

Abstract
Electrospray ionization of methanolic solutions of p-hydroxyphenacyl derivatives HO-C6H4-C(O)-CH2-X (X = leaving group) provides abundant signals for the deprotonated species which are assigned to the corresponding phenolate anions −O-C6H4-C(O)-CH2-X. Upon collisional activation in the gas phase, these anions inter alia undergo loss of a neutral “C8H6O2” species concomitant with formation of the corresponding anions X−. The energies required for the loss of neutral roughly correlate with the gas phase acidities of the conjugate acids (HX). Extensive theoretical studies performed for X = CF3COO in order to reveal the energetically most favorable pathway for the formation of neutral “C8H6O2” suggest three different routes of similar energy demands, involving a spirocyclopropanone, epoxide formation, and a diradical, respectively.
Keywords: collisional activation, Favorskii rearrangement, p-hydroxyphenacyl compounds, mass spectrometry
Introduction
p-Hydroxyphenacyl derivatives HO-C6H4-C(O)-CH2-X (1) bearing a suitable leaving group X undergo an efficient photoreaction in water to yield the phenylacetic acid (2) via initial formation of a biradical (3), followed by ring closure to a cyclopropanone (4), and subsequent hydrolysis to 2. Overall, the sequence 1 → 2 corresponds to a photo-Favorskii rearrangement. A competing reaction, the decarbonylation to the quinomethane 5, also takes place, followed by hydration to yield the corresponding p-hydroxybenzyl alcohol 6 (Scheme 1).1
Scheme 1.

Photo-induced Favorskii rearrangement of p-hydroxyphenacyl derivatives.
Owing to its high photoefficiency and compatibility with aqueous solvents, the reaction is of potential interest for the photorelease of compounds for medical or biological purposes2 and has therefore been the subject of intense research in the last years.3,4 While transient absorption spectroscopy has provided deep insight into the photochemical reaction,1–4 the precise mechanism of the complex rearrangement is still a matter of debate.5
Herein, we report the results of some mass spectrometric experiments which deal with free −O-C6H4-C(O)-CH2-X ions in the gas phase.6,7 Consideration of the unsolvated ion represents, of course, a major change when comparison is made with condensed-phase chemistry in many respects. Moreover, in the studies described below we use collisional heating, rather than the photoexcitation applied in Scheme 1. Most important in the specific context is that water has been shown to play a critical role in the photocleavage of the title compounds in solution, whereas the gaseous ions investigated below are unsolvated. Notwithstanding these differences, the gas-phase experiments can allow for the determination of the intrinsic reaction mechanism as well as their energetics and to directly compare these with theoretical results.8 Our original intention was actually to explore the gas-phase chemistry of the ionized compounds (either cations or anions) in order to find a suitable system for probing the Favorskii-type photochemistry that occurs in solution in a “gas-phase photochemical experiment”.9 It was found, however, that simple deprotonation to the corresponding phenolate ion and their subsequent collision-induced fragmentation reactions bear an analogy with the formal Favorskii-type process observed in solution-phase photolysis. Furthermore, regardless of their possible relevance to condensed-phase processes, the fragmentations of the phenolate anions are of interest on their own.
Results and discussion
Upon collision-induced dissociation (CID) of the mass-selected ions in an ion-trap mass spectrometer, each of the p-hydroxyphenacyl derivatives studied that bears a reasonably good anionic leaving group showed dissociation according to reaction (1).
| (1) |
The mass spectrometric experiments reported here do not reveal any information about the nature of the neutral species “C8H6O2”, which does not even need to be an intact entity. A more definitive assignment by mass spectrometric means would require more sophisticated methods,10,11 which cannot be applied in this particular case due to instrumental limitations. By reference to the already existing mechanistic knowledge summarized in Scheme 1, the “C8H6O2” species could thus correspond to either the biradical 3, the cyclopropanone 4, the quinomethane 5 as the product of decarbonylation, or as other alternatives, such as the oxirane 7 or the ketene 8, which can be formed via hydrogen migration following a Favorskii-type rearrangement. These structural alternatives are shown in Scheme 2; note that the location of the radical centers in 3 is only formal and the spin is likely to be located within the aromatic ring system.12
Scheme 2.

Relevant isomeric structures of the intermediate “C8H6O2” species (structure 5 has lost CO).
When comparisons are made with condensed-phase experiments, it is important to note that water is not involved in these gas-phase studies. Though traces of water are present as a background in the ion-trap mass spectrometer and react with the primary product ions at extended timescales, the energizing collisions in the CID experiments occur with the helium bath gas. Therefore, the subsequent hydrolysis reactions of neutral “C8H6O2” to phenylacetic acid 2 or of the quinomethane 5 to 6 do not play a role for the gas-phase fragmentations.
As an example, the ion abundances in the energy-resolved CID spectra of mass selected −O-C6H4-C(O)-CH2-OP(O)(OEt)2 are summarized in Figure 1. At low collision energies, no fragmentation takes place. Starting from an appearance energy of about AE = 2.4 eV, the formation of −OP(O)(OEt)2 concomitant with loss of neutral C8H6O2 is observed and then rapidly gains in intensity. Slightly above 3 eV collision energy, the parent ion nearly disappears. With a branching ratio (BR) of a 2 % for X = OP(O)(OEt)2, the loss of the corresponding acid (i.e. HOP(O)(OEt)2) with concomitant formation of an ion with m/z 133 (i.e. deprotonated C8H6O2) is observed. Within the experimental error the fragmentation thresholds of these two channels are identical, which might hint at the proposed mechanism (see below). Of particular note is the relatively steep rise of the fragmentation yield with increasing collision energy, indicating that the fragmentation reaction involved in reaction (1) is not subject to any pronounced entropic or kinetic restrictions that are likely to arise from atom- or group transfer reactions, more complex structural rearrangements, or changes in spin state.
