

Abstract
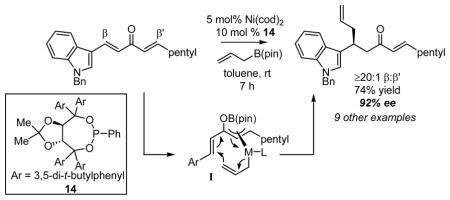
The nickel-catalyzed enantioselective addition of allylboronic acid pinacol ester - allylB(pin) – is described. This reaction is highly effective with dialkylidene ketones and favors the allylation of the benzylidene site in non-symmetric substrates. The reaction appears to proceed by conversion of the dialkylidene ketone substrate to an unsaturated π-allyl complex (I), followed by reductive elimination. Enantioselectivities range from 91–94% ee for a range of substrates when chiral ligand 14 is employed.
1. Introduction
Conjugate addition of allyl groups to activated alkenes is an important method for the construction of organic compounds. Not only does this transformation establish a new carbon-carbon bond and result in the creation of a new stereocenter, it also provides a malleable product that contains carbonyl and olefin functional groups. The conjugate addition of allyl nucleophiles is a well known process that can be accomplished with silicon,1 tin,2 and copper reagents.3 More recently, the synthetic utility of allyltantalum,4 allylbarium,5 and allylindium6 reagents in conjugate allylation has been recognized. However, asymmetric variants of these processes have not been achieved. For control of absolute facial selectivity chiral auxiliaries are required, and this approach has been examined with allyltributylstannane,7 allyltrimethylsilane,8 and allyl Grignard reagents,9 in conjunction with stoichiometric amounts of copper or a Lewis acid. Significantly, despite intense research focused toward enantioselective metal-catalyzed conjugate addition processes,10 a catalytic asymmetric conjugate addition of allyl nucleophiles to α,β-unsaturated carbonyl electrophiles has yet to emerge.
In a previous communication, we reported that Ni and Pd complexes can catalyze the addition of allylboronic acid pinacol ester (allylB(pin), 2, Scheme 1) to dialkylidene ketones (i.e. 1, Scheme 1).11 While allylboronates typically undergo 1,2-allylation reactions with carbonyl electrophiles, in the presence of a transition metal catalyst, dialkylidene ketones participate in 1,4-conjugate addition reactions. Preliminary studies suggest that this reaction proceeds by a mechanism involving oxidative addition of a Lewis acid-activated enone to the transition metal catalyst (to provide II, Scheme 1),12 followed by transmetalation (II→III) and 3,3′ reductive elimination13,14 from unsaturated π-allyl complex III; the latter elementary process was calculated to proceed with an extremely small activation barrier (for M=Pd, ΔG‡=1.52 kcal/mol). These preliminary studies also documented an example of asymmetric catalysis with dibenzylidene acetone as substrate and a phosphoramidite-derived Pd complex as catalyst. Herein, we describe an effective Ni(0) catalyst system that allows enantioselective and chemoselective conjugate allylation of non-symmetric dialkylidene ketones. Also described are a model for chemoselectivity and mechanistic aspects of this reaction process.
Scheme 1.

2. Results and Discussion
2.1 Reaction Development and Substrate Scope
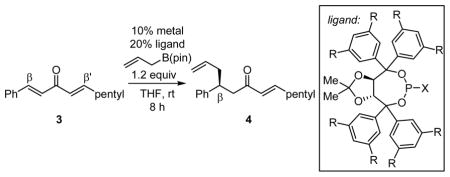
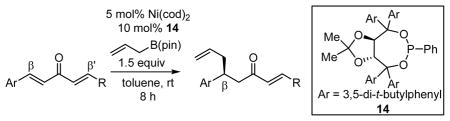
With the goal of developing a general catalytic enantioselective conjugate allylation, a collection of chiral phosphorous ligands was surveyed in the conjugate allylation of non-symmetric dialkylidene ketone 3 (Table 1). TADDOL-derived phosporamidites,15 phosphonites, and phosphites were initially explored. For comparison purposes, the reaction with Ni(cod)2 and PCy3 is presented in entry 1; this catalyst favors allylation at the alkylidene site. The catalyst derived from Pd2(dba)3 and phosphoramidite 5, an effective combination with dba as substrate, displayed low chemoselectivity with non-symmetric substrate 3 (entry 2). Alternatively, moderate chemoselectivity was observed with Ni(cod)2 and phosphoramidite 5 (entry 7), an observation which stimulated further investigation of related ligands. While a variety of TADDOL-derived phosphoramidites and phosphites furnished moderate enantioselection and/or chemoselection, phosphonite ligand 14 (entry 12) provided a unique combination of high enantioselection and high chemoselectivity with non-symmetric substrate 3. Notably, the sense of chemoselectivity with all the heteroatom-derived phosphorous ligands, including 14, was opposite to that observed with PCy3 (entry 1). Thus, the present catalyst system provides a means for construction of benzylic hydrocarbon stereocenters through catalytic asymmetric conjugate allylation.
