

Abstract
Anchorage to a compliant extracellular matrix (ECM) and contact with neighboring cells impose important constraints on the proliferation of epithelial cells. How anchorage and contact dependence are inter-related and how cells weigh these adhesive cues alongside soluble growth factors to make a net cell cycle decision remain unclear. Here, we show that a moderate 4.5-fold stiffening of the matrix reduces the threshold amount of epidermal growth factor (EGF) needed to over-ride contact inhibition by over 100-fold. At EGF doses in the range of the dissociation constant (Kd) for ligand binding, epithelial cells on soft matrices are contact inhibited with DNA synthesis restricted to the periphery of cell clusters. By contrast, on stiff substrates, even EGF doses at sub-Kd levels over-ride contact inhibition, leading to proliferation throughout the cluster. Thus, matrix stiffening significantly sensitizes cells to EGF, enabling contact-independent spatially uniform proliferation. Contact inhibition on soft substrates requires E-cadherin, and the loss of contact inhibition upon matrix stiffening is accompanied by the disruption of cell–cell contacts, changes in the localization of the EGF receptor and ZO-1, and selective attenuation of ERK, but not Akt, signaling. We propose a quantitative framework for the epigenetic priming (via ECM stiffening) of a classical oncogenic pathway (EGF) with implications for the regulation of tissue growth during morphogenesis and cancer progression.
Key words: Cancer, Contact inhibition, Matrix stiffness, Proliferation, Mechanotransduction
Introduction
Contact inhibition of proliferation is a hallmark of normal epithelial cells. By contrast, cancer cells over-ride this key constraint and proliferate in a contact-independent manner, leading to tumor formation (Hanahan and Weinberg, 2000). Contact inhibition is enforced in a rich microenvironment that includes conflicting mitogenic stimuli, such as soluble growth factors. Antagonistic interactions between growth factors and cell–cell contact are mediated through several mechanisms involving the atypical cadherin, Fat (protocadherin Fat 1), the ERM family proteins, Merlin and Expanded, the Hippo–YAP pathway and interactions between cadherins and growth factor receptors (Curto et al., 2007; Hamaratoglu et al., 2006; Lampugnani et al., 2003; Lampugnani et al., 2006; Yin and Pan, 2007).
We recently demonstrated that this crosstalk has quantitative implications for contact inhibition in a microenvironment that includes the mitogen EGF (epidermal growth factor) (Kim et al., 2009). Cell–cell contact does not act as an autonomous switch and is titrated against the level of EGF to determine the net effect on cell proliferation. Only when the level of EGF is below a threshold amount does cell–cell contact inhibit proliferation, leading to a spatial pattern in proliferation in epithelial cell clusters. Furthermore, this threshold is a tuneable property. Enhancing cell–cell interactions either specifically by overexpressing E-cadherin or non-specifically by crowding cells in a micropatterned region elevates the EGF threshold. These quantitative features of contact inhibition are captured in a state diagram model (supplementary material Fig. S1).
The state diagram model provides a quantitative framework for the contact dependence of cell proliferation. Cell cycle progression, however, is regulated by cell adhesion not only to its neighbors, but also to the ECM. In non-transformed cells, adhesion to the ECM is required for a full mitogenic response to growth factor stimulation (Lee and Juliano, 2004). The loss of ECM-dependent proliferation leads to anchorage-independent proliferation, another hallmark of cancer cells (Assoian, 1997). However, how anchorage-dependent and contact-dependent proliferation are inter-related remains to be elucidated. This issue is particularly relevant in many physiological contexts in which epithelial cells are exposed to soluble growth factors while adhered to both an underlying ECM and to neighboring cells. How does the three-way crosstalk among cell–cell contact, ECM and growth factors quantitatively affect cell cycle regulation? How does the ECM factor into or modify the state diagram model?
To begin to examine these questions, we focused on a physiologically significant property of the ECM: its mechanical compliance. Changes in ECM stiffness are associated with disease progression. A prominent example is the stiffening of the ECM during cancer progression and its role in metastasis and disruption of tissue architecture (Butcher et al., 2009; Levental et al., 2009). Matrix stiffness is now broadly appreciated to affect several types of cell behavior, including cell cycle activity (Klein et al., 2009), stem cell differentiation (Engler et al., 2006) and cell migration (Pelham and Wang, 1997). However, how cells evaluate ECM compliance in the context of other environmental cues, such as cell–cell contact and soluble growth factors, remains unclear. This question is particularly important with regard to cell cycle activity because its regulation involves crosstalk between the ECM, cell–cell contacts and soluble growth factors.