Figure 1.

Breakdown graph for the CID of the mass-selected phenolate ion −O-C6H4-C(O)-CH2-OP(O)(OEt)2 ( ) as a function of the collision energy (in eV) to yield the fragments −OP(O)(OEt)2 (
) as a function of the collision energy (in eV) to yield the fragments −OP(O)(OEt)2 ( , m/z 153) and C8H5O2− (
, m/z 153) and C8H5O2− ( , m/z 133). Both fragmentation channels have the same appearance energy of AE = (2.4 ± 0.2) eV. In addition, the graph shows the characteristic energy E1/2 at which 50 % of the parent ion fragments and the appearance energy AE derived from linear extrapolation of the slope at E1/2 to the baseline.
, m/z 133). Both fragmentation channels have the same appearance energy of AE = (2.4 ± 0.2) eV. In addition, the graph shows the characteristic energy E1/2 at which 50 % of the parent ion fragments and the appearance energy AE derived from linear extrapolation of the slope at E1/2 to the baseline.
As detailed in the introduction, the title compounds undergo a very effective photolytic cleavage in the condensed phase. Given the fact that the energies of about 2 eV required for fragmentation of the isolated ions in the gas phase are much lower than the energy of the mid-UV photons (about 4 eV) typically used in photodeprotection, two obvious questions arise.
Could the reaction be initiated by a photo-induced deprotonation of the phenol, followed by a nucleophilic attack starting from the phenoxide ion to displace the leaving group and form 4 or 7?
Can intersystem crossing of thermally excited phenoxide ions serve as a plausible explanation for the facile fragmentation of gaseous −O-C6H4-C(O)-CH2-X ions?
Similar to the example shown in Figure 1, we investigated several other −O-C6H4-C(O)-CH2-X ions made by ESI of the corresponding p-hydroxyphenacyl derivatives in the negative ion mode. For all ions investigated, the CID breakdown behavior can be reproduced reasonably well with simple sigmoid fits and the relevant branching ratios and appearance energies are summarized in Table 1.
Table 1.
Summarized appearance energies (AE)a and branching ratios (BR) in the dissociation of mass-selected −O-C6H4-C(O)-CH2-X ions together with the gas-phase acidities (ΔHacid in kJ mol−1) of the conjugate acids of the leaving groups.b The last column gives the major “other” fragments (see supplementary material).
| Xc | ΔHacid(HX) | AE(X−) | BR(X−) | AE(others) | BR(others) | others |
|---|---|---|---|---|---|---|
| TsO1b | 1298 | 1.94 | 100 | |||
| MsO1b | 1343 | 1.84 | 100 | |||
| CF3COO | 1355 | 2.19 | 99 | 2.24 | 1 | m/z 133 |
| (PhO)2(O)PO1e,3c | 2.25 | 100 | ||||
| (EtO)2(O)PO3e | 1387 | 2.43 | 98 | 2.46 | 2 | m/z 133 |
| p-CF3C6H4O | 1410 | 2.82 | 74 | 2.81 | 26 | severald |
| p-MeOC6H4COO | 1426 | 2.91 | 90 | 2.90 | 10 | m/z 133 |
| CH3COO3c,4c | 1436 | 2.52 | 100 | - CH2CO | ||
| HCOO | 1449 | 2.18 | 100 | - CO | ||
| H2N(CH2)3COO2b | 1451 | 2.65 | 100 | - C4H7NO | ||
| C6H5O | 1463 | 3.04 | 64 | 2.99 | 36 | m/z 133 |
| p-MeOC6H4O | 1466 | 3.30 | 4 | 3.03 | 97 | severale |
The relative error of the AEs with regard to the quality of the fits is below ± 0.05 eV in all cases, whereas the absolute error is estimated as ± 0.2 eV.
Values taken from the NIST database in kJ mol−1. Values in italics are estimated from analogous compounds which were in the database.
The references refer to the preparation of the various precursors; the synthesis of new derivatives is described in the Supplementary Information.
According to the mass differences observed, the majority of these side reactions are due to losses of HF (Δm = −20), phenol (Δm = −94), and HX (Δm = −162).
According to the mass differences observed, these major fragmentation reactions are due to losses of CH3• (Δm = −15), CH3• + phenol (Δm = −109), CH3OC6H4O• (Δm = −123), and “C9H8O3” (Δm = −164).
Interestingly, all fragmentation channels for an ion with a given X start at the same appearance energies (within the experimental error), suggesting that the threshold values are subject to a common, kinetically-controlled step (e.g. the barrier of a Favorskii-type process). In this context, we note that the appearance energies were derived via linear extrapolations of sigmoid fits of the fragment ion abundances to the energy axis. While this is not the most elegant approach and, furthermore, we are assuming energy-independent branching ratios where multiple fragmentations are observed, the data do provide a semi-quantitative comparison of the effect of the leaving group on the fragmentation process. This is illustrated by the reasonable correlation between the appearance energies for the formation of X− and the gas-phase acidities of the conjugate acids (HX) in Figure 2. It should be noted, however, that some of the literature ΔHacid(HX) values are associated with considerable error bars (e.g. ± 28 kJ mol−1 for MsOH) and that several others had to be estimated.
Figure 2.
