

Summary
Background
Although epilepsy affects most patients with tuberous sclerosis complex (TSC), little is known about the natural history of epilepsy in this genetic disease.
Methods
A retrospective chart review of all patients with TSC seen between January 2002 and October 2008. Charts were reviewed for a history of infantile spasms (IS), seizure other than IS, refractory epilepsy, Lennox-Gastaut syndrome (LGS), anticonvulsant medication use, ages of seizure onset, last seizure, last clinic visit, clinical seizure phenotype(s), cognitive impairment, and genetic mutation.
Results
Two hundred ninety-one patients were included. Among these patients, 37.8% had a history of IS; 85.2% had a history of seizure; 54.1% developed multiple seizure types, not including IS; 63.2% had seizure onset in the first year of life; and 12.1% of adults without a seizure history developed epilepsy. Of epilepsy patients, 62.5% developed refractory epilepsy and 33.5% achieved epilepsy remission; 37.5% of these patients achieved medication freedom. IS was a risk factor for refractory epilepsy (p<0.0001) and LGS (p<0.0001). History of seizure, IS, age at seizure onset, and refractory epilepsy each correlated with poor cognitive outcome (p<0.0001). Epilepsy remission correlated with better cognitive outcome (p<0.0001). TSC2 was a risk factor for IS and epilepsy; patients without an identified mutation were more likely to achieve remission.
Conclusion
Most patients with TSC develop epilepsy and most develop multiple seizure types. Onset typically occurs in the first year of life; however, adults remain at risk. Although refractory epilepsy is common, many patients achieve seizure control. Many features of seizure history are predictive of cognitive and epilepsy outcome.
Keywords: Epilepsy, Infantile spasms, Outcome, Genetics, Tuberous sclerosis complex
Tuberous sclerosis complex (TSC) is an autosomal dominant, multiorgan disease with widely variable expression. TSC is estimated to occur in at least 1 in 6,000 live births, although true incidence is likely higher due to undetected mild cases (Webb & Osborne, 1995; Crino et al., 2006). Approximately 85% of patients with TSC are found to have a mutation in one of two genes, TSC1, encoding hamartin, or TSC2, encoding tuberin (Dabora et al., 2001; Sancak et al., 2005). Protein products of these genes have been shown to form a heterodimer (TSC1–TSC2 complex) that inhibits the mammalian target of rapamycin (mTOR) signaling cascade (Astrinidis & Henske, 2005; Kwiatkowski & Manning, 2005). Inadequate suppression of the mTOR pathway results in dysgenic lesions in multiple organ systems, including cortical tubers, radial glial bands, subependymal nodules, and subependymal giant cell tumor formation in the fetal and developing brain (Crino, 2004; Sarbassov et al., 2005; Sabatini, 2006).
Epilepsy is the most common neurologic symptom in patients with TSC, and a significant source of morbidity and mortality (Shepherd et al., 1991; Thiele, 2004; Holmes et al., 2007). Although epilepsy is known to affect the majority of patients with TSC, reports of the prevalence of epilepsy and infantile spasms in varied, smaller TSC populations have been inconsistent (Webb et al., 1991, 1996; Roach et al., 1998; Jozwiak et al., 2000; Cross, 2005; Crino et al., 2006; Devlin et al., 2006). Furthermore, there have been few studies evaluating the natural history of epilepsy in TSC (Webb et al., 1996).
In order to better characterize and understand epilepsy in TSC, we sought to describe the prevalence of epilepsy and infantile spasms in our large pediatric and adult TSC clinic. We further set out to describe in detail the natural history of epilepsy in TSC, including age of onset, frequency of multiple seizure types, evolution to refractory epilepsy, prevalence of Lennox-Gastaut syndrome, and likelihood of achieving seizure-freedom and medication-freedom. We further evaluated the relationships among epilepsy history, cognitive outcome, and genetic mutation in our patient population.
Methods
We performed a retrospective chart review of all patients seen at Herscot Center for tuberous sclerosis complex at Massachusetts General Hospital between January 2002 and October 2008. Patients were included if they met clinical criteria for definite TSC (Roach et al., 1998). Two hundred ninety-one patients were included.