Results
Substratum compliance affects spatial patterns in proliferation and contact inhibition
To investigate the interplay between ECM compliance, cell–cell contact and growth factors, we studied EGF-mediated proliferation of non-transformed, contact-dependent epithelial cells grown on mechanically compliant substrata. Madin-Darby canine kidney (MDCK) epithelial cells formed 2D multicellular clusters when cultured on collagen-coated polyacrylamide gels of varying stiffness (7–31 kPa) and identical adhesion ligand composition (supplementary material Fig. S2). On the most compliant substratum (7 kPa), treatment with 100 ng/ml EGF induced BrdU uptake exclusively at the periphery of clusters (Fig. 1Ai,B). Increased substratum stiffness eliminated this spatial pattern in proliferation (Fig. 1Aiii), with interior and peripheral cells exhibiting equivalent levels of BrdU uptake (Fig. 1B).
Fig. 1.

Substratum compliance affects spatial patterns in cell-cycle activity and contact inhibition of proliferation. (A) BrdU incorporation (green) and DAPI staining (blue) were assessed in serum-starved MDCK cells seeded on collagen-coated gels of varying stiffness following 16 hours of treatment with 100 ng/ml EGF. (B) The graph shows the percentage of peripheral and interior cells undergoing DNA synthesis. Values are mean ± s.d. (n=2–5). *P<0.01 (Student's t-test) for comparison of the percentage of BrdU incorporation among peripheral versus interior cells. The lines connecting bars denote the pair of data points being compared in the Student's t-test. (C) Downregulation of E-cadherin eliminates the spatial pattern in proliferation on soft substrates. MDCK cells grown on soft substrates (7 kPa) were transfected with control siRNA or siRNA against E-cadherin and the percentage of peripheral and interior cells incorporating BrdU was quantified following EGF stimulation as in A. The extent of E-cadherin knockdown was determined by western blot. Equal loading was confirmed by probing for actin. Values are mean ± s.d. (n=2). *P<0.01 (Student's t-test). Scale bars: 100 μm.
The proliferation pattern on soft substrates is consistent with contact inhibition of proliferation of interior cells, whereas cells at the periphery are not surrounded by neighbors and undertake DNA synthesis. To test directly whether the observed pattern was in fact due to contact inhibition, we examined the effect of diminishing cell–cell interactions by downregulating E-cadherin expression using siRNA. Compared with a control construct, transfection with siRNA targeting E-cadherin significantly reduced E-cadherin expression and eliminated the spatial pattern in proliferation on soft substrates (Fig. 1C). These results demonstrate that E-cadherin-mediated cell–cell contact is involved in inhibiting proliferation of the interior cells, leading to a spatial pattern in proliferation. By contrast, on stiffer substrates, cell–cell contact is not sufficient to halt proliferation of interior cells, leading to uniform cell cycle activity across the cluster.
Substratum compliance quantitatively modulates the threshold level of EGF needed to override contact inhibition
Our initial experiments suggest that soft substrates favor contact inhibition, whereas stiff substrates promote contact-independent proliferation. However, this interpretation is based on observations at a single supra-saturating dose of EGF. We have shown previously that epithelial cells transition between contact-inhibited and contact-independent proliferation when the amount of EGF crosses a critical threshold level (supplementary material Fig. S1). Thus, we reasoned that it might be important to evaluate the effect of substratum compliance on contact inhibition at different levels of EGF.
We examined cell cycle activity in clusters of non-transformed human mammary epithelial cells (MCF-10A) cultured on substrates of different stiffness over a broad range of EGF concentration. On soft substrata, at the low and intermediate EGF concentrations (0.01 and 1 ng/ml, respectively), peripheral cells proliferated with a higher propensity than interior cells, exhibiting a spatially patterned, contact inhibited mode of proliferation (Fig. 2Ai,ii; quantification in Fig. 2B). When the EGF concentration was increased above 10 ng/ml, the proliferation pattern diminished such that an equal fraction of interior and peripheral cells incorporated BrdU when stimulated with 100 ng/ml EGF (Fig. 2B). These results reveal that soft surfaces permit both contact-inhibited and contact-independent proliferation. Thus, contact dependence is not only regulated by substratum stiffness, but also depends on whether the level of EGF is above or below the threshold (in this case ~10 ng/ml EGF).