Table 1.
Substrate Survey for Asymmetric Conjugate Allylation.
 | |||||||
|---|---|---|---|---|---|---|---|
| entry | metal | ligand | R | X | β:β′a | % yieldb | % eec |
| 1 | Ni(cod)2 | PCy3 | 1:7.5 | 82 | |||
| 2d | Pd2(dba)3 | 5 | t-Bu | NMe2 | 1:1.6 | 74 | 33(96) |
| 3 | Ni(cod)2 | 6 | H | NMe2 | 3.8:1 | 36 | 44(75) |
| 4 | Ni(cod)2 | 7 | H | N(CH2)4 | 4.3:1 | 31 | 14 |
| 5 | Ni(cod)2 | 8 | H | N(CH2)5 | 2.2:1 | 57 | −3 |
| 6 | Ni(cod)2 | 9 | Me | NMe2 | >20:1 | 53 | 44 |
| 7 | Ni(cod)2 | 5 | t-Bu | NMe2 | 4.7:1 | 29 | 69 |
| 8 | Ni(cod)2 | 10 | t-Bu | N(CH2)5 | 2.5:1 | 24 | 47 |
| 9 | Ni(cod)2 | 11 | t-Bu | OEt | 16:1 | 62 | −3 |
| 10 | Ni(cod)2 | 12 | H | Ph | 3.6:1 | 49 | 28 |
| 11 | Ni(cod)2 | 13 | Me | Ph | >20:1 | 50 | −15 |
| 12 | Ni(cod)2 | 14 | t-Bu | Ph | 20:1 | 69 | 88 |
Chemoselectivity determined by 1H NMR.
Yield of conjugate addition product after silica gel chromatography.
Enantioselectivity of β isomer, number in parentheses is for the β′ isomer.
2.5 mol% Pd2(dba)3, 6 mol% ligand 5, and reaction for 24 hours.
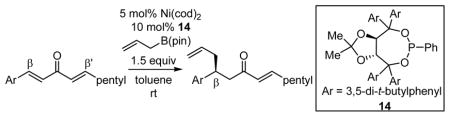
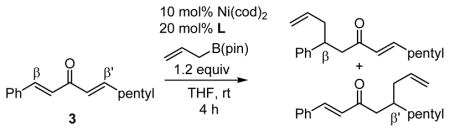
After further optimization of the reaction conditions (decreased catalyst loading, solvent optimization), an analysis of the substrate scope for the Ni-catalyzed asymmetric conjugate allylation was undertaken. As shown in Table 2, the reaction exhibits high enantioselection regardless of the nature of the arylidene group, and generally favors chemoselective allylation at the arylidene site with Ni(cod)2 and phosphonite 14. Electron-deficient arenes were efficient in terms of reaction rate, however, chemoselectivity was lower for these substrates (entries 2 and 6). Electron-rich arenes reacted slower and therefore required longer reaction times, however, chemoselectivities were better for these substrates (entries 3, 9, and 10). Substitution at the ortho position of the substrate dramatically increased the chemoselectivity, but these substrates were allylated more slowly (entries 4, 5 and 8). In addition, substrates bearing functionalized aromatic groups common in natural products and drug targets were effective in the reaction (entries 9 and 10).
Table 2.