Patient charts were reviewed for genetic mutation, history of infantile spasms, history of seizure other than infantile spasms, age at seizure onset (in 1-month intervals), age at last seizure (in 1-month intervals), age at last clinic appointment (in 1-month intervals), history of epilepsy, clinical seizure phenotype(s), refractory epilepsy, history of Lennox-Gastaut syndrome (LGS), epilepsy surgery history, anticonvulsant medication history including discontinuation, and cognitive impairment. Seizure types were classified by an experienced epileptologist strictly according to clinical semiology provided by direct observation in clinic or by description by a witness.
For the purpose of this article, epilepsy was defined as at least two clinical seizures not including infantile spasms, which were considered separately in our analyses. Refractory epilepsy was defined as uncontrolled seizures after at least three first-line anticonvulsant medication trials. Patients were considered seizure-free if they were without clinical seizures for at least 1 year, using the last clinic visit documenting seizure status as the endpoint of follow-up. Definite LGS was defined as characteristic generalized slow spike and wave pattern on electroencephalography (EEG), at least three generalized seizure phenotypes (atypical absence, myoclonic, generalized tonic–clonic, and/or atonic), and cognitive impairment. Probable LGS was defined as two of the above criteria, with insufficient data available for review for the third category. Genetic testing of the TSC1 and TSC2 genes, including detection of large DNA deletions and rearrangements of the TSC2 gene, was performed at Athena Diagnostics (Worcester, MA, U.S.A.) or the MGH Neurogenetic Diagnostic Laboratory (Boston, MA, U.S.A.). Patients for whom genetic testing was inconclusive were classified as no mutation identified (NMI).
Formal neuropsychological evaluations were available for review in 126 patients. When an intelligence quotient (IQ) was not obtained, a developmental quotient (DQ) was calculated by dividing the mental age of the child by his or her chronological age and multiplying by 100. Those with an IQ or DQ<70 were considered cognitively impaired. If formal neuropsychological testing was not performed, intellectual status was determined by an experienced pediatric neurologist based on developmental examination, school performance, activities of daily living, and language abilities. If cognitive function was unknown, patients were excluded from analysis.
Statistical analyses were performed using logistic regression, chi-square analyses, and Fisher’s exact test. All reported p-values used two-tailed tests of significance with α set at 0.05.
The institutional review board of the Massachusetts General Hospital approved this study.
Results
Descriptive statistics of epilepsy
Two hundred ninety-one patients with definite clinical TSC were included. Two hundred forty-eight patients had a history of at least one seizure (85.2%). Of these patients, 246 developed epilepsy (99.2%). Two patients (0.8%) had only a single seizure (3.75 years and 21.2 years of follow-up). One hundred fifty-five of 248 patients (62.5%) developed refractory epilepsy.
One hundred ten of 291 patients (37.8%) had a history of infantile spasms. Only four patients with a history of infantile spasms (3.6%) did not develop another seizure type, and two of these patients are currently only 2 years of age, thus limiting follow-up. Of patients with a history of infantile spasms, 83 of 110 (75.4%) developed refractory epilepsy compared with 72 of 181 patients (39.8%) without a history of infantile spasms (p<0.0001).
Of the 248 patients with a history of seizure, seizure types were documented for 237 (95.6%) (Fig. 1). Excluding infantile spasms, 126 of 237 patients (53.2%) had more than one seizure type. At least one seizure phenotype with a clear focal onset clinically (simple partial or complex partial seizures with or without secondary generalization) was documented in 221 of 237 patients (93.2%).
Figure 1.

CPS, complex partial seizures (includes CPS with secondary generalization); IS, infantile spasms; GTC, generalized tonic–clonic seizures; SPS, simple partial seizures (includes SPS with secondary generalization); ESES, electrographic status epilepticus of sleep.
Age at seizure onset was known for 231 patients (Fig. 2). The most common age at seizure onset was 3 months (average 29.0 months, median 7 months, range 1 day to 45.4 years). Seizure onset occurred within the first 4 weeks of life in 15 patients (6.5%), before 6 months of age in 106 patients (45.8%), by the end of the first year of life in 146 patients (63.2%), and before 3 years of age in 188 patients (82.1%). First seizure occurred after age 13 years in five patients (2.2%). Among 33 TSC adults without a history of seizure, four (12.1%) subsequently developed epilepsy.
Figure 2.