Fig. 2.

Substratum stiffening reduces the EGF threshold needed to transition from contact-inhibited to contact-independent proliferation. MCF-10A cells plated on soft (7 kPa) and stiff (31 kPa) substrates coated with fibronectin were serum starved and stimulated with the indicated doses of EGF or left untreated. (A) BrdU uptake (green) and DAPI staining (blue) were assessed 22 hours after EGF treatment. (B) The fractions of interior and peripheral cells incorporating BrdU were quantified, and the ratio of these two fractions is plotted as a function of EGF concentration. Values are mean ± s.d. (n=2–3), ANOVA followed by post-hoc tests *,**P<0.05. Scale bar: 100 μm.
A key question is whether this EGF threshold is sensitive to substratum compliance. That is, does changing the substratum stiffness quantitatively shift the transition point between contact inhibition and contact-independent proliferation? To address this question, we repeated the EGF dose study, now using stiffer substrates. As with the soft surface, at low EGF concentrations (0.001 and 0.01 ng/ml), BrdU uptake was concentrated at the periphery of clusters (Fig. 2Aiv,B). However, in contrast to the soft surface, an intermediate level of EGF (0.1–1 ng/ml) was sufficient to eliminate the spatial pattern in cell cycle activity (Fig. 2Av,B).
These results demonstrate that substratum stiffening (from 7 to 31 kPa) quantitatively reduces the EGF threshold from 10 to 0.1 ng/ml in MCF-10A cells. Thus, proliferation is not simply contact inhibited on soft substrates and contact independent on stiff substrates. Rather, matrix stiffening reduces the EGF threshold needed to induce contact independence, thereby quantitatively facilitating the loss of contact inhibition.
We corroborated this quantitative effect of substratum compliance in another epithelial system, MDCK cells. As discussed above, MDCK cells were contact inhibited on soft substrates (7 kPa) even at a supra-saturating dose of EGF (100 ng/ml). Thus, the threshold EGF was too high to attain contact-independent growth on these substrates (supplementary material Fig. S3). However, on substrates of intermediate stiffness (17 kPa), MDCK cells underwent a clear transition from contact-inhibited to contact-independent growth at a threshold of approximately 1 ng/ml EGF. Thus, substrate stiffening reduces the EGF threshold to a physiologically accessible level. Finally, upon stiffening the substratum further to 31 kPa, cells exhibited contact-independent proliferation for all EGF concentrations, revealing that the threshold EGF has diminished below the range tested in our experiments.
Taken together, these results in MCF-10A and MDCK epithelial cells demonstrate that substratum stiffening quantitatively modulates contact inhibition by reducing the EGF threshold needed to shift cells from contact-inhibited to contact-independent proliferation. In addition, the results show that epithelial cell systems can exhibit different sensitivities to substratum compliance. Over the same range of substratum compliance (7–31 kPa), the EGF threshold shifted two orders of magnitude in MCF-10A cells. In MDCK cells, the effect extended even beyond the range of EGF concentrations used in our experiments. The differential sensitivity to substratum compliance might arise from differences in adhesion structures. For example, MCF-10A cells lack Crumbs3, which is required for tight junction formation and maintaining epithelial cell polarity (Fogg et al., 2005).
Substratum compliance affects the maturation of cell–cell contacts
Recent evidence suggests that cell–cell contacts are mechanically coupled to cell–matrix adhesions. The spatial distribution of traction forces exerted by cells on the ECM depends on cell–cell interactions mediated by cadherins (Nelson et al., 2005). A tug-of-war model has been presented by which cell-generated traction on the ECM enables a cell to disengage from its adhesion to a neighboring cell during in vitro cell scatter (de Rooij et al., 2005). Consistent with this model, the formation of three-dimensional (3D) cell aggregates is hampered on stiff substrates where cell–matrix adhesions are larger and more intense than on soft substrates (Guo et al., 2006). Indeed, stiffening 3D protein gels by increasing the concentration of ECM proteins enhances cell–matrix interactions while disrupting cell–cell adhesions (Paszek et al., 2005). However, it is unclear whether the disruptive effect of ECM stiffening on cell–cell contacts applies only when cells organize into 3D aggregates or can be achieved even in a 2D monolayer cluster of cells that sit on a compliant ECM.