Scope of Ni-Catalyzed Enantioselective Conjugate Allylation.
 | |||||
|---|---|---|---|---|---|
| entry | product | time (h) | β:β′a | yield (%)b | % eec |
| 1 | 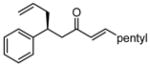 |
7 | 17:1 | 77 | 93 |
| 2 | 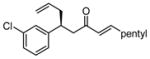 |
7 | 6.5:1 | 81 | 93 |
| 3 | 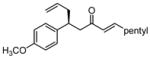 |
12 | 20:1 | 66 | 91 |
| 4 | 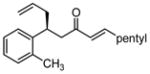 |
12 | ≥20:1 | 66 | 93 |
| 5 | 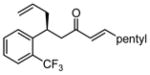 |
48 | 32:1 | 45 | 94 |
| 6 | 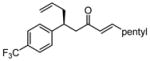 |
7 | 7.0:1 | 78 | 94 |
| 7 | 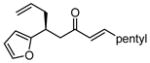 |
12 | 5.1:1 | 52 | 93 |
| 8 | 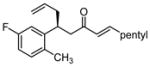 |
12 | 49:1 | 81 | 92 |
| 9 | 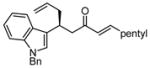 |
12 | ≥20:1 | 74 | 92 |
| 10 | 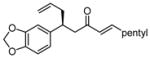 |
12 | 21:1 | 60 | 92 |
Determined by GC or 1H NMR analysis of the unpurified reaction mixture.
Isolated yield of purified material. Value is an average of two experiments.
Determined by chiral GC or SFC analysis.
Manipulation and cleavage processes of the activating alkylidene auxiliary merit mention. As depicted in Scheme 2, the alkylidene unit provides a convenient handle for alkene metathesis16 and a ring-closing version was readily accomplished with the NHC-derived Hoveyda-Grubbs catalyst17 to give cyclohexenone 15. Alternatively, regioselective Baeyer-Villiger oxidation was accomplished by treatment of the conjugate allylation product with bis(trimethylsilyl)peroxide in the presence of Lewis acid.18 This transformation selectively provided an intermediate enol ester which might be readily converted to a variety of functional groups including the derived carboxylic acid 16 as is shown.
Scheme 2.

2.2 Mechanistic Aspects (a) Probe Substrates
The nickel-catalyzed conjugate addition of allylB(pin) to enones requires alkylidene activation. As depicted in Scheme 3 (equation 3), there was <5% conversion when benzylidene acetone was used as the substrate in the reaction. Even when the reaction with benzylidene acetone was carried out at higher temperature, the 1,4 allylation product was not observed, the only product being that derived from 1,2 allylation of the ketone. Notably, the rate and chemoselectivity of the benzylidene acetone allylation reaction at high temperature were nearly identical in the presence or absence of catalyst, further suggesting the requirement for alkylidene activation of the catalytic reaction. Considering that the catalyst derived from Ni(cod)2 and PCy3 favors allylation at the alkylidene site (Table 1, entry 1), the substrate in equation 4 was examined; again, no reaction occurs without benzylidene activation.
Scheme 3.

The enhanced reactivity of dialkylidene ketones relative to simple enones is consistent with the proposed requirement that the reaction proceed by 3,3′ reductive elimination of an unsaturated Ni bis(allyl) complex (III, Scheme 1). As mentioned in the introduction, DFT calculations are consistent with this conjecture. To experimentally substantiate the relevance of this elementary step, the experiments in equations 5 and 6 (Scheme 4) were undertaken. When substrate 17, possessing an internal phosphine ligand, was treated with allylB(pin) and 10 mol% Ni(cod)2, the conjugate allylation product was isolated as a single constitutional isomer wherein allyl addition occurred distal to the phosphine directing group. The control experiment with substrate 18 (equation 6) highlights the significant enhancement in chemoselectivity that the tethered phosphine imparts on the catalytic allylation, in its absence a 1.9:1 mixture of constitutional isomers – favoring allylation proximal to the benzoate – was observed.
Scheme 4.

To verify whether the tethered phosphine in compound 17 directs the metal to the proximal or distal site in derived π complexes, the structure of a 1:1:1 mixture comprised of 17, Ni(cod)2, and PPh3 was examined (Scheme 5). While x-ray quality crystals could not be obtained, NMR analysis of the mixture in solution was informative. Proton and carbon assignments were made by 1H, 13C, COSY, NOESY and HSQC spectroscopic analysis. Upon treatment with Ni(cod)2 and PPh3, the hydrogens at C3 and C4 exhibit an upfield shift of Δδ 3.01 ppm and Δδ 2.45 ppm, respectively. Likewise the carbon resonances for C3 and C4 move to higher field by Δδ 71.6 ppm and Δδ 60.2 ppm. In contrast, the hydrogens at C6 and C7 exhibit less substantial perturbation (Δδ 1.01 ppm and Δδ <0.05 ppm). Similarly, the carbon resonances for C6 and C7 are relatively unaffected, showing Δδ 0.6 ppm and Δδ 1.8 ppm, respectively. In addition to its effect on the alkene hydrogens, the metal association affects the chemical shift of the hydrogens at C2 and C1. The proton resonance at C2 experiences a substantial upfield shift (Δδ 0.76 ppm) and the protons at C1 in the metal complex have clearly become diastereotopic, one moving 0.72 ppm downfield and the other 0.44 ppm upfield. Collectively, the spectroscopic changes observed in substrate 17, upon addition of nickel and PPh3, are consistent with the Ni binding to the alkene adjacent to the directing group. Other spectroscopic features are also consistent with the formulation in Scheme 5; the 31P NMR shows two doublet resonances at 41.21 ppm and 28.59 ppm (J=32 Hz).