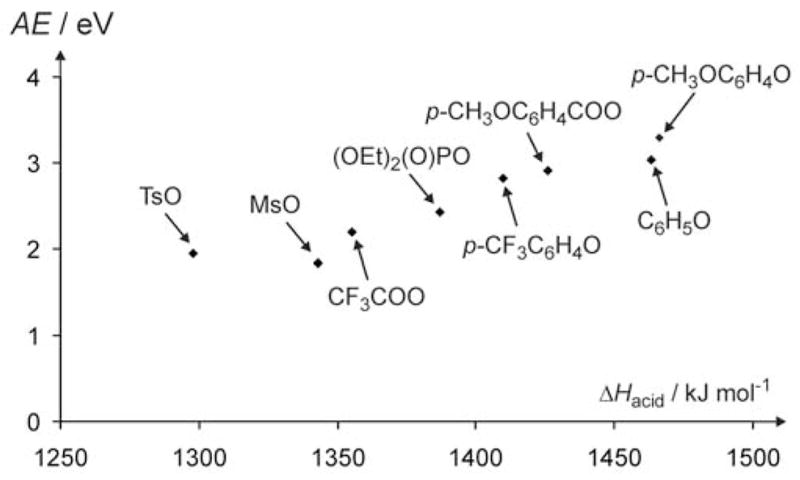
Correlation of the experimentally determined appearance energies (in eV) for the formation of X− upon CID of mass-selected−O-C6H4-C(O)-CH2-X ions and the gas-phase acidities of the conjugate acids (HX).
From this correlation we can roughly estimate that the occurrence of reaction (1) for the simple carboxylates (i.e. X = HCOO, CH3COO, and H2N(CH2)3COO) should have onsets at about 3 eV. Obviously, the lower thresholds of their competitive and specific fragmentations (i.e. loss of CO for formate, loss of ketene for acetate, and loss of C4H7NO for γ-aminobutyrate) suppress the formation of X− via reaction (1).13
To gain more insight into the reaction mechanism and the possible neutral products (Scheme 3), we have investigated reaction (1) for X = CF3COO by means of density functional theory, where all energies are given relative to the deprotonated starting compound [1-H]− with X = CF3COO. For an initial pre-optimization, B3LYP calculations with the small 6–31+G* were performed, while the discussion refers to the results obtained with cc-pVTZ basis sets. The choice of X = CF3COO for the computational study was due to the good quality of the experimental data and negligible side reactions in conjunction with the moderate size and the absence heavier elements like phosphorus or sulfur.
Scheme 3.

Possible neutral “C8H6O2” products for loss of X− upon CID of −O-C6H4-C(O)-CH2-X and their notation in the theoretical studies including the spin states (X = CF3COO); ISC stands for intersystem crossing between the spin states.
We have first explored possible fragmentations of [1-H]− in the singlet ground state. Two possible scenarios are proposed. The elimination of the CF3COO− can be assisted by the closing of either an oxirane ring or a cyclopropanone ring. The first reaction pathway is associated with an energy barrier of 0.99 eV (TS 11/19) and leads to a van der Waals complex 19. The break-up of the complex 19 yields the oxirane product 17 concomitant with free CF3COO−. The concurrent fragmentation of [11-H]− leads via a larger energy barrier of 1.39 eV (TS 11/110) to a van der Waals complex 110, which after elimination of CF3COO−, yields the spirodione 14. The spirodione can further undergo a facile decarbonylation via the transition state TS 14/15 which is only 0.37 eV higher in energy and provide the enone product 15. Interestingly, both primary “C6H8O2” products 14 and 17 are almost isoenergetic and the barriers en route to these neutral fragments are lower than the exit channels themselves, i.e. there are no barriers in excess of the overall reaction endothermicities. Accordingly, the fragmentation is likely to be under thermochemical control and hence 14 and 17 both should be formed during the dissociation of [1-H]−.
We explored the possible involvement of the biradical fragment 33. The formation of the biradical (1[1-H]− → 33 + X−)14 requires only slightly more energy (2.10 eV, Table 2) than those of the singlet products 14 and 17 (Figure 3). Given that the calculated energy demands for the formations of 33, 14, as well as 17 are all within the range of the experimental estimate of AE = (2.2 ± 0.2) eV for X = CF3COO (Table 1), the apparent CID threshold from experiment cannot be used as a criterion to distinguish these channels. A reasonable agreement between the computed and experimentally found endothermicities associated with the formation of CF3COO− also lends further support the calibration of the energy scale used in the CID experiments.15
Table 2.
Calculated energetics (relative energies at 0 K in eV) of −O-C6H4-C(O)-CH2-OOCCF3 and its possible fragments at the B3LYP/cc-pVTZ level of theory.
| Erel | |
|---|---|
| 1[1-H]− | 0.00a |
| 33 + CF3COO− | 2.10 |
| 14 + CF3COO− | 1.99 |
| TS 33/35 + CF3COO− | 2.80 |
| TS 14/15 + CF3COO− | 2.36 |
| TS 14/17 + CF3COO− | 3.01 |
| TS 14/18 + CF3COO− | 3.48 |
| 15 + CO + CF3COO− | 0.85 |
| 35 + CO + CF3COO− | 2.59 |
| 17 + CF3COO− | 2.02 |
| 18 + CF3COO− | 1.39 |
| 18′ + CF3COO− | 0.30 |
Total energy: −985.384563 Hartree.
Figure 3.