To address this question, we examined the subcellular localization of E-cadherin and tight junction protein ZO-1 in MDCK cells. The most striking observation involved the regulation of nuclear localization of ZO-1 (Fig. 3A,B). On the soft substratum, ZO-1 was localized to cell–cell junctions among the growth-arrested cells in the interior of the cluster, and only peripheral cells exhibited nuclear and cytoplasmic ZO-1 localization. By contrast, on the stiff substratum, significant nuclear ZO-1 was observed in all cells in the cluster. This pattern in nuclear localization of ZO-1 was highly correlated with the proliferation patterns on soft and stiff substrates (Fig. 1).
Fig. 3.

Matrix stiffness affects the molecular organization of adhesion structures at cell–cell contacts. MDCK cells cultured on soft (7 kPa) and stiff (31 kPa) substrata were serum starved and immunostained for ZO-1 (green) and E-cadherin (red). Nuclei were co-stained with Hoechst 33342 (blue). (A) Heat maps show the relative abundance of ZO-1 in the epithelial clusters integrated across the z-stacks. (B) Merged x-y images (top) were generated by averaging the pixel intensity across the z-stacks. White lines in the merged image (black arrows) indicate the planes for which x-z views (C) were generated. Scale bar: 10 μm. The line graphs show the quantification of relative intensity of ZO-1 and E-cadherin staining along the x-axis at a fixed y-axis position indicated by the white lines in B. Arrowheads indicate the location of cell–cell contacts.
Concomitant with the nuclear localization of ZO-1, substratum stiffening disrupted the localization of E-cadherin and ZO-1 to cell–cell junctions (Fig. 3C). On soft substrates, E-cadherin and ZO-1 were strongly localized to the basolateral and apical regions of cell–cell junctions, respectively, indicative of mature cell–cell contacts (Balkovetz et al., 1997). This localization to cell–cell contacts was most evident in interior cells, where the intensity of E-cadherin and ZO-1 staining at cell–cell interfaces exceeded the levels of these proteins within the cell body (see quantification in Fig. 3C). By contrast, on stiff substrates, the fraction of cellular E-cadherin and ZO-1 at cell–cell junctions diminished and the intensity of this staining was at or below the levels within the cell body. This disruption of E-cadherin and ZO-1 localization to cell–cell contacts was also observed on glass substrates (supplementary material Fig. S4), demonstrating that regardless of whether the stiff substratum is gel-based or glass, the localization of cell–cell adhesion proteins to intercellular contacts is attenuated. Furthermore, consistent with these results, cells grown on glass exhibited a uniform distribution of BrdU uptake, matching the proliferation phenotype on stiff gels (supplementary material Fig. S5). Thus, substratum stiffening, even in a 2D context, disrupts cell–cell junctions at a molecular level by diminishing the localization of key adhesion proteins to contact sites.
In addition to the reduction of E-cadherin and ZO-1 localization at cell–cell contacts, we observed qualitative changes in cell spreading due to matrix stiffening (Fig. 3A). Others have shown that isolated cells spread more extensively on stiffer substrates (Discher et al., 2005; Paszek et al., 2005). A similar trend is observed here in the context of multicellular clusters. MDCK cells on soft surfaces establish a mean spread area of 250 μm2; by contrast, cells on stiff substrates spread to a significantly greater extent, achieving a mean spread area of 630 μm2 (supplementary material Fig. S6A). These results suggest a correlation between spreading and the proliferation phenotype on compliant surfaces.
To begin to separate the role of cell spreading and cell–cell contacts in the regulation of the proliferation phenotype, we carried out experiments to knock down E-cadherin. Knockdown of E-cadherin expression using siRNA eliminated contact inhibition and promoted uniform proliferation on soft gels (Fig. 1C). We asked whether the effect of E-cadherin knockdown on the proliferation phenotype is accompanied by changes in cell spreading or occurs independently of cell shape changes.