Scheme 5.

The propensity for the phosphine in 17 to direct Ni coordination to the adjacent alkene, combined with the chemoselectivity observed for the Ni-catalyzed allylation of 17, is highly suggestive of a reaction that proceeds by the proposed 3,3′ reductive elimination process. As shown in Scheme 6, it is tenable that upon directed oxidative formation of π-allyl 19, transmetalation occurs to give 20. Consistent with the proposed low-energy barrier for 3,3′ reductive elimination, 20 transfers the allyl group to the olefin that is distal to the phosphine directing group providing 21.
Scheme 6.

Experiments undertaken with isotopically labeled boronate 22 were also revealing. As depicted in equations 7 and 8 (Scheme 7), the isotope label in 22 was scrambled between the internal and terminal allyl sites when the reaction was carried out with Ni(cod)2/PCy3 catalyst, but was retained at the internal site when Pd2(dba)3/PCy3 was employed. Considering that transmetalation of allyl groups is known to proceed by an SE′-type mechanism,19 it stands to reason that with Pd as catalyst, a rapid 3,3′ reductive elimination leads to the product which is deuterated at the internal site (Scheme 8). The isotope scrambling observed with Ni catalysts can be ascribed to isomerization of the intermediate allyl complex 24, perhaps through a structure such as 26 (Scheme 8).
Scheme 7.

* note: 22 contaminated with 9% vinylB(pin)
Scheme 8.

2.2 Mechanistic Aspects (b) Chemoselectivity
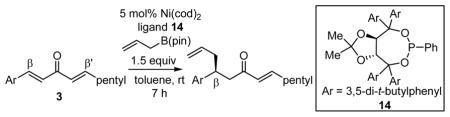
As noted above, the hexylidene substituent activates the adjacent arylidene for conjugate allylation. Examination of other alkylidene groups revealed a correlation between alkylidene size and both stereo- and chemoselectivity. When the pentyl group of the alkylidene was replaced with a methyl group (Table 3, entry 1), the chemoselectivity for arylidene allylation increased (17:1 → ≥20:1), however, a corresponding decrease in reaction enantioselectivity was noted. When the pentyl group was replaced with a larger cyclohexyl substituent (entry 4), the opposite outcome was observed: chemoselectivity was diminished and the enantioselectivity was enhanced. This outcome is consistent with the above mechanistic hypothesis: enlarging the alkylidene substituent causes increased π-allyl formation at the arylidene site which leads to more alkylidene allylation product. The same outcome was observed as the size of the arylidene substituent was enhanced: in the series 2-furyl, phenyl, ortho-tolyl, ortho-trifluoromethylphenyl, the selectivity for arylidene allylation increased from 5.1:1 to 32:1 (Table 1, entries 5, 3, 6 and 7, respectively).
Table 3.
Correlation between Substrate Structure and Chemoselectivity.
 | |||||
|---|---|---|---|---|---|
| entry | Ar | R | β:β′a | % yieldb | % eec |
| 1 | Ph | Me | ≥20:1 | 60 | 67 |
| 2 | Ph | Et | ≥20:1 | 82 | 83 |
| 3 | Ph | n-pentyl | 17:1 | 77 | 93 |
| 4 | Ph | Cy | 7:1 | 76 | 96 |
| 5 | 2-furyl | n-pentyl | 5.1:1 | 52 | 93 |
| 6 | o-tol | n-pentyl | ≥20:1 | 66 | 93 |
| 7 | o-CF3Ph | n-pentyl | 32:1 | 45 | 94 |
Chemoselectivity determined by 1H NMR or GC analysis of the unpurified reaction mixture.
Yield of conjugate addition product after silica gel chromatography. Value is an average of two experiments in each case.