B3LYP/cc-pVTZ singlet potential energy surface for the fragmentation and rearrangements of [1-H]−. Energies are given in eV and refer to 0 K (the energies in brackets are obtained at B3LYP/6–31+G* level of theory). Only the most relevant channels are shown; for others see Table 2.
The major difference among the possible products concerns their spin multiplicities. Given that the reactant ion 1[1-H]− is a singlet, formation of 33 would require a spin flip along the reaction coordinate (“two-state reactivity”),16 whereas formation of 14 and 17 can proceed on a single spin surface as depicted in Figure 4. Though it is often implied that effective spin changes require the presence of heavy elements and/or transition metals, some rather simple CHO compounds have been shown to obey two-state scenarios in their dissociation (e.g. CH3O+, C2O2, and C2O42+).17,18 Hence, the spin argument cannot be used to a priori exclude the high-spin product 33.19 If we consider the singlet and triplet potential energy curves calculated along the elongated C–C bond of the spirodione and the shortened the C–C bond of the biradical (Figure 4), we find that the crossing of both curves is located about 0.5 eV above the respective minima. Another aspect, perhaps unusual for synthetic chemists, is that structure 3 does exist only as a triplet and structure 4 only as a singlet, in that neither 13 nor 34 are minima on the respective potential energy surface. Intersystem crossing of 33 to the singlet surface is thus associated with a ring closure to afford 14 and the reverse process corresponds to ring opening (Scheme 3).
Figure 4.
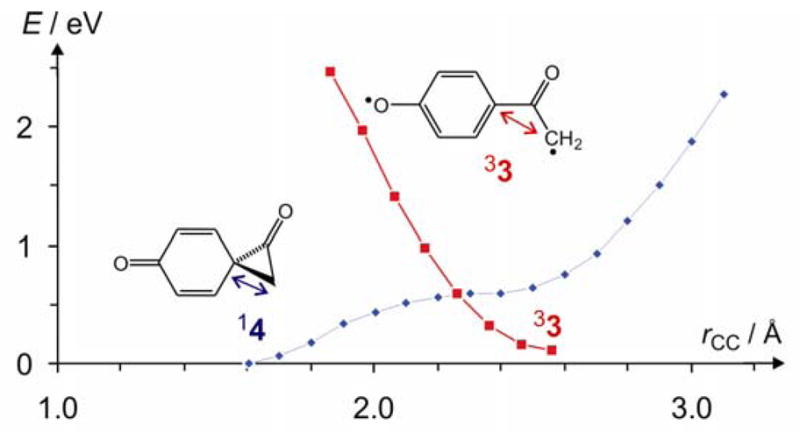
Estimated surface crossing (energies in eV) between the opening spirodione (blue, singlet) and the closing diradical (red, triplet) derived from B3LYP/6–31+G* geometry optimizations with fixed length of the (opening/closing) C–C bond.
The high strain energy of the cyclopropanone 14 prompted us to probe its rearrangement via hydrogen migration. Though the resulting ketene 18 is significantly more stable than 33 as well as 14 (Table 2), the energy barrier resulting from the associated transition structure TS 14/18 (Erel = 3.48 eV) excludes this as a relevant intermediate in the experiment. Likewise, a tautomer of 18, (4-hydroxyphenyl) ketene 18′, is rather stable, but not accessible in a direct rearrangement from either 33 or 14. Furthermore, decarbonylation of the diradical 3 and the spiro compound 14 that affords quinomethane 5 in either spin state is quite favorable if the singlet products are formed, but the barriers involved (TS 33/35, Erel = 2.80 eV and TS 14/15, Erel = 2.36 eV) appear too large to account for the experimental findings.
Correlating the experimental data (apparent thresholds and energy behavior) as well as the theoretical findings (minima, barriers, and spin states), we conclude that close to the threshold reaction (1) for X = CF3COO can lead to the cyclopropanone 14 or the oxirane 17 with nearly equal probability. At elevated energies the triplet biradical 33 may also contribute. We note in passing that B3LYP/cc-pVTZ predicts the gas-phase acidity of CF3COOH in the correct range (1371 kJ mol−1 compared to the experimental value of 1355 ± 12 kJ mol−1).
