholar]
- 51.Huang CC, Couch GS, Pettersen EF, Ferrin TE. Chimera: an extensible molecular modeling application constructed using standard components. Pacific Symposium on Biocomputing. 1996;1:724. [Google Scholar]
- 52.Trifonov EN, Sussman JL. The pitch of chromatin DNA is reflected in its nucleotide sequence. Proc Natl Acad Sci USA. 1980;77:3816–3820. doi: 10.1073/pnas.77.7.3816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hagerman PJ. Sequence dependence of the curvature of DNA: a test of the phasing hypothesis. Biochemistry. 1985;24:7033–7037. doi: 10.1021/bi00346a001. [DOI] [PubMed] [Google Scholar]
- 54.Koo HS, Wu HM, Crothers DM. DNA bending at adenine·thymine tracts. Nature. 1986;308:501–506. doi: 10.1038/320501a0. [DOI] [PubMed] [Google Scholar]
- 55.Hagerman PJ. Sequence-directed curvature of DNA. Nature. 1986;321:449–450. doi: 10.1038/321449a0. [DOI] [PubMed] [Google Scholar]
- 56.Ulanovsky LE, Trifonov EN. Estimation of wedge components in curved DNA. Nature. 1987;326:720–722. doi: 10.1038/326720a0. [DOI] [PubMed] [Google Scholar]
- 57.Schultz SC, Shields GC, Steitz TA. Crystal structure of a CAP-DNA complex: the DNA is bent by 90°. Science. 1991;253:1001–1007. doi: 10.1126/science.1653449. [DOI] [PubMed] [Google Scholar]
- 58.Parkinson G, Wilson C, Gunasekera Ebright YW, Ebright RE, Berman HM. Structure of the CAP-DNA complex at 2.5 Å resolution: a complete picture of the protein-DNA interface. J Mol Biol. 1996;260:395–408. doi: 10.1006/jmbi.1996.0409. [DOI] [PubMed] [Google Scholar]
- 59.Richmond TJ, Davey CA. The structure of DNA in the nucleosome core. Nature. 2003;423:145–150. doi: 10.1038/nature01595. [DOI] [PubMed] [Google Scholar]
- 60.Calladine CR, Drew HR, Luisi BF, Travers AA. Understanding DNA: The Molecule and How It Works. Chpt 4. San Diego, CA: Elsevier Academic Press; 2004. [Google Scholar]
- 61.Olson WK, Srinivasan AR, Marky NL, Balaji VN. Theoretical probes of DNA conformation examining the B leads to Z conformational transition. Cold Spring Harb Symp Quant Biol. 1983;47:229–241. doi: 10.1101/sqb.1983.047.01.028. [DOI] [PubMed] [Google Scholar]
- 62.Harvey SC. DNA structural dynamics: longitudinal breathing as a possible mechanism for the B->Z transition. Nucleic Acids Res. 1983;11:4867–4878. doi: 10.1093/nar/11.14.4867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ha SC, Lowenhaupt K, Rich A, Kim YG, Kim KK. Crystal structure of a junction between B-DNA and Z-DNA reveals two extruded bases. Nature. 2005;437:1183–186. doi: 10.1038/nature04088. [DOI] [PubMed] [Google Scholar]
- 64.Nowakowski J, Shim PJ, Stout CD, Joyce GF. Alternative conformations of a nucleic acid four-way junction. J Mol Biol. 2000;300:93–102. doi: 10.1006/jmbi.2000.3826. [DOI] [PubMed] [Google Scholar]
- 65.Klein DJ, Schmeing TM, Moore PB, Steitz TA. The kink-turn: a new RNA secondary structure motif. EMBO J. 2001;20:4214–4221. doi: 10.1093/emboj/20.15.4214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Matsumoto A, Olson WK. Sequence-dependent motions of DNA: a normal mode analysis at the base-pair level. Biophys J. 2002;83:22–41. doi: 10.1016/S0006-3495(02)75147-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chen S, Vojtechovsky J, Parkinson GN, Ebright RH, Berman HM. Indirect readout of DNA sequence at the primary-kink site in the CAP-DNA complex: DNA binding specificity based on energetics of DNA kinking. J Mol Biol. 2001;314:63–74. doi: 10.1006/jmbi.2001.5089. [DOI] [PubMed] [Google Scholar]
- 68.Nissen P, Ippolito JA, Ban N, Moore PB, Steitz TA. RNA tertiary interactions in the large ribosomal subunit: the A-minor motif. Proc Natl Acad Sci USA. 2001;98:4899–4903. doi: 10.1073/pnas.081082398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wimberly BT, Guymon R, McCutcheon JP, White SW, Ramakrishnan V. A detailed view of a ribosomal active site: the structure of the L11-RNA complex. Cell. 1999;97:491–502. doi: 10.1016/s0092-8674(00)80759-x. [DOI] [PubMed] [Google Scholar]
- 70.Zoll J, Tessari M, Van Kuppeveld FJM, Melchers WJG, Heus HA. Breaking pseudo-twofold symmetry in the poliovirus 3′-UTR Y-stem by restoring Watson-Crick base pairs. RNA. 2007;13:781–792. doi: 10.1261/rna.375607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Davey CA, Sargent DF, Luger K, Mäder AW, Richmond TJ. Solvent mediated interactions in the structure of the nucleosome core particle at 1.9 Å resolution. J Mol Biol. 2002;319:1097–1113. doi: 10.1016/S0022-2836(02)00386-8. [DOI] [PubMed] [Google Scholar]
- 72.Westbrook J, Ito N, Nakamura H, Henrick K, Berman HM. PDBML: the representation of archival macromolecular structure data in XML. Bioinformatics. 2005;7:988–992. doi: 10.1093/bioinformatics/bti082. [DOI] [PubMed] [Google Scholar]