Enantioselectivity of major isomer.
Along with a correlation between substrate steric properties and chemoselectivity, a rough correlation between ligand basicity and chemoselectivity was observed. As depicted in Table 4, the Lewis basic phosphine PCy3 favors allylation of the alkylidene site of non-symmetric substrate 3. With achiral ligands, when the basicity of the ligand structure decreased, as measured by the CO stretching frequency of derived trans-L2Rh(CO)Cl complex,20,21 the selectivity for alkylidene allylation decreased (Table 4). Comparing entries 4 and 5 suggests that steric effects may be less important than electronic effects: the Tolman cone angles of P(O-o-t-BuPh)3 and P(OPh)3 are 175° and 128° respectively,22 yet the chemoselectivity was almost identical and appears to better follow the ligand basicity correlation. Notably, these generalizations do not appear to apply to chiral ligands, where chemoselectivity is not clearly related to either electronic or steric features. For instance, ligand 9 gives predominant addition to the arylidene, whereas the reaction employing electronically similar ligand PPh3 is relatively non-selective. The fact that there is a non-linear correlation between the size of remote ligand substituents and chemoselectivity (compare entries 3, 6 and 7, in Table 1) suggests that catalyst-substrate interactions, distant from the metal center, can have a significant effect on selectivity.
Table 4.
Correlation between Ligand Basicity and Chemoselectivity.
 | ||||
|---|---|---|---|---|
| entry | L | νCO (cm−1)a | β:β′b | % yieldc |
| 1 | PCy3 | 1943 | 1:7.5 | 82 |
| 2 | P(NMe2)3 | 1964 | 1:2.5 | 82 |
| 3 | PPh3 | 1979 | 1:2.4 | 49 |
| 4 | P(O-o-tBuPh)3 | 2012 | 1:1.5 | 40 |
| 5 | P(OPh)3 | 2016 | 1:1.1 | 55 |
| 6c | 9 | 1984 | ≥20:1 | 53 |
Values are for the derived trans-L2Rh(CO)Cl complex in CH2Cl2 solvent. See reference 21 for values.
Chemoselectivity determined by 1H NMR analysis of the unpurified reaction mixture.
Yield of conjugate addition product after silica gel chromatography. Value is an average of two experiments in each case.
A working model for chemoselectivity is depicted in Scheme 9. The data above is most readily accommodated by considering the stability of the π-allyl complexes: both small alkylidene substituents and large arylidene groups enhance the amount of π-allyl complex at the alkylidene site. Subsequent 3,3′ elimination transfers the allyl group to the arylidene unit and leads to an enhanced β:β′ ratio. The opposite outcome is observed with large alkylidene groups and small arylidene groups. Additionally, alkylidene allylation is increasingly favored with electron-rich ligands. This outcome may be rationalized by assuming again that π-allyl stability is product determining, and that with an electron-rich metal the π-allyl bearing the electron-accepting aryl ring is favored. Arguably, the aryl group may facilitate dπ-π* donation in the precursor LnNi π complex, similar to the dba-effect23 recognized in cross-coupling reactions, and thereby favor π-allyl formation at the arylidene site. This contention is supported by observations with electronically modified substrates in the presence of chiral ligand 14: the para-methoxy phenyl subsitutent provides more arylidene allylation (Table 2, entry 3) relative to the electron withdrawing para-trifluoromethyl phenyl derivative (Table 2, entry 6).
Scheme 9.

2.2 Mechanistic Aspects (c) Catalyst Composition
The concentration of ligand relative to the metal catalyst has a significant impact on the reaction outcome. As depicted in Table 5, in the absence of ligand, 1,2 allylation of the enone was the predominant product and the 1,4 addition occurred exclusively at the alkylidene site (entry 1). Addition of 0.5 equivalents of ligand, relative to nickel, was sufficient to reverse the 1,2:1,4 selectivity providing a 5:1 ratio favoring conjugate allylation; further, the addition of ligand reversed the chemoselectivity of the conjugate addition product and favored the arylidene site by 6.6:1. The fact that pronounced ligand acceleration24 operates was revealed by noting a significant enhancement in extent of reaction (82% versus 38% conversion) and high enantioselection for the arylidene allylation product (95% ee), even in the presence of excess Ni(cod)2 relative to ligand. A further increase in ligand:metal ratio (1:1) embellished the trend initiated by the first data point, and a ligand:metal ratio >1:1 did not offer any additional improvement.