In addition, some phenacyl compounds with differently substituted aromatic moieties were probed in order to elucidate possible electronic effects. The influence of methoxy substitution was examined for X = (EtO)2(O)PO, where AE(X−) = 2.43 eV was obtained for the unsubstituted parent p-hydroxyphenacyl compound (Table 1), whereas the 2-methoxy- and for 3,5-dimethoxy phenacyl ions gave AEs of 2.42 eV and 2.34 eV, respectively. These differences are, however, within the experimental error margins of about ± 0.05 eV in the relative and ± 0.2 eV in the absolute CID thresholds. For cyano-substituted phenacyl derivatives with X = CH3COO as potential leaving groups, again, no formation of X− was observed and loss of ketene prevailed instead with thresholds of 2.90 eV for the 2-cyano and 2.96 eV for the 3-cyano compared to only 2.52 eV for the all-H compound. The emerging trends for substitutions of the phenacyl core can by and large be attributed to mere thermodynamic effects operating on the respective reactant anions [1-H]−. Thus, donors such as methoxy substituents, are expected to decrease the electron affinity of the neutral counterpart [1-H]• and thus also decrease the stability of the [1-H]− anion. However, in the products resulting from dissociation of [1-H]− according to reaction (1), the substituents in the phenacyl core end up in the neutral product and mere electron-withdrawing or -donating effects thus become much less important. Hence, for a given X, the dissociation threshold is likely to be determined more by the thermochemical properties of the precursor ion [1-H]−. Consistent with this line of reasoning are the significantly elevated AEs of the cyano-substituted derivatives, where the electron withdrawing substituents primarily stabilize the precursor [1-H]−, whereas the corresponding effect on the neutral product of reaction (1) is much smaller. In fact, these findings parallel the photochemistry in the condensed phase. In a comparative investigation of a broad range of substituent effects that included F, MeO, CN, CO2R, CONH2, and CH3 on the photochemical rearrangements of p-hydroxyphenacyl esters, there was little effect on either the rate or the quantum efficiency for the photo-Favorskii rearrangement and the release of the acid leaving group or on the lifetimes of the reactive triplet state.20
Conclusions
Collision-induced dissociation of mass-selected p-hydroxyphenacyl derivatives as their conjugate bases, i.e. −O-C6H4-C(O)-CH2X, with various leaving groups X were probed by means of ion-trap mass spectrometry. The losses of X− concomitant with formal generation of “C8H6O2” have surprisingly small thresholds and occur with high efficiencies once the reaction endothermicity is provided in the collision experiments for most leaving groups X. Simple carboxylates such as X = HCOO or CH3COO, however, follow different fragmentation pathways. Detailed computational studies using density functional theory for X = CF3COO reveal a competition of two pathways and two spin surfaces for the loss of trifluoroacetate from the conjugate base of p-hydroxyphenacyl. In fact, we face a situation in which C–X bond heterolysis leads to cyclopropanone 14 and oxirane 17 on the singlet surface and to the biradical 33 for triplet spin. The energy demands of about 2 eV for all three pathways are too close to each other to allow an assignment of the preferred route with regard to the errors in both theory and experiment. Nevertheless, crossing between 33 and 14 is expected to be facile at ambient temperatures, and structure 4 may be regarded as a direct precursor for the formation of p-hydroxyphenylacetic acid and quinomethane 5 that hydrolyzes to 6.
Last, but not least, let us phenomenologically try to extrapolate the gas-phase data to the condensed phase. At first, it is obvious that the phenoxide group will experience a significant stabilization upon solvation, such that the activation energy associated with C X bond heterolysis is expected to be larger in solution. Next, several rearrangements which are relatively difficult in the gas phase, in particular the tautomerizations 4 → 8 and 8 → 8′, will proceed much more efficiently in protic solvents. In fact, an intimate interaction of water molecules with intermediates such as 4 may very much facilitate the interconversion to derivatives of phenylacetic acid. In future studies, we will try to address the latter aspect by investigation of the gas-phase chemistry of partially solvated ions derived from p-hydroxyphenacyl precursors using not only CID, but also experiments at elongated reaction times in the presence of water and possibly also spectroscopic methods.9
Experimental and computational details
The measurements were performed with an ion-trap mass spectrometer (IT-MS) which has been described elsewhere21 by electrospray ionization of dilute methanolic solutions of the p-hydroxyphenacyl derivatives HO-C6H4-C(O)-CH2-X. In brief, our IT-MS bears a conventional ESI source consisting of the spray unit (typical flow rate 5 μl/min., typical spray voltage 5 kV) with nitrogen as a sheath gas, followed by a heated transfer capillary (kept at 200 °C), a first set of lenses which determines the soft- or hardness of ionization by variation of the degree of collisional activation in the medium-pressure regime,22–24 two transfer octopoles, and a Paul ion-trap with ca. 10−3 mbar helium for ion storage, manipulation, and a variety of MSn experiments.21,25 In the ESI source, the sample solution is evaporated at room temperature in the presence of a nitrogen stream and assistance by a kV voltage. The spray then passes through a high-temperature zone (typically operated at 200 °C) that facilitates droplet shrinkage and propels the spray into the differential pumping system. The nascent ions are then transferred to the mass spectrometer. For detection, the ions are ejected from the trap to an electron multiplier. Note that in the standard mode of operation, the IT-MS used has a low-mass cut-off at m/z 50.13 In the present experiments, the instrument was operated in negative ion mode, the corresponding deprotonated ions −O-C6H4-C(O)-CH2-X (X = leaving group) were mass-selected, and then subjected to collision-induced dissociation (CID) by application of an excitation AC voltage to the end caps of the trap to induce collisions of the isolated ions with the helium buffer gas. In a CID experiment, the ion of interest is gradually excited kinetically and allowed to collide with helium as a non-reactive collision partner which causes ion fragmentations. Note that rearrangements themselves are “invisible” in standard MS because these are not associated with mass changes. When the CID experiments are conducted at variable collision energies, quantitative reaction thresholds can be derived.26 In the present IT-MS experiments, we used an ion-excitation period of 20 ms, for which we have recently introduced a phenomenological linear conversion factor for the extraction of approximate appearance energies (AEs) based on comparisons with reference molecules of known bond strengths,15 i.e. Ag(CH3OH)+, Ag(CH3OH)2+, and Fe(C5H5)2+ as suggested by O’Hair and coworkers27 as well as Cu(C5H5N)2+.28 In brief, the AEs are derived from a sigmoid fit of the fragment ion abundances as a function of the collision energy.29 We note that the agreement between the AE derived experimentally and the theoretical prediction for X = CF3COO (see below) can be regarded as an additional test for the conversion scheme from relative to absolute collision energies in IT-MS and its applicability to gaseous anions.15 Furthermore, it is important to recognize that the threshold energies are determined by the highest point in energy the system has to pass prior to fragmentation and AEs can thus reflect either thermodynamic or kinetic control. Due to the presence of some residual water in the ion trap, some very reactive ionic species tend to rapidly associate with this background water during the time of trapping.30 These adducts are summed accordingly into the primary fragments.
