

Abstract
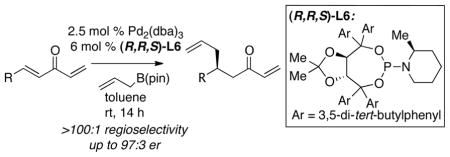
The use of unsaturated methylidene ketones in catalytic conjugate allylations allows for a significant expansion in substrate scope and, with appropriate chiral ligands, occurs in a highly enantioselective fashion.
Catalytic enantioselective conjugate addition of organometallic reagents to unsaturated carbonyls is an important method in asymmetric synthesis. These reactions are usually accomplished under the aegis of late transition metal catalysts and occur with a broad variety of organometallic reagents.1 While significant successes have been recorded with aryl, alkyl, vinyl, and alkynyl nucleophiles, reactions of allylmetal reagents are much less developed. In recent studies, we have addressed this limitation and have introduced an enantio- and regioselective catalytic conjugate addition of allylB(pin) (2, Scheme 1) to arylidene alkylidene ketones (1).2 Mechanistic studies suggest that this reaction proceeds by way of Lewis acid-induced oxidative addition of the metal to the less hindered alkylidene enone (to give I), followed by transmetallation and 3,3′-reductive elimination3 via transition state II.4 Thus for substrate 1, the hexylidene group functions as an activator for allylation of an aryl-substituted enone. While this strategy is effective, it leaves a gap in technology for the selective allylation of alkyl-substituted enones.5 In this manuscript, we address this issue and describe a general strategy that is effective for the regio- and enantioselective allylation of both aromatic and aliphatic substituted enones.
Scheme 1.

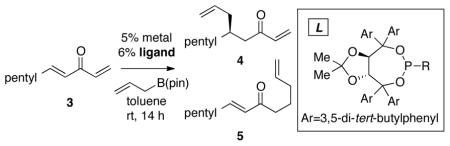
Under the paradigm described above, it appeared plausible that a general catalytic allylation of both aryl- and alkyl-substituted enones might arise from reactions of methylidene-substituted enones such as 3 (Table 1). In this scenario, oxidative addition of the transition metal to the less hindered methylidene site would be favored, and subsequent 3,3′ reductive elimination would deliver the allyl group to the adjacent internal prochiral alkene. An initial experiment probing this strategy was conducted with Taddol-derived phosphonite ligand L16 (Taddol = trans-4,5-Bis(diphenylhydroxymethyl)-2,2-dimethyldioxolane), the most effective ligand for conjugate allylation of arylidene alkylidene ketones (1). In the event, conjugate allylation of methylidene substrate 3 (Table 1) in the presence of 5 mol % Ni(cod)2, 6 mol % ligand L1 and 1.2 equiv of allylB(pin), provided addition product 4 with high regioselectivity and moderate enantiocontrol (entry 1). This encouraging result prompted further study. Examination of ligand structure L1 with Pd2(dba)3 (entry 2) revealed that the regioselectivity was slightly enhanced with Pd relative to Ni; however, the enantioselectivity was diminished. To improve enantioselection a number of other Taddol-derived phosphorus ligands were examined, and a selection of results is collected in Table 1. Some generalizations can be made: first, Pd-based catalysts provide higher regioselectivity than their Ni-based counterparts with the Pd catalysts most often delivering >20:1 ratio of regioisomeric conjugate allylation products (cf., entry 1 with 2; entry 3 vs. 4). Second, with phosphonite ligands, the Ni-catalyzed reaction occurs with higher enantioselection than does the Pd-catalyzed process (cf., entry 1 and 2). However, with phosphoramidite ligands, the opposite is true, and Pd catalysts offer superior levels of stereocontrol (cf., entry 5 and 6).
Table 1.
Catalytic Conjugate Allylation of Methylidene Ketone 3 with Chiral Taddol-Derived Ligandsa
 | ||||||
|---|---|---|---|---|---|---|
| entry | L | R | metal | 4:5 | % yield | er 4 |
| 1 | L1 | Ph | Ni(cod)2 | 14:1 | 21 | 77:23 |
| 2 | L1 | Ph | Pd2(dba)3 | 17:1 | 22 | 48:52 |
| 3 | L2 | NMe2 | Ni(cod)2 | 2.4:1 | 32 | 51:49 |
| 4 | L2 | NMe2 | Pd2(dba)3 | 20:1 | 15 | 66:34 |
| 5 | L3 | N(CH2)5 | Ni(cod)2 | 1.5:1 | 44 | 50:50 |
| 6 | L3 | N(CH2)5 | Pd2(dba)3 | 35:1 | 10 | 75:25 |
| 7 | L4 | N(CH2)6 | Pd2(dba)3 | 63:1 | 50 | 76:24 |
| 8 | L5 | NBn2 | Pd2(dba)3 | >100:1 | 14 | 75:25 |
Reactions were quenched by addition of H2O. Regioisomer ratio and enantiomer ratio were determined by GLC analysis.