Table 5.
Effect of Ligand:Metal Ratio.
 | |||||
|---|---|---|---|---|---|
| entry | mol% (R,R)-14 | 1,4:1,2 | β:β′a | % convb | % ee (β)c |
| 1 | 0 | 1:1.4 | 1:>50 | 38 | |
| 2 | 2.5 | 5:1 | 6.6:1 | 82 | 95 |
| 3 | 5 | >20:1 | 17:1 | >95 | 95 |
| 4 | 7.5 | >20:1 | 17:1 | >95 | 94 |
| 5 | 10 | >20:1 | 17:1 | >95 | 93 |
Chemoselectivity determined by GC analysis of the unpurified reaction mixture.
Yield of conjugate addition product after silica gel chromatography. Value is an average of two experiments in each case.
Enantioselectivity of major isomer.
To learn more about the nature of the catalytic reaction, nonlinear effects were examined. As depicted in Figure 1, there was a subtle negative non-linear effect when the reaction enantioselectivity and ligand enantiopurity were compared. Importantly, the chemoselectivity also changed in this series of experiments, decreasing from 27:1 β:β′ when racemic 14 was used, to 17:1 β:β′ when ligand 14 of >99% ee was employed. Along with a change in selectivity, it was also determined that the reaction with racemic catalyst is faster than the reaction with enantiopure catalyst (Figure 2).
Figure 1.

Correlation between product enantiomeric purity, chemoselectivity, and ligand enantiomeric purity. Dashed line is for a linear correlation between % ee product and % ee ligand.
Figure 2.

Comparison of reaction rates in the presence of enantiomerically enriched ligand and racemic ligand.
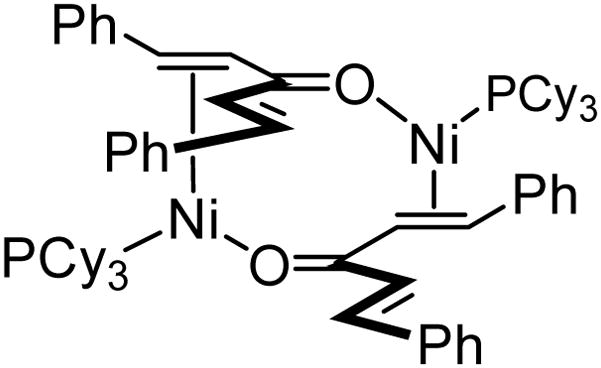
Given the propensity for Pd to bind two monodentate phosphines when bound to dibenzylideneacetone,25 it is conceivable that the non-linear effects described above might arise due to the intermediacy of a (substrate)ML2 complex. Examination of the catalyst structure by NMR provided an alternate rationale for the effect of catalyst enantiomeric purity on reaction outcome. When PCy3 was added to Ni(cod)2 in a 1:1 ratio in THF-d8, no observable reaction occurs. However, when dibenzylidene acetone was added in an equimolar proportion, 31P NMR indicated clean formation of a new complex characterized by a singlet resonance at 38.19 ppm. Importantly, when two equivalents of PCy3 were employed, only one equivalent of the ligand was incorporated into the new structure, the remainder was uncomplexed as determined by 31P NMR. The 1H NMR spectrum of the 1:1:1 complex revealed upfield-shifted olefinic resonances. Integration was consistent with only one alkene participating in coordination to the metal, but not the other. A structural formulation such as that in Scheme 10, may account for the spectral properties of the complex. Similar dimeric Ni-enone complexes have recently been isolated by Kurosawa as a product of the reaction between Ni(0), PCy3 and cyclopropyl ketones, and they are spectroscopically similar to the 1:1:1 complex described above.26 Thus, in the case of substrate 3 and chiral ligand 14 of varying enantiomeric purity, it is tenable that dimeric [(14)Ni-enone]2 structures are involved; either allylB(pin) induced oxidative addition, or dimer dissociation, occurs at different rates and with different chemoselectivity for the heterochiral versus the homochiral complexes.
Scheme 10.
3. Conclusion
In conclusion, we have documented the first catalytic enantioselective conjugate allylation reactions that may be applied to a range of activated aromatic enones. Experiments implicate the intermediacy of dimeric metal-ligand-substrate complexes and a catalytic cycle that operates by oxidative π-allyl formation, followed by 3,3′ reductive elimination. Studies that focus on the application of these concepts to related reactions are in progress.