Ab initio calculations were performed using the Gaussian03 package31 at the B3LYP level of theory with the cc-pVTZ basis set.32 The minimum geometries were obtained from ab initio optimization and further checked by frequency analysis with the same method and basis set. At this level of theory, all stationary points discussed had only positive frequencies and thus correspond to genuine minima. Harmonic frequency analysis was also used to obtain thermochemical data. All calculations refer to the gaseous state in that additional solvation, aggregation etc. is deliberately not included in order to match the present experimental conditions.
Supplementary Material
Acknowledgments
This work was supported by the Czech Academy of Sciences (Z40550506), the European Research Council (AdG HORIZOMS), the Ministry of Education of the Czech Republic (MSM0021620857), and the National Institute of health (RO1GM72910).
Footnotes
Supplementary information available. Synthesis of new pHP esters and their spectroscopic properties, breakdown graphs of all ions, additional data about ion fragmentations, and the Cartesian coordinates of the computed structures are available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Conrad PG, Givens RS, Hellrung B, Rajesh CS, Ramseier M, Wirz J. J Am Chem Soc. 2000;122:9346.Givens RS, Heger D, Hellrung B, Kamdzhilov Y, Mac M, Conrad PG, Cope E, Lee JI, Mata-Segreda JF, Schowen RL, Wirz J. J Am Chem Soc. 2008;130:3307. doi: 10.1021/ja7109579.Stensrud KF, Heger D, Šebej P, Wirz J, Givens RS. Photochem Photobiol Sci. 2008;7:614. doi: 10.1039/b719367j.Stensrud K, Noh J, Kandler K, Wirz J, Heger D, Givens RS. J Org Chem. 2009;74:5219. doi: 10.1021/jo900139h.See also: Park CH, Givens RS. J Am Chem Soc. 1997;119:2453.
- 2.(a) Givens RS, Goeldner M, editors. Dynamic Studies in Biology: Phototriggers, Photoswitches, and Caged Biomolecules. Wiley; New York: 2005. [Google Scholar]; (b) Givens RS, Weber JJF, Jung AH, Park C-H. In: Methods in Enzymology on “Caged Compounds: Chemistry, Instrumentation, and Applications”. Marriott G, editor. Vol. 291. 1998. p. 1. [Google Scholar]
- 3.(a) Ma CS, Chan WS, Kwok WM, Yuo P, Phillips DL. J Phys Chem B. 2004;108:9264. [Google Scholar]; (b) Ma CS, Zuo P, Kwok WM, Chan WS, Kan JTW, Toy PH, Phillips DL. J Org Chem. 2004;69:6641. doi: 10.1021/jo049331a. [DOI] [PubMed] [Google Scholar]; (c) Ma CS, Kwok WM, Chan WS, Zuo P, Kan JTW, Toy PH, Phillips DL. J Am Chem Soc. 2005;127:1463. doi: 10.1021/ja0458524. [DOI] [PubMed] [Google Scholar]; (d) Zuo P, Ma CS, Kwok WM, Chan WS, Phillips DL. J Org Chem. 2005;70:8661. doi: 10.1021/jo050761q. [DOI] [PubMed] [Google Scholar]; (e) Ma CS, Kwok WM, Chan WS, Du Y, Kan JTW, Toy PH, Phillips DL. J Am Chem Soc. 2006;128:2558. doi: 10.1021/ja0532032. [DOI] [PubMed] [Google Scholar]; (f) Chen XB, Ma CS, Kwok WM, Guan XG, Du Y, Phillips DL. J Phys Chem A. 2006;110:12406. doi: 10.1021/jp064490e. [DOI] [PubMed] [Google Scholar]; (g) Wirz J. Chimia. 2007;61:638. [Google Scholar]; (h) Chen XB, Ma CS, Kwok WM, Guan XG, Du Y, Phillips DL. J Phys Chem B. 2007;111:11832. doi: 10.1021/jp073529s. [DOI] [PubMed] [Google Scholar]; (i) Ma CS, Kwok WM, Chan WS, Du Y, Zuo P, Kan JTW, Toy PH, Phillips DL. Curr Sci. 2009;97:202. [Google Scholar]; (j) Cao Q, Guan X, George MW, Phillips DL, Ma CS, Kwok WM, Li MD, Du Y, Sun XZ, Xue JD. Faraday Disc. 2010;145:171. [Google Scholar]
- 4.Zabadal M, Pelliccioli AP, Klan P, Wirz J. J Phys Chem A. 2001;105:10329. doi: 10.1021/ja016088d.Klan P, Pelliccioli AP, Pospisil T, Wirz J. Photochem Photobiol Sci. 2002;1:920. doi: 10.1039/b208171g.See also: Zhang K, Corrie JET, Munasinghe VRN, Wan P. J Am Chem Soc. 1999;121:5625.
- 5.Bucher G, Phillips DL, Schröder D, Carpenter B, Guthrie JP, Shaik S, Tahara T, Warshel A, Michl J, Williams I, Ashfold MNR, Pandey R, Meech SR, Misra R, Henchman RH, Hutchings GJ, Buurma NJ, Bain CD, Glowacki D, George MW, Pantos GD, Cramer CJ, Lloyd-Jones GC, Hunter CA, Varley LM, Hillman AR, Webb S, Philp D, Page MI. Faraday Disc. 2010;145:255. [Google Scholar]
- 6.For previous work on Favorskii-type rearrangements in the gas phase, see: Dua S, Pollnitz AP, Bowie JH. J Chem Soc Perkin Trans 2. 1993:2235.See also: Han IS, Kim CK, Kim CK, Lee BS, Lee I. J Comp Chem. 1997;18:1773.