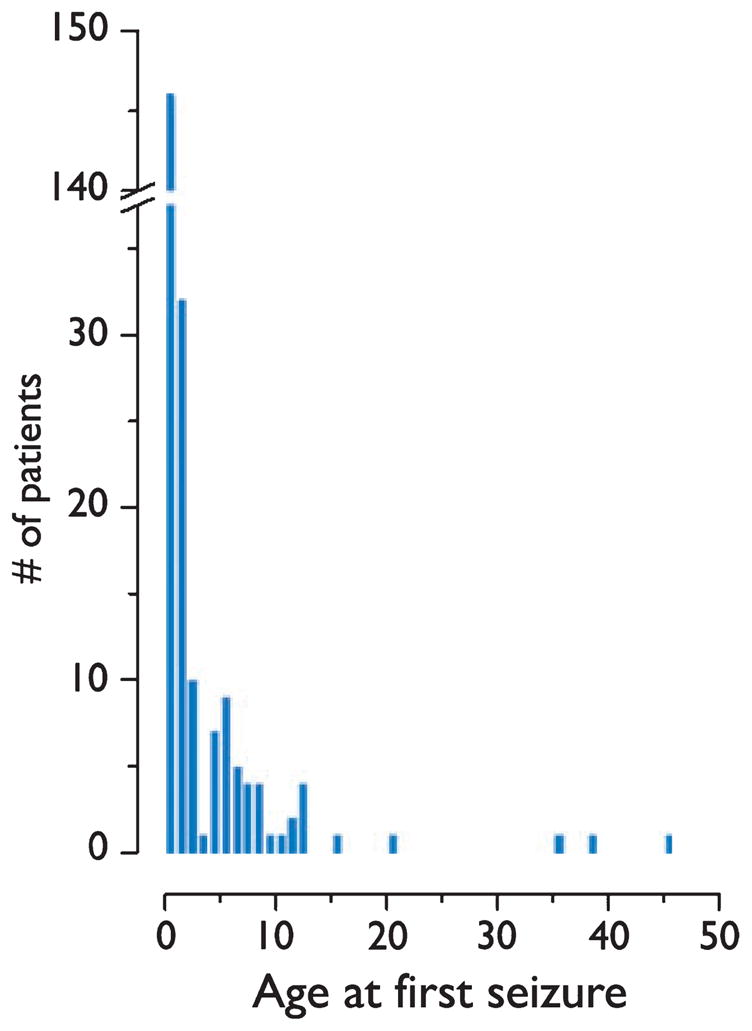
Distribution of age at seizure onset among 231 patients with tuberous sclerosis complex.
Of patients with epilepsy, clinical follow up was available for 242. Eighty-one (33.5%) of 242 patients were seizure-free for at least 12 months at last clinic visit. These patients had clinical follow-up of an average of 11.8 years after last seizure [standard deviation (SD) 11.4 years, median 6.9 years, mode 4.0 years, range 12 months to 51.6 years]. Of 81 seizure-free patients, 33 patients underwent attempted medication wean and 30 (37.5%) of 81 remained seizure-free off medication. Average age of seizure-freedom was 13 years 0 months (SD 16.3 months, mode 8 years, range 8 months to 56.5 years).
Of 155 patients with refractory epilepsy, 29 (18.7%) subsequently achieved epilepsy remission. Ten (25.6%) of 39 patients with refractory epilepsy who underwent epilepsy surgery achieved seizure freedom and 19 (16.4%) of 116 patients who did not undergo epilepsy surgery became seizure-free. This comparison was not statistically significant (p=0.24). One patient with medication refractory epilepsy achieved seizure freedom 3 months after placement of a vagus nerve stimulator (VNS) and has subsequently remained seizure-free with combination anticonvulsant treatment and VNS for 4 years and 3 months. No patients included in this study achieved seizure freedom while on dietary therapy for epilepsy.
Of 248 patients with a history of seizure, 15 patients (6.0%) developed LGS. Fifty-three further patients (21.4%) had probable LGS. Of patients with a history of infantile spasms, 43 of 110 (39.1%) developed definite or probable LGS, compared with 25 (13.8%) of 181 patients without a history of infantile spasms (p<0.0001).
Relationship of epilepsy to cognitive outcome
Results are shown in Table 1. Of 256 patients whose cognitive function was documented, 120 (46.9%) were cognitively normal. Formal neuropsychological testing results with estimated IQ or DQ was available for 126 patients (mean 68.5, SD 32.37, range 7–137).
Table 1.