Quantification of cell spread area revealed that a reduction of E-cadherin expression does not affect cell spreading (supplementary material Fig. S6B). These results demonstrate that although stiffening the matrix can cause changes in cell spreading, E-cadherin-mediated cell–cell contacts have a direct role in regulating contact inhibition and spatial patterns in proliferation. Thus, even though cell spreading remains unaffected, manipulating the level of E-cadherin has a direct impact on the proliferation phenotype. These findings, however, do not rule out the possibility that substratum compliance regulates cell proliferation at least partly through effects on cell spreading. For example, cell spreading might operate in parallel with or upstream of cell–cell contacts to regulate the proliferation phenotype.
Enhanced contact maturation on soft substrates selectively affects signalling through ERK but not through Akt
During the maturation of confluent epithelial cell monolayers, membrane-associated EGFR shifts to cell–cell junctions and its trafficking and signaling properties are altered (Curto et al., 2007). Thus, we hypothesized that the disruptive effect of substratum stiffening on cell–cell contacts might regulate EGFR. To test this hypothesis, we first examined the effect of substratum stiffening on EGFR subcellular localization. On a soft substratum, EGFR was highly localized to the basolateral membrane compartments at which stable E-cadherin-mediated adherens junctions formed (Fig. 4A). By contrast, on stiffer substrata, EGFR was more evenly distributed among apical and basal membranes without colocalizing with E-cadherin.
Fig. 4.

Substratum compliance affects subcellular localization of EGFR and selectively regulates phosphorylation of ERK, but not Akt. (A) MCF-10A cells cultured on soft (7 kPa) and stiff (31 kPa) substrata were serum starved and immunostained for EGFR (green) and E-cadherin (red). Nuclei were co-stained with Hoechst 33342 (blue). Merged x-y images (top) and the x-z views (bottom) were generated and labeled as in Fig. 3B. Scale bar: 10 μm. (B) Serum-starved MDCK cells on soft and stiff substrates were stimulated with 100 ng/ml EGF for 15 minutes. Phosphorylation ERK and Akt (green) was assessed by immunofluorescence. Nuclei were labeled by DAPI staining. The bar graphs show the relative intensities of phosphorylated ERK and Akt in peripheral and central cells. Signaling intensities are reported relative to the amount of signals in peripheral cells Values are mean ± s.d. (n=3). *P<0.01 (Student's t-test), for comparison of results indicated by the line. Scale bar: 50 μm.
To determine whether this change in EGFR sequestration affects downstream signaling, we assayed the phosphorylation of extracellular signal-regulated kinase (ERK) and Akt following EGF stimulation of MDCK clusters grown on soft and stiff substrates (Fig. 4B). The level of phosphorylated ERK was distinctly lower in the interior cells on the soft surface. This spatial pattern in ERK phosphorylation corresponds to the observed growth patterns (Fig. 1). However, on stiffer substrates, the ERK signal was homogeneous across the cell cluster, consistent with uniform growth pattern. By contrast to ERK signaling, Akt signaling was uniform within cell clusters regardless of substratum compliance (Fig. 4B). Thus, substratum stiffening selectively enhances EGF-mediated ERK signaling, but not Akt signaling en route to promoting contact-independent, spatially uniform proliferation.
Discussion
Substratum stiffening promotes quantitative, progressive loss of contact inhibition: implications for cancer and morphogenesis
Here, we elucidate the quantitative interplay between three classes of environmental cues – ECM, cell–cell contacts and soluble growth factors – in regulating cell proliferation. We show that the mechanical compliance of the ECM works synergistically with EGF signaling to regulate contact inhibition of proliferation. Matrix stiffening reduces the EGF threshold that an epithelial system must cross to over-ride contact inhibition of proliferation.
The quantitative effect of ECM stiffening has two key features that have implications for cancer and developmental morphogenesis. First, the magnitude of the effect is significant. Increasing the elastic modulus by 4.5-fold reduces the threshold EGF 100-fold in MCF-10A cells (Fig. 2B), thereby significantly reducing the EGF amplification needed to shift from contact-inhibited to contact-independent proliferation. Matrix stiffening is a widely observed phenomenon in cancer progression (Butcher et al., 2009; Levental et al., 2009). Our data suggest that even early, moderate changes in the mechanical properties of the microenvironment can quantitatively sensitize epithelial cells to EGF and prime the system closer to contact-independent proliferation. In the context of morphogenesis, the ECM is actively remodeled through the deposition, crosslinking and cleavage of ECM proteins (Wiseman and Werb, 2002). ECM remodeling can lead to local differences in the mechanical compliance of the matrix. Our results suggest that such spatial differences in ECM stiffness would be amplified into significant spatial heterogeneity in EGF responsiveness, thereby sensitizing tissue growth in specific locations during morphogenetic processes, such as tubulogenesis and branching.