- 7.For review of anion rearrangements in the gas phase, see: Eichinger PCH, Dua S, Bowie JH. Int J Mass Spectrom Ion Processes. 1993;133:1.Ahmad MR, Kass SR. Austr J Chem. 2003;56:453.
- 8.(a) Alcami M, Mó O, Yañez M. Mass Spectrom Rev. 2001;20:195. doi: 10.1002/mas.10005. [DOI] [PubMed] [Google Scholar]; (b) Schwarz H. Int J Mass Spectrom. 2004;237:75. [Google Scholar]; (c) Roithová J, Schröder D. Coord Chem Rev. 2009;253:666. [Google Scholar]; (d) Roithová J, Schröder D. Chem Rev. 2010;110:1170. doi: 10.1021/cr900183p. [DOI] [PubMed] [Google Scholar]
- 9.For a promising example towards UV photochemistry in the gas phase, see: Scuderi D, Maitre P, Rondino F, Le Barbu-Debus K, Lepere V, Zehnacker-Rentien A. J Phys Chem A. 2010;114:3306. doi: 10.1021/jp9094497.
- 10.Neutralization-reionization mass spectrometry has been used to characterize the neutral products of ion/molecule reactions in the gas phase, see: Schröder D, Sülzle D, Hrušák J, Böhme DK, Schwarz H. Int J Mass Spectrom Ion Processes. 1991;110:145.Schröder D, Schwarz H. Helv Chim Acta. 1992;75:1281.Schröder D, Müller J, Schwarz H. Organometallics. 1993;12:1972.Schröder D, Schwarz H, Polarz S, Driess M. Phys Chem Chem Phys. 2005;7:1049. doi: 10.1039/b415078c.
- 11.For a neutralization-reionization study of the related acetolactone, see: Schröder D, Goldberg N, Zummack W, Schwarz H, Poutsma JC, Squires RR. Int J Mass Spectrom Ion Processes. 1997;165/166:71.
- 12.(a) Roithová J, Schröder D, Schwarz H. Angew Chem Int Ed. 2005;44:3092. doi: 10.1002/anie.200461783. [DOI] [PubMed] [Google Scholar]; (b) Roithová J, Schröder D. Int J Mass Spectrom. 2007;267:134. [Google Scholar]; (c) Milko P, Roithová J, Schröder D, Lemaire J, Schwarz H, Holthausen MC. Chem Eur J. 2008;14:4318. doi: 10.1002/chem.200800052. [DOI] [PubMed] [Google Scholar]; (d) Milko P, Roithová J, Tsierkezos NG, Schröder D. J Am Chem Soc. 2008;130:7186. doi: 10.1021/ja801079r. [DOI] [PubMed] [Google Scholar]
- 13.With respect to the detection of formate and acetate as ionic fragments, we note that the IT-MS has a default low mass cut-off at m/z 50. However, independently generated CH3COO− could be detected and handled without problems and in the low-mass operation mode, also HCOO− can be detected. In fact, traces of CH3COO− at m/z 59 were observed upon CID of the acetate (X = CH3COO in Table 1), but the abundance was much less than a percent of the other fragments and no quantitative analysis of the energy dependence was possible.
- 14.Although formation of 3 can occur directly from the anion, it is important to distinguish this formally heterolytic bond cleavage (i.e. −O-C6H4-C(O)-CH2-X → −O-C6H4-C(O)-CH2+ + X−) from the more energy demanding homolytic bond cleavage of the neutral phenacyl compound (i.e. HO-C6H4-C(O)-CH2-X → HO-C6H4-C(O)-CH2• + X•) often invoked in photolysis.
- 15.(a) Šrogl J, Hyvl J, Révész A, Schröder D. Chem Comun. 2009:3463. doi: 10.1039/b904403e. [DOI] [PubMed] [Google Scholar]; (b) Tsierkezos NG, Buchta M, Holý P, Schröder D. Rap Commun Mass Spectrom. 2009;23:1550. doi: 10.1002/rcm.4035. [DOI] [PubMed] [Google Scholar]; (c) Schröder D, Ducháčková L, Jušinski I, Eckert-Maksić M, Heyda J, Tůma L, Jungwirth P. Chem Phys Lett. 2010;490:14. [Google Scholar]; (d) Révész A, Milko P, Žabka J, Schröder D, Roithová J. J Mass Spectrom. 2010;45:1246. doi: 10.1002/jms.1793. [DOI] [PubMed] [Google Scholar]; (e) Zins EL, Pepe C, Schröder D. J Mass Spectrom. 2010;45:1253. doi: 10.1002/jms.1847. [DOI] [PubMed] [Google Scholar]
- 16.Schröder D, Shaik S, Schwarz H. Acc Chem Res. 2001;33:139. doi: 10.1021/ar990028j.Schwarz H. Int J Mass Spectrom. 2004;237:75.Harvey JN, Poli R, Smith KM. Coord Chem Rev. 2003;238:347.Shaik S, Hirao H, Kumar D. Acc Chem Res. 2007;40:532. doi: 10.1021/ar600042c.See also: Yarkony DR. J Phys Chem. 1996;100:18612.