Supplementary Material
Acknowledgments
Support by the NIGMS (GM-64451) and Merck Research Laboratories is appreciated. JDS acknowledges an ACS Nelson J. Leonard/Organic Syntheses fellowship and a John LaMattina Graduate Fellowship.
Footnotes
Supporting Information: Characterization and procedures. This material is free of charge via the internet at http://pubs.acs.org.
References
- 1.(a) Hosomi A, Sakurai H. J Am Chem Soc. 1977;99:1673. [Google Scholar]; (b) Hayashi M, Mukaiyama T. Chem Lett. 1987:289. [Google Scholar]; (c) Lee PH, Lee K, Sung SY, Chang S. J Org Chem. 2001;66:8646. doi: 10.1021/jo0105641. and references therein. [DOI] [PubMed] [Google Scholar]
- 2.Hosomi A, Iguchi H, Endo M, Sakurai H. Chem Lett. 1979:977. [Google Scholar]
- 3.(a) House HO, Wilkins JM. J Org Chem. 1978;43:2443. [Google Scholar]; (b) Lipshutz BH, Hackmann C. J Org Chem. 1994;59:7437. [Google Scholar]
- 4.Shibata I, Kano T, Kanazawa N, Fukuoka S, Baba A. Angew Chem Int Ed. 2002;41:1389. doi: 10.1002/1521-3773(20020415)41:8<1389::aid-anie1389>3.0.co;2-d. [DOI] [PubMed] [Google Scholar]
- 5.Yanagisawa A, Habaue S, Yasue K, Yamamoto H. J Am Chem Soc. 1994;116:6130. [Google Scholar]
- 6.(a) Lee PH, Ahn H, Lee K, Sunga SY, Kimb S. Tetrahedron Lett. 2001;42:37. [Google Scholar]; (b) Lee PH, Seomoon D, Kim S, Nagaiah K, Damle SV, Lee K. Synthesis. 2003:2189. [Google Scholar]
- 7.Williams DR, Mullins RJ, Miller NA. Chem Commun. 2003:2220. doi: 10.1039/b305159e. [DOI] [PubMed] [Google Scholar]
- 8.(a) Hon YS, Chen FL, Huang YP, Lu TJ. Tetrahedron: Asymm. 1991:879. [Google Scholar]; (b) Wu MJ, Wu CC, Lee PC. Tetrahedron Lett. 1992;33:2547. [Google Scholar]
- 9.(a) Williams DR, Kissel WS, Li JJ. Tetrahedron Lett. 1998;39:8593. [Google Scholar]; (b) Schneider C, Reese O. Synthesis. 2000:1689. [Google Scholar]
- 10.(a) Tomioka K, Nagaoka Y. In: Comprehensive Asymmetric Catalysis. Jacobsen EN, Pfaltz A, Yamamoto H, editors. Vol. 3. Springer-Verlag; Berlin: 1999. p. 1105. [Google Scholar]; (b) Krause N, Hoffmann-Röder A. Synthesis. 2001. p. 171. [Google Scholar]; (c) Feringa BL, Naasz R, Imbos R, Arnold LA. In: Modern Organocopper Chemistry. Krause N, editor. Wiley-VCH; Weinham: 2002. p. 224. [Google Scholar]; (d) Christoffers J, Koripelly G, Rosiak A, Rossle M. Synthesis. 2007;9:1279. [Google Scholar]
- 11.Sieber JD, Shubin L, Morken JP. J Am Chem Soc. 2007;129:2214. doi: 10.1021/ja067878w. [DOI] [PubMed] [Google Scholar]
- 12.For related oxidative additions with Ni and Pd, see: Brookhart M, Young GJ. J Chem Soc, Chem Comm. 1974:205.Johnson JR, Tully PS, Mackenzie PB, Sabat M. J Am Chem Soc. 1991;113:6172.Grisso BA, Johnson JR, Mackenzie PB. J Am Chem Soc. 1992;14:5160.Grennberg H, Gogoll A, Baeckvall JE. Organometallics. 1993;12:1790.Ogoshi S, Yoshida T, Nishida T, Morita M, Kurosawa H. J Am Chem Soc. 2001;123:1944. doi: 10.1021/ja0036099.Morita M, Inoue K, Ogoshi S, Kurosawa H. Organometallics. 2003;22:5468.Ogoshi S, Morita M, Kurosawa H. J Am Chem Soc. 2003;125:9020. doi: 10.1021/ja0361042.Morita M, Inoue K, Yoshida T, Ogoshi S, Kurosawa H. J Organomet Chem. 2004;689:894.