A second key observation is that the profound quantitative effect of ECM stiffening on the EGF threshold can be concealed entirely if one observes the system only at the phenotypic level. For example, when the EGF concentration is 0.01 ng/ml, increasing the elastic modulus from 7 to 31 kPa does not affect the spatial pattern in proliferation in MCF-10A cell clusters (Fig. 2). Although the phenotype is unchanged, a significant quantitative effect accrues because the threshold amount of EGF needed to induce contact-independent proliferation drops from 10 to 0.1 ng/ml. This phenotypically latent, quantitative loss in contact inhibition has intriguing implications for the multi-hit model of cancer progression. By latently shifting epithelial cells closer to a transition to contact-independent proliferation, the epigenetic event of ECM stiffening might have no morphologically perceptible effect on tumor initiation but could sensitize non-transformed cells to respond more readily to an amplification of the classical oncogenic EGF pathway. This early role of ECM stiffening in quantitatively enabling the loss of contact inhibition and tumor formation might complement the role of ECM stiffening in advancing metastasis (Paszek et al., 2005).
When evaluating the physiological implications of the threshold EGF concentrations, it is meaningful to consider the dissociation constant (Kd) for EGF binding to EGFR. The Kd for EGF–EGFR binding is in the range of 1–10 ng/ml (Dahmane et al., 1996; French et al., 1995). The important observation in this work is that stiffening the matrix shifts the threshold EGF dose from near the Kd to significantly below the Kd. For example, in MCF-10A cells, the threshold EGF is 10 ng/ml on soft substrates and drops to 0.1 ng/ml on stiff substrates (the effect is even more profound in MDCK cells). Hence, even near-Kd doses of EGF (i.e. ~50% of the receptors occupied) are unable to break contact inhibition on a soft matrix, which keeps interior cells from proliferating. However, when the stiffness of the matrix increases, even sub-saturating doses are adequate to induce proliferation throughout the cluster. Thus, substratum stiffness has a pivotal role in setting the EGF threshold level relative to the Kd. In this manner, the softness of the matrix provides a remarkable regulatory constraint by enabling contact inhibition to overcome significant occupancy of EGF receptors. Stiffening of the matrix, a phenomenon observed in numerous cancer contexts, would relax contact inhibition such that cell proliferation is triggered at levels well below Kd values.
The quantitative interplay that we have elucidated between mechanical and biomolecular cues in the microenvironment can be captured in our state diagram model of contact inhibition. Stiffening of the ECM shifts a non-transformed epithelial cell system leftward on the state diagram, bringing it closer to the transition line to contact independence (Fig. 5). This leftward shift captures the observation that ECM stiffening disrupts cell–cell contacts (Fig. 3) and reduces the EGF threshold (Fig. 2). Thus, graded changes in ECM stiffness promote progressive, quantitative loss in the degree of contact inhibition, even without an apparent phenotypic effect (i.e. cells remain in the contact-inhibited state). Furthermore, this model captures the observation that cell types differ in their sensitivity to ECM stiffness (supplementary material Fig. S3C). Our findings provide a quantitative framework for gauging the joint effect of soluble growth factors, cell–cell contacts and ECM compliance on cell proliferation, with implications for the regulation of tissue growth during morphogenesis and cancer progression.
Fig. 5.