Relationship of epilepsy history to cognitive outcome
| Cognitively impaired (%) | Cognitively normal (%) | |
|---|---|---|
| History of seizurea | ||
| Yes | 131 (60.9) | 84 (39.1) |
| No | 5 (12.2) | 36 (87.8) |
| Infantile spasmsa | ||
| Yes | 73 (74.4) | 25 (25.6) |
| No | 62 (39.2) | 96 (60.8) |
| Age of seizure onseta | ||
| <3 years | 114 (32.1) | 54 (67.9) |
| ≥3 years | 7 (80.6) | 30 (19.4) |
| Refractory epilepsya | ||
| Yes | 104 (73.2) | 38 (26.8) |
| No | 28 (37.8) | 46 (62.2) |
| Epilepsy remissiona | ||
| Yes | 32 (43.3) | 42 (56.7) |
| No | 99 (69.8) | 43 (30.2) |
Denotes statistically significant comparison.
Of patients with a history of seizure, 131 of 215 (60.9%) were cognitively impaired compared with 5 of 41 patients (12.2%) without a history of seizure (p<0.0001). Of patients with a history of infantile spasms, 73 of 98 (74.4%) were cognitively impaired, compared with 62 of 158 patients (39.2%) without a history of infantile spasms (p<0.0001).
Cognitive outcome and IQ were significantly related to age of seizure onset (logistic regression, p=0.004 and p=0.000; Table 2). When patients with a history of infantile spasms were removed from analysis, the relationship between age at seizure onset and IQ remained significant (logistic regression, p=0.006). There was no difference in cognitive outcome between patients ≤6 months of age at seizure onset compared with those between 7 and 12 months of age (p=0.16; Fig. 2). Notably, 6 (33.3%) of 18 patients with seizure onset at ≤1 month of age had normal cognitive outcome. Three of these cognitively normal patients also had a history of infantile spasms. Patients with seizure onset before 3 years of age were more likely to be cognitively impaired than patients with seizure onset after 3 years of age (p < 0.0001). There was no relationship between cognitive outcome and age of seizure onset for patients <3 years of age (logistic regression, p = 0.165).
Table 2.
Age at seizure onset of 231 patients with TSC and cognitive outcome
| Age (years) | Cognitively normal (%)a | Cognitively impaired (%)a | Cognitive status unknown | Total (N = 231) (%) |
|---|---|---|---|---|
| <1 | 39 (29.5) | 93 (70.5) | 14 | 146 (63.7) |
| 1 | 12 (42.9) | 16 (57.1) | 4 | 32 (14.0) |
| 2 | 3 (37.5) | 5 (62.5)b | 2 | 10 (4.4) |
| 3 | 1 (100) | 0 (0) | 0 | 1 (0.4) |
| 4 | 6 (100) | 0 (0) | 1 | 7 (3.1) |
| 5 | 6 (75) | 2 (25) | 1 | 9 (3.9) |
| 6 | 4 (100) | 0 (0) | 1 | 5 (2.2) |
| 7 | 3 (75) | 1 (25) | 0 | 4 (1.7) |
| 8 | 2 (66.7) | 1 (33.3) | 1 | 4 (1.7) |
| 9 | 1 (100) | 0 (0) | 0 | 1 (0.4) |
| 10 | 1 (100) | 0 (0) | 0 | 1 (0.4) |
| 11 | 1 (100) | 0 (0) | 1 | 2 (0.9) |
| 12 | 2 (50) | 2 (50) | 0 | 4 (1.7) |
| 13 | 0 (0) | 0 (0) | 0 | 0 (0) |
| 14 | 0 (0) | 0 (0) | 0 | 0 (0) |
| 15 | 0 (0) | 0 (0) | 1 | 1 (0.4) |
| 16 | 0 (0) | 0 (0) | 0 | 0 (0) |
| 17 | 0 (0) | 0 (0) | 0 | 0 (0) |
| >18 | 3 (75) | 1 (25) | 0 | 4 (1.7) |
Percentage of patients (among those with known cognitive status).
Patients with seizure onset prior to age 3 were significantly more likely to have cognitive impairment compared to patients with seizure onset after age 3 (p < 0.0001).
TSC, tuberous sclerosis complex.
Of 142 patients with a history of refractory epilepsy 104 (73.2%) were cognitively impaired compared with 28 of 74 patients (37.8%) without a history of refractory epilepsy (p < 0.0001). Of 74 patients who were seizure-free, 42 (56.7%) were cognitively normal compared with 43 of 142 (30.2%) who were not seizure-free (p < 0.0001).