The observations above stimulated further examination of Pd catalysts in the presence of phosphoramidite ligands. While small cyclic and acyclic dialkylamine derived phosphoramidite ligands generally furnished the conjugate allyation product in the 50% ee range, when substitution was added to the piperidine ring of phosphoramidite ligand L3, a significant enhancement in selectivity was observed. Notably, this effect is stereoisomer-specific: (R,R,S)-L6 delivered the conjugate allylation product in 93:7 enantiomer ratio whereas (S,S,S)-L6 provided the product in a 28:72 er (Scheme 2). Note that the turnover in enantioselection when the enantiomeric Taddol ligand is used in these experiments suggests that the Taddol backbone is the dominant enantiocontrolling element of the ligand with the piperidine substituent serving to embellish selectivity.
Scheme 2.

Enantioselective Conjugate Allylation of 3 with Diastereoisomeric Ligands.
As data in Scheme 2 reveals, the enantioselectivity of conjugate allylation with ligand (R,R,S)-L6 was quite good; however, the reaction yield was suboptimal. 1H NMR analysis of the reaction mixture after neutral aqueous work-up revealed a side-product that exhibited resonances corresponding to the boron enolate. The surprising reluctance of the putative enolate to undergo protonation was overcome by acidification during the reaction work-up: treatment with saturated ammonium chloride or glacial acetic acid delivered the reaction product in improved yields.
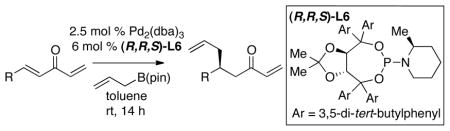
With an effective ligand for the asymmetric conjugate allylation of methylidene-activated enones and an effective experimental protocol, a study of the substrate scope was commenced. As depicted in Table 2, the simple pentyl-substituted substrate is converted to the allylation product with high levels of stereoinduction and in good yield (entry 1). This reaction constitutes the first catalytic enantioselective conjugate allylation of an acyclic aliphatic enone. Other aliphatic enones also participate in the reaction and it was shown that those bearing an aromatic ring (entry 2), those bearing branched substituents (entries 3 and 6), and substrates with oxygenation in the sidechain (entries 4–6) are also tolerated. Importantly, silyl ethers and more Lewis basic benzyl ethers are both processed with similar levels of stereoinduction, thereby indicating the absence of any complicating interaction between coordinating functional groups and boron-substituted catalytic intermediates. Also of note, aromatic enones can also react with good yield and stereoselection (entries 7–9), suggesting that the methylidene-based substrate design indeed offers a general solution to the conjugate allylation problem.
Table 2.
Catalytic Asymmetric Conjugate Allylation of Methylidene Ketones in the Presence of (R,R,S)-L6.a
 | ||||
|---|---|---|---|---|
| entry | product | regiob | % yieldc | erd |
| 1 |  |
>100:1 | 59 | 93:7 |
| 2 | 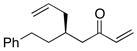 |
>100:1 | 79 | 94:6 |
| 3 | 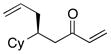 |
>100:1 | 57 | 95:5 |
| 4 | >100:1 | 91 | 91:9 | |
| 5 | 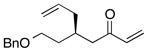 |
>100:1 | 53 | 91:9 |
| 6e | 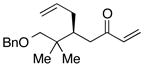 |
>100:1 | 81 | 96:4 |
| 7 | 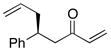 |
>100:1 | 80 | 95:5 |
| 8 | 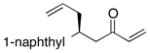 |
>100:1 | 37 | 96:4 |
| 9 | 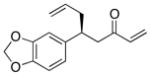 |
>100:1 | 76 | 95:5 |
Unless otherwise noted, the reaction was carried out at room temperature for 14 h and quenched with saturated NH4Cl(aq) or acetic acid.
Regioselectivity determined by uncalibrated GLC analysis in comparison to authentic materials.
Isolated yield after purification. Value is an average of two or more experiments.
Enantiomer ratio determined by GLC, SFC or HPLC analysis.
Experiment for 48 h and quenched with glacial acetic acid at 45 °C for 3 h.
The functional group pattern that results from the conjugate allylation of methylidene ketones allows transformation of the products in a variety of ways and may offer shortened enantioselective routes to important compounds. For example, silyl-protected cyclohexenone 11 (Scheme 3) is a critical intermediate in a recent approach to stenine natural products.7 As depicted in Scheme 3, an effective enantioselective synthesis of 11 can be achieved in five steps from 1-butyn-4-ol. Following silyl protection and reductive iodination with Schwartz reagent and I2,8 the alkenyl iodide 8 was isolated in good yield. Subsequent carbonylative Stille coupling9 provided conjugate allylation substrate 9.10 Employing the conditions described in Table 2 the allylation product 10 was obtained and ring-closing metathesis with the Hoveyda-Grubbs second generation catalyst11 provided cyclohexenone 11. This five step route to 11 compares favorably with the non-enantioselective eight step route used previously.
Scheme 3.

Synthesis of Cyclohexenone 11.