- 13.(a) Méndez M, Cuerva JM, Gómez-Bengoa E, Cárdenas DJ, Echavarren AM. Chem Eur J. 2002;8:3620. doi: 10.1002/1521-3765(20020816)8:16<3620::AID-CHEM3620>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]; (b) Cárdenas DJ, Echavarren AM. New J Chem. 2004;28:338. [Google Scholar]; (c) Ariafard A, Lin Z. J Am Chem Soc. 2006;128:13010. doi: 10.1021/ja063944i. [DOI] [PubMed] [Google Scholar]
- 14.For Ni and Pd-catalyzed conjugate additions that operate by oxidative addition/transmetalation/reductive elimination, as opposed to transmetalation/migratory insertion, see: (a) reference 12 c, e. Hirano K, Yorimitsu H, Oshima K. Org Lett. 2007:1541. doi: 10.1021/ol070288y.
- 15.Review of these ligands in catalysis: Feringa BL. Acc Chem Res. 2000;33:346. doi: 10.1021/ar990084k.Select examples, hydrogenation: ven den Berg M, Minnard AJ, Schudde EP, van Esch J, de Vries AHM, de Vries JG, Feringa BL. J Am Chem Soc. 2000;122:11539.Hydrosilation: Jensen JF, Svendsen BY, la Cour TV, Pedersen HL, Johannsen M. J Am Chem Soc. 2002;124:4558. doi: 10.1021/ja025617q.Hydroboration: Ma MFP, Li K, Zhou Z, Tang C, Chan ASC. Tetrahedron: Asymm. 1999;10:3259.
- 16.Grubbs RH, editor. Handbook of Metathesis. Wiley-VCH; Weinheim, Germany: 2003. [Google Scholar]
- 17.Garber SB, Kingbury JS, Gray BL, Hoveyda AH. J Am Chem Soc. 2000;122:8168. [Google Scholar]
- 18.Göttlich R, Yamakoshi K, Sasai H, Shibasaki M. Synlett. 1997:971. [Google Scholar]
- 19.(a) Hayashi T, Konishi M, Kumada M. J Chem Soc Chem Commun. 1983:736. [Google Scholar]; (b) Hatanaka Y, Goda K, Hiyama T. Tetrahedron Lett. 1994;35:1279. [Google Scholar]; (c) Hiyama T, Matsuhashi H, Fujita A, Tanaka M, Hirabayashi K, Shimizu M, Mori A. Organometallics. 1996;15:5762. [Google Scholar]; (d) Yamamoto Y, Takada S, Miyaura N. Chem Lett. 2006;35:704. [Google Scholar]
- 20.(a) Vastag S, Heil B, Markó L. J Mol Cat. 1979;5:189. [Google Scholar]; (b) Ohgomori Y, Yoshida SI, Watanabe Y. J Chem Soc, Dalton Trans. 1987:2969. [Google Scholar]
- 21.For the values in Table 3, see: Entries 1–3: Otto S, Roodt A. Inorg Chim Acta. 2004;357:1.Entry 4: Fernández E, Ruiz A, Claver C, Castillón S, Polo A. Organometallics. 1998;17:2857.Entry 5: ref 20a. Entry 6: Burks HE, Liu S, Morken JP. J Am Chem Soc. 2007;129:8766. doi: 10.1021/ja070572k.
- 22.Tolman CA. Chem Rev. 1977;77:313. [Google Scholar]
- 23.(a) Mace Y, Kapdi AR, Fairlamb JS, Jutand A. Organometallics. 2006;25:1795. [Google Scholar]; (b) Fairlamb IJS, Kapdi AR, Lee AF. Org Lett. 2004;6:4435. doi: 10.1021/ol048413i. [DOI] [PubMed] [Google Scholar]
- 24.Berrisford DJ, Bolm C, Sharpless KB. Angew Chem Int Ed. 1995;34:1059. [Google Scholar]
- 25.(a) Amatore C, Jutland A. Coord Chem Rev. 1998;178–180:511. [Google Scholar]; (b) Amatore C, Jutland A, Meyer G. Inorg Chim Acta. 1998;273:76. [Google Scholar]
- 26.Ogoshi S, Nagata M, Kurosawa H. J Am Chem Soc. 2006;128:5350. doi: 10.1021/ja060220y. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.