Relationship of epilepsy to genetic mutation
Results are shown in Table 3. Of 291 patients with TSC, 231 underwent genetic testing: 123 (53.2%) were found to have a TSC2 mutation, 60 (26.0%) a TSC1 mutation, and 48 NMI (20.8%).
Table 3.
Relationship of epilepsy prognosis to genetic mutation
| TSC1 (%) | TSC2 (%) | NMI (%) | |
|---|---|---|---|
| History of seizurea | 51/60 (85) | 112/123 (91.0) | 34/48 (70.8) |
| Refractory epilepsy | 27/51 (52.9) | 76/112 (67.9) | 20/34 (58.8) |
| Epilepsy remissiona | 17/50 (34.0) | 34/122 (27.9) | 18/31 (54.5) |
Denotes statistically significant comparison.
Of 123 patients with a TSC2 mutation, 112 (91.0%) had a history of seizure compared with 51 of 60 patients (85%) with a TSC1 mutation and 34 of 48 patients (70.8%) with NMI (p=0.004). Subgroup analysis revealed that patients with TSC2 were more likely to have a history of seizure compared with patients with NMI (p=0.001), but not TSC1 (p = 0.22).
Of patients with a history of seizure, 76 of 112 patients (67.9%) with a TSC2 mutation developed refractory epilepsy compared with 27 of 51 patients (52.9%) with a TSC1 mutation and 20 of 34 patients (58.8%) with NMI (p = 0.169).
Of 123 patients with a TSC2 mutation, 69 (56.1%) had a history of infantile spasms compared with 6 of 60 patients (10%) with a TSC1 mutation and 12 of 48 patients (25.0%) with NMI (p < 0.0005). Subgroup analysis revealed that patients with TSC2 were more likely to have a history of IS compared with patients with TSC1 (p < 0.0005) or NMI (p < 0.0005). Patients with NMI were more likely to have a history of IS compared with patients with TSCI (p = 0.021).
Among patients with clinical follow-up available, 18 of 33 patients (54.5%) with NMI became seizure-free compared with 34 of 122 patients (27.9%) with TSC2 mutation and 17 of 50 patients (34.0%) with TSC1 mutation (p = 0.016). On subgroup analysis, patients with NMI were more likely to become seizure-free than patients with TSC2 (p = 0.004) but not patients with TSC1 patients (p = 0.064), although there was a trend.
Discussion
Previous data obtained from small cohorts of patients seen in neurology clinics estimate the prevalence of epilepsy in TSC to be 93–96% (Jozwiak et al., 2000; Devlin et al., 2006). A small, unbiased genetic linkage study estimated a prevalence of 62% (Webb et al., 1991). In a large cohort of 291 patients with TSC seen in a specialty clinic, we found that 85.2% have a history of seizure and 84.5% met criteria for clinical epilepsy. The high prevalence of infantile spasms in TSC is a consistent observation, although estimates of previous studies in varied populations vary from 27–96% (Pampiglione & Moynahan, 1976; Webb et al., 1996; Jozwiak et al., 2000; Curatolo et al., 2001; O’Callaghan et al., 2004). In this large cohort, we found a history of infantile spasms in 37.8%. It is routine in our clinic to screen and treat identified family members with subclinical disease, and our estimates include both symptomatic probands as well as subclinical family members. However, these results are still likely overestimated due to referral bias. Unavailable remote clinical histories in newly referred adult patients may contribute an underestimation of infantile spasms.
In patients with TSC, the likelihood of developing epilepsy after presenting with a single seizure is nearly 100%, and, therefore, strong consideration should be given to treat with anticonvulsant medication after first seizure. Even among known epilepsy patients, frequent surveillance for new paroxysmal events is necessary, as nearly all patients with infantile spasms develop another seizure phenotype and more than half of epilepsy patients develop multiple seizure types. Classic seizures with focal onset are seen in most patients with TSC; however, many patients also had seizure phenotypes that appeared clinically generalized at onset. The number of patients with true primary generalized seizures was likely overestimated, as clinical data was used for seizure characterization rather than ictal EEG recordings. Given the focal neuropathologies found in TSC, it is likely that most seizures are partial in onset, with or without rapid secondary generalization.
Similar to previous reports, we found that nearly two-thirds of patients had seizure onset within the first year of life (Webb et al., 1996; Gomez, 1999; Jozwiak et al., 2000). Furthermore, nearly 80% of patients had seizure-onset within the first 3 years of life. Despite the strong propensity for patients with TSC to develop epilepsy in childhood, we found that >12% of adult patients without a previous history of seizure subsequently developed epilepsy, indicating that patients with TSC remain at significantly increased risk of epilepsy throughout their lifetime.
The cognitive impact of each infantile spasms and refractory epilepsy has been shown previously and is supported here (Webb et al., 1996; Goh et al., 2005; Winterkorn et al., 2007; Jansen et al., 2008a). In our cohort, nearly two-thirds of epilepsy patients with TSC develop refractory epilepsy compared with 23.2% reported in the general epilepsy population (Berg et al., 2006). We have further shown that age of seizure-onset correlates with cognitive outcome independent of infantile spasms. Previous work in a smaller population found that seizure onset in the first year was associated with poor cognitive outcome (Jansen et al., 2008b). In our cohort, children with seizure-onset before 3 years of age were at increased risk of cognitive impairment; however, those with seizure onset at <1 year were not at further increased risk.
Although epilepsy can be difficult to manage in TSC, one-third of patients achieved epilepsy remission, including nearly 20% of patients with a history of refractory epilepsy. The majority of patients in whom epilepsy remission was achieved were cognitively normal. Although few patients in this study achieved seizure freedom on dietary therapy or with VNS, these nonpharmacologic options have been found to significantly reduce the frequency of seizures in this patient population and should be considered (Kossoff et al., 2005; Jansen et al., 2007; Major & Thiele, 2008). Subsequent to the termination of this study, we do have one patient with medication refractory epilepsy who has been seizure-free for >10 months on low-glycemic index dietary treatment.
A prior study evaluating 15 children with TSC found that more than one-fourth experienced seizure relapse after anticonvulsant discontinuation (Sparagana et al., 2003). In our population, nearly one-third of seizure-free patients were also medication-free, and only 3 of 33 (9.1%) of patients experienced recurrent seizure after attempted medication taper. This discrepancy may be due to differences in the populations studied, as both adults and children were included in our study, as well as differences in clinical practice, given that we routinely require at least 1 year without clinical seizures and improved or resolved EEG abnormalities prior to attempted medication wean. Carefully selected, well-controlled patients should be considered for medication taper even in this high-risk population.
Previous work evaluating genotype–phenotype relationships in patients with TSC1 and TSC2 have similarly found that patients with TSC2 mutations experience higher frequency of seizures than those with TSC1 mutations (Dabora et al., 2001; Jansen et al., 2008a). We have further shown that the risk of infantile spasms is greatest among patients with TSC2 mutations followed by those with NMI. Furthermore, NMI patients with epilepsy were most likely to achieve seizure freedom. It is likely that at least some NMI patients are TSC1 or TSC2 mosaics, which could explain the mixed clinical features seen in this group. It has been postulated that some retention of function of the tuberin GTPase activating protein (GAP) domain could account for the decreased clinical severity seen in TSC1 patients. However, previous work has not found a relationship between infantile spasms and deletions predicted to impact the GAP domain among TSC2 patients (Jansen et al., 2008a). Further work in animal and cell models measuring the functional activity of the hamartin–tuberin complex as it relates to genotype is needed.
In summary, in a large population of patients with TSC attending a tertiary care center, epilepsy remains a major source of morbidity, particularly for young children. Careful surveillance and prompt and aggressive treatment of epilepsy remains critical to minimize, and potentially prevent, the adverse impacts of uncontrolled seizures in this at-risk population.
Acknowledgments
This study was supported by the Carol and James Herscot Center for Tuberous Sclerosis Complex.
We confirm that we have read the Journal’s position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
Footnotes
Disclosure: None of the authors has any conflicts of interest to report.