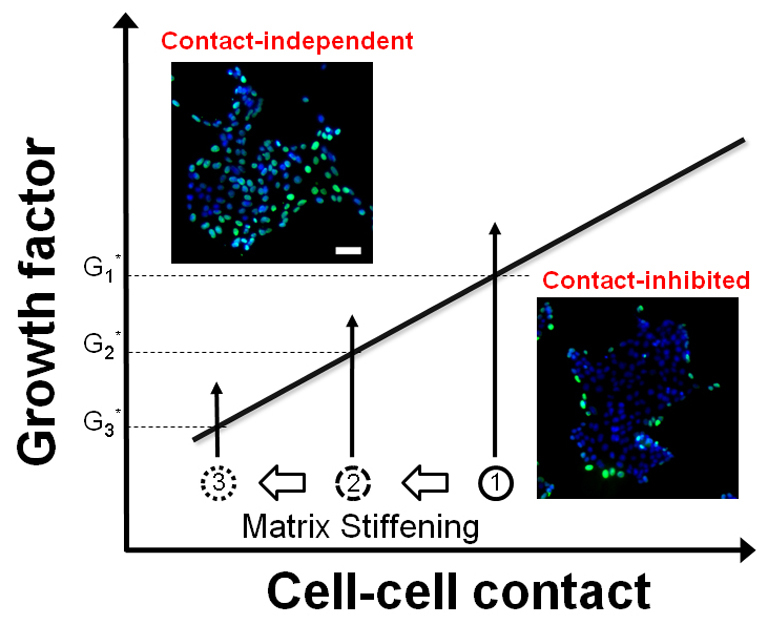
ECM stiffening promotes quantitative, progressive attenuation of contact inhibition of proliferation. The effect of matrix stiffening is depicted in a state diagram model of contact inhibition. The y-axis denotes the relative EGF concentration, and the x-axis conceptually denotes the level of cell-cell contact. This work would suggest that a useful metric of the extent of cell–cell contact might be the spatial enrichment of E-cadherin and ZO-1 at cell–cell contacts, but other possibilities include the level of E-cadherin expression or the area of cell–cell contact. Matrix stiffening quantitatively shifts normal cells closer to the transition line to contact-independence (1→2→3). Although such perturbations might not have a phenotypic effect (i.e. cells remain contact-inhibited), they have a quantitative, measurable effect on the threshold amount of EGF (G1*→G2*→G3*) needed to induce contact-independent growth. Insets show fluorescence images of BrdU (green) and DAPI (blue) stains of representative epithelial clusters in the contact-inhibited and contact-independent modes of proliferation. Scale bar: 50 μm.
The quantitative effect of substratum compliance on the EGF threshold involves the regulation of cell–cell contacts
The effect of substratum compliance on cell–matrix adhesions is well established. Substratum compliance affects traction forces that isolated cells and multicellular clusters generate on the underlying substratum and the size and content of integrin-mediated focal adhesions (Discher et al., 2005; Guo et al., 2006). Elevated traction forces and integrin-mediated signaling in rigid microenvironments promote proliferation (Klein et al., 2009; Paszek et al., 2005). Here, we show that the effect of matrix stiffening on proliferation has intriguing spatial features with consequences for patterning growth of multicellular structures. In a 2D multicellular cluster, matrix stiffness affects the effectiveness with which E-cadherin-mediated cell–cell contact inhibits proliferation of interior cells. Thus, matrix stiffness influences whether cell proliferation is confined to the periphery of cell clusters or occurs uniformly throughout the cluster. These effects of matrix stiffness on EGF-mediated proliferation involve the regulation of EGFR localization and downstream signaling pathways. EGFR is sequestered to mature cell–cell contacts in cell clusters grown on soft substrates. This sequestration corresponds to reduced EGFR internalization and the attenuation of ERK signaling, but not Akt signalling, among central cells in the cluster.
These spatial patterns in proliferation, EGFR localization and ERK signaling correspond to the effect of matrix stiffening on cell–cell contacts. Substratum stiffening disrupts cell–cell contacts by perturbing the localization of E-cadherin and ZO-1 at cell–cell contacts. Cells on stiff substrates exhibit distinct nuclear localization of ZO-1 that correlates with the uniform, contact-independent mode of proliferation. By contrast, on soft substrates, nuclear localization of ZO-1 was observed only in the peripheral cells of a cluster, correlating with the spatial pattern in proliferation. This modulation of nuclear localization of ZO-1 by substratum compliance might be mechanistically involved in cell cycle regulation. ZO-1 has been observed to shift from the nucleus to cell–cell junctions during the maturation of confluent MDCK monolayers (Gottardi et al., 1996), and this event sequesters a transcription factor, ZONAB, out of the nucleus, preventing it from transcribing genes required for cell cycle activity (Balda et al., 2003; Tsapara et al., 2006). The pronounced effect of matrix stiffness on the nuclear localization of ZO-1 seems to be specific because the nuclear localization of β-catenin, another intracellular protein