- 17.(a) Aschi M, Harvey JN, Schalley CA, Schröder D, Schwarz H. J Chem Soc, Chem Commun. 1998:531. [Google Scholar]; (b) Schröder D, Heinemann C, Schwarz H, Harvey JN, Dua S, Blanksby SJ, Bowie JH. Chem Eur J. 1998;4:2550. [Google Scholar]; (c) Roithová J, Ricketts CL, Schröder D, Price SD. Angew Chem Int Ed. 2007;46:9316. doi: 10.1002/anie.200704286. [DOI] [PubMed] [Google Scholar]; (d) Feixas F, Ponec R, Schröder D, Roithová J, Fišer J, Price SD. J Phys Chem A. 2010;114:6681. doi: 10.1021/jp1020559. [DOI] [PubMed] [Google Scholar]
- 18.For selected other examples of two-state scenarios with only 2nd row elements, see: Janaway GA, Zhong ML, Gatev GG, Chabinyc ML, Brauman JI. J Am Chem Soc. 1997;119:11697.Hu J, Hill BT, Squires RR. J Am Chem Soc. 1997;119:11699.Yarkony DR. J Phys Chem A. 1998;102:5305.Cui Q, Morokuma K, Bowman JM, Klippenstein SJ. J Chem Phys. 1999;110:9469.Griesbeck A, Abe M, Bondock S. Acc Chem Res. 2004;37:919. doi: 10.1021/ar040081u.Contreras R, Galvan M, Oliva M, Safont VS, Andres J, Guerra D, Aizman A. Chem Phys Lett. 2008;457:216.
- 19.For solvent effects on the spin inversion of carbenes, see: Hess GC, Kohler B, Likhotvorik I, Peon J, Platz MS. J Am Chem Soc. 2000;122:8087.Wang J, Kubicki J, Peng H, Platz MS. J Am Chem Soc. 2008;130:6604. doi: 10.1021/ja711385t.
- 20.Givens RS, Stensrud K, Conrad PG, II, Yousef AL, Perera C, Senadheera SN, Heger D, Wirz J. Can J Chem. 2011 doi: 10.1139/V10-143. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tintaru A, Roithová J, Schröder D, Charles L, Jušinski I, Glasovac Z, Eckert-Maksić M. J Phys Chem A. 2008;112:12097. doi: 10.1021/jp805897f. [DOI] [PubMed] [Google Scholar]
- 22.Cech NB, Enke CG. Mass Spectrom Rev. 2001;20:362. doi: 10.1002/mas.10008. [DOI] [PubMed] [Google Scholar]
- 23.Schröder D, Weiske T, Schwarz H. Int J Mass Spectrom. 2002;219:729. [Google Scholar]
- 24.Trage C, Diefenbach M, Schröder D, Schwarz H. Chem Eur J. 2006;12:2454. doi: 10.1002/chem.200500718. [DOI] [PubMed] [Google Scholar]
- 25.O’Hair RAJ. Chem Commun. 2006:1469. doi: 10.1039/b516348j. [DOI] [PubMed] [Google Scholar]
- 26.(a) Armentrout PB. Int J Mass Spectrom. 2000;200:219. [Google Scholar]; (b) Ervin KM. Chem Rev. 2001;101:391. doi: 10.1021/cr990081l. [DOI] [PubMed] [Google Scholar]; (c) Narancic S, Bach A, Chen P. J Phys Chem A. 2007;111:7006. doi: 10.1021/jp072092l. [DOI] [PubMed] [Google Scholar]; (d) Armentrout PB, Ervin KM, Rodgers MT. J Phys Chem A. 2008;112:10071. doi: 10.1021/jp805343h. [DOI] [PubMed] [Google Scholar]
- 27.O’Hair RAJ, Vrkic AK, James PF. J Am Chem Soc. 2004;126:12173. doi: 10.1021/ja048476w. [DOI] [PubMed] [Google Scholar]
- 28.(a) Rodgers MT, Stanley JR, Amunugama R. J Am Chem Soc. 2000;122:10969. [Google Scholar]; (b) Rannulu NS, Rodgers MT. J Phys Chem A. 2007;111:3465. doi: 10.1021/jp066903h. [DOI] [PubMed] [Google Scholar]; (c) Than S, Maeda H, Irie M, Itoh S, Kikukawa K, Mishima K. J Phys Chem A. 2007;111:5988. doi: 10.1021/jp071664z. [DOI] [PubMed] [Google Scholar]
- 29.Schröder D, Engeser M, Brönstrup M, Daniel C, Spandl J, Hartl H. Int J Mass Spectrom. 2003;228:743. [Google Scholar]
- 30.(a) Jagoda-Cwiklik B, Jungwirth J, Rulíšek L, Milko P, Roithová J, Lemaire J, Maitre P, Ortega JM, Schröder D. ChemPhysChem. 2007;8:1629. doi: 10.1002/cphc.200700196. [DOI] [PubMed] [Google Scholar]; (b) Milko P, Roithová J, Schröder D, Lemaire J, Schwarz H, Holthausen MC. Chem Eur J. 2008;14:4318. doi: 10.1002/chem.200800052. [DOI] [PubMed] [Google Scholar]; (c) Tyo EC, Castleman AW, Jr, Schröder D, Milko P, Roithová J, Ortega JM, Cinellu MA, Cocco F, Minghetti G. J Am Chem Soc. 2009;131:13009. doi: 10.1021/ja902773b. [DOI] [PubMed] [Google Scholar]
- 31.Gaussian 03, Revision E.01. Gaussian, Inc; Wallingford CT: 2004. [Google Scholar]
- 32.Dunning TH., Jr J Chem Phys. 1989;90:1007. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.