References
- Astrinidis A, Henske EP. Tuberous sclerosis complex: linking growth and energy signaling pathways with human disease. Oncogene. 2005;24:7475–7481. doi: 10.1038/sj.onc.1209090. [DOI] [PubMed] [Google Scholar]
- Berg AT, Vickrey BG, Testa FM, Levy SR, Shinnar S, DiMario F, Smith S. How long does it take for epilepsy to become intractable? A prospective investigation. Ann Neurol. 2006;60:73–79. doi: 10.1002/ana.20852. [DOI] [PubMed] [Google Scholar]
- Crino PB. Molecular pathogenesis of tuber formation in tuberous sclerosis complex. J Child Neurol. 2004;19:716–725. doi: 10.1177/08830738040190091301. [DOI] [PubMed] [Google Scholar]
- Crino PB, Nathanson KL, Petri Henske E. The tuberous sclerosis complex. N Engl J Med. 2006;355:1345–1356. doi: 10.1056/NEJMra055323. [DOI] [PubMed] [Google Scholar]
- Cross JH. Neurocutaneous syndromes and epilepsy—issues in diagnosis and management. Epilepsia. 2005;46:17–23. doi: 10.1111/j.1528-1167.2005.00353.x. [DOI] [PubMed] [Google Scholar]
- Curatolo P, Seri S, Verdecchia M, Bombardieri R. Infantile spasms in tuberous sclerosis complex. Brain Dev. 2001;23:502–507. doi: 10.1016/s0387-7604(01)00300-x. [DOI] [PubMed] [Google Scholar]
- Dabora SL, Jozwiak S, Franz DN, Roberts PS, Nieto A, Chung J, Choy YS, Reeve MP, Thiele E, Egelhoff JC, Kaspryzk-Obara J, Domanska-Pakiela D, Kwiatkowski DJ. Mutational analysis in a cohort of 224 tuberous sclerosis patients indicates increased severity of TSC2, compared with TSC1, disease in multiple organs. Am J Hum Genet. 2001;68:64–80. doi: 10.1086/316951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devlin LA, Shepherd CH, Crawford H, Morrison PJ. Tuberous sclerosis complex: clinical features, diagnosis, and prevalence within Northern Ireland. Dev Med Child Neurol. 2006;48:495–499. doi: 10.1017/S0012162206001058. [DOI] [PubMed] [Google Scholar]
- Goh S, Kwiatkowski DJ, Dorer DJ, Thiele EA. Infantile spasms and intellectual outcomes in children with tuberous sclerosis complex. Neurology. 2005;65:235–238. doi: 10.1212/01.wnl.0000168908.78118.99. [DOI] [PubMed] [Google Scholar]
- Gomez MR. Natural history of cerebral tuberous sclerosis. In: Gomez MR, Sampson JR, Whittemor VH, editors. Tuberous Sclerosis Complex: Developmental Perspectives in Psychiatry. Oxford University Press; New York: 1999. pp. 29–46. [Google Scholar]
- Holmes GL, Stafstrom CE Tuberous Sclerosis Study Group. Tuberous sclerosis complex and epilepsy: recent developments and future challenges. Epilepsia. 2007;48:617–630. doi: 10.1111/j.1528-1167.2007.01035.x. [DOI] [PubMed] [Google Scholar]
- Jansen FE, van Huffelen AC, Algra A, van Nieuwenhuizen O. Epilepsy surgery in tuberous sclerosis: a systematic review. Epilepsia. 2007;48:1477–1484. doi: 10.1111/j.1528-1167.2007.01117.x. [DOI] [PubMed] [Google Scholar]
- Jansen FE, Braams O, Vinken KL, Algra A, Anbeck P, Jennekens-Schinkel A, Halley D, Zonnenberg BA, van den Ouweland A, van Huffelen AC, van Nieuwenhuizen O, Nellist M. Overlapping neurologic and cognitive phenotypes in patients with TSC1 or TSC2 mutation. Neurology. 2008a;70:908–915. doi: 10.1212/01.wnl.0000280578.99900.96. [DOI] [PubMed] [Google Scholar]
- Jansen FE, Vincken KL, Algra A, Anbeek P, Braams O, Nellist M, Zonnenberg BA, Jennekens-Schinkel A, van den Ouweland A, Halley D, van Huffelen AC, van Nieuwenhuizen O. Cognitive impairment in tuberous sclerosis complex is a multifactorial condition. Neurology. 2008b;17:916–923. doi: 10.1212/01.wnl.0000280579.04974.c0. [DOI] [PubMed] [Google Scholar]