In conclusion, we have described a highly regioselective and enantioselective conjugate allylation of unsaturated ketones. The orthogonal reactivity of the allyl group and the methylidene ketone bodes well for the utility of these reaction products in organic synthesis. Further studies in this regard are in progress.
Supplementary Material
Acknowledgments
We are grateful to the NIH (NIGMS GM-64451) for support of this work.
Footnotes
Supporting Information Available. Complete experimental procedures and characterization data (1H and 13C NMR, IR, and mass spectrometry). This material is free of charge via the internet at http://pubs.acs.org.
References
- 1.Reviews: (a) Krause N, Hoffmann-Röder A. Synthesis. 2001:171.Christoffers J, Koripelly G, Rosiak A, Rössle M. Synthesis. 2007:1279.Alexakis A, Bäckvall JE, Krause N, Pàimes O, Diéguez M. Chem Rev. 2008;108:2796. doi: 10.1021/cr0683515.Harutyunyan SR, den Hartog T, Geurts K, Minnard AJ, Feringa BL. Chem Rev. 2008;108:2824. doi: 10.1021/cr068424k.Hawner C, Alexakis A. Chem Commun. 2010;46:7295. doi: 10.1039/c0cc02309d.
- 2.Sieber JD, Liu S, Morken JP. J Am Chem Soc. 2007;129:2214. doi: 10.1021/ja067878w.Sieber JD, Morken JP. J Am Chem Soc. 2008;130:4978. doi: 10.1021/ja710922h.For a related process, see: Zhang P, Morken JP. J Am Chem Soc. 2009;131:12550. doi: 10.1021/ja9058537.
- 3.(a) Méndez M, Cuerva JM, Gómez-Bengoa E, Cárdenas DJ, Echavarren AM. Chem Eur J. 2002;8:3620. doi: 10.1002/1521-3765(20020816)8:16<3620::AID-CHEM3620>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]; (b) Cárdenas DJ, Echavarren AM. New J Chem. 2004;28:338. [Google Scholar]
- 4.For other catalytic reactions that appear to proceed by 3,3′ reductive elimination, see: Keith JA, Behenna DC, Mohr JT, Ma S, Marinescu SC, Oxgaard J, Stoltz BM, Goddard WA., III J Am Chem Soc. 2007;129:11876. doi: 10.1021/ja070516j.Bao M, Nakamura H, Yamamoto Y. J Am Chem Soc. 2001;123:759. doi: 10.1021/ja003718n.Lu S, Xu Z, Bao M, Yamamoto Y. Angew Chem Int Ed. 2008;47:4366. doi: 10.1002/anie.200800529.Ariafard A, Lin Z. J Am Chem Soc. 2006;128:13010. doi: 10.1021/ja063944i.
- 5.For another catalytic enantioselective conjugate allylation, see: Shizuka M, Snapper ML. Angew Chem Int Ed. 2008;47:5049. doi: 10.1002/anie.200800628.For non-enantioselective catalytic version, see: Shaghafi MB, Kohn BL, Jarvo ER. Org Lett. 2008;10:4743. doi: 10.1021/ol801830h.
- 6.For a review of Taddol-derived phosphonites and phosphoramidites in catalysis: Teichert JF, Feringa BL. Angew Chem Int Ed. 2010;49:2486. doi: 10.1002/anie.200904948.Feringa BL. Acc Chem Res. 2000;33:346. doi: 10.1021/ar990084k.Select examples, hydrogenation: ven den Berg M, Minnard AJ, Schudde EP, van Esch J, de Vries AHM, de Vries JG, Feringa BL. J Am Chem Soc. 2000;122:11539.Hydrosilation: Jensen JF, Svendsen BY, la Cour TV, Pedersen HL, Johannsen M. J Am Chem Soc. 2002;124:4558. doi: 10.1021/ja025617q.Hydroboration: Ma MFP, Li K, Zhou Z, Tang C, Chan ASC. Tetrahedron Asymm. 1999;10:3259.Cycloaddition: Yu RT, Rovis T. J Am Chem Soc. 2006;128:12370. doi: 10.1021/ja064868m.
- 7.Zhu L, Lauchli R, Loo M, Shea KJ. Org Lett. 2007;9:2269. doi: 10.1021/ol070397c. [DOI] [PubMed] [Google Scholar]
- 8.Labinger JA, Schwartz J. Angew Chem Int Ed Engl. 1976;15:333. [Google Scholar]
- 9.Goure WF, Wright ME, Davis PD, Labadie SS, Stille JK. J Am Chem Soc. 1984;106:6417. [Google Scholar]
- 10.(a) Statsuk AV, Liu D, Kozmin SA. J Am Chem Soc. 2004;126:9546. doi: 10.1021/ja046588h. [DOI] [PubMed] [Google Scholar]; (b) Marjanovic J, Kozmin SA. Angew Chem Int Ed Engl. 2007;46:9010. doi: 10.1002/anie.200702440. [DOI] [PubMed] [Google Scholar]
- 11.Garber SB, Kingsbury JS, Gray BL, Hoveyda AH. J Am Chem Soc. 2000;122:8168. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.