- Jozwiak S, Shwarz RA, Janniger CK, Bielicka-Cymerman J. Usefulness of diagnostic criteria of tuberous sclerosis complex in pediatric patients. J Child Neurol. 2000;15:652–659. doi: 10.1177/088307380001501003. [DOI] [PubMed] [Google Scholar]
- Kossoff EH, Thiele EA, Pfeifer HH, McGrogan JR, Freeman JM. Tuberous sclerosis complex and the ketogenic diet. Epilepsia. 2005;46:1684–1686. doi: 10.1111/j.1528-1167.2005.00266.x. [DOI] [PubMed] [Google Scholar]
- Kwiatkowski DJ, Manning BD. Tuberous sclerosis: a GAP at the crossroads of multiple signaling pathways. Hum Mol Genet. 2005;14(2):R251–R258. doi: 10.1093/hmg/ddi260. [DOI] [PubMed] [Google Scholar]
- Major P, Thiele EA. Vagus nerve stimulation for intractable epilepsy in tuberous sclerosis complex. Epilepsy Behav. 2008;13:357–360. doi: 10.1016/j.yebeh.2008.04.001. [DOI] [PubMed] [Google Scholar]
- O’Callaghan FJ, Harris T, Joinson C, Bolton P, Noakes M, Presdee D, Renowden S, Shiell A, Martyn CN, Osborne JP. The relation of infantile spasms, tubers and intelligence in tuberous sclerosis complex. Arch Dis Child. 2004;89:530–533. doi: 10.1136/adc.2003.026815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pampiglione G, Moynahan EJ. The tuberous sclerosis syndrome: clinical and EEG studies in 100 children. J Neurol Neurosurg Psychiatry. 1976;39:666–673. doi: 10.1136/jnnp.39.7.666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roach ES, Gomez MR, Northrup H. Tuberous sclerosis complex consensus conference: revised diagnostic criteria. J Child Neurol. 1998;13:624–628. doi: 10.1177/088307389801301206. [DOI] [PubMed] [Google Scholar]
- Sabatini DM. mTOR and cancer: insights into a complex relationship. Nat Rev Cancer. 2006;6:729–734. doi: 10.1038/nrc1974. [DOI] [PubMed] [Google Scholar]
- Sancak O, Nellist M, Goedbloed M, Elfferich P, Wouters C, Maat-Kievit A, Zonnenberg B, Verhoef S, Halley D, van den Ouweland A. Mutational analysis of the TSC1 and TSC2 genes in a diagnostic setting: genotype–phenotype correlations and comparison of diagnostic DNA techniques in tuberous sclerosis complex. Eur J Hum Genet. 2005;13:731–741. doi: 10.1038/sj.ejhg.5201402. [DOI] [PubMed] [Google Scholar]
- Sarbassov DD, Ali SM, Sabatini DM. Growing roles for the mTOR pathway. Curr Opin Cell Biol. 2005;17:596–603. doi: 10.1016/j.ceb.2005.09.009. [DOI] [PubMed] [Google Scholar]
- Shepherd CW, Gomez MR, Lie JT, Crowson CS. Causes of death in patients with tuberous sclerosis. Mayo Clin Proc. 1991;66:792–796. doi: 10.1016/s0025-6196(12)61196-3. [DOI] [PubMed] [Google Scholar]
- Sparagana SP, Delgado MR, Batchelor LL, Roach S. Seizure remission and antiepileptic drug discontinuation in children with tuberous sclerosis complex. Arch Neurol. 2003;60:1286–1289. doi: 10.1001/archneur.60.9.1286. [DOI] [PubMed] [Google Scholar]
- Thiele EA. Managing epilepsy in tuberous sclerosis complex. J Child Neurol. 2004;19:680–686. doi: 10.1177/08830738040190090801. [DOI] [PubMed] [Google Scholar]
- Webb DW, Fryer AE, Osborne JP. On the incidence of fits and mental retardation in tuberous sclerosis. J Med Genet. 1991;28:395–397. doi: 10.1136/jmg.28.6.395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webb DW, Osborne JP. Tuberous Sclerosis. Arch Dis Child. 1995;72:471–474. doi: 10.1136/adc.72.6.471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webb DW, Fryer AE, Osborne JP. Morbidity associated with tuberous sclerosis: a population study. Dev Med Child Neurol. 1996;38:146–155. doi: 10.1111/j.1469-8749.1996.tb12086.x. [DOI] [PubMed] [Google Scholar]
- Winterkorn EB, Pulsifer MB, Thiele EA. Cognitive prognosis of patients with tuberous sclerosis complex. Neurology. 2007;68:62–64. doi: 10.1212/01.wnl.0000250330.44291.54. [DOI] [PubMed] [Google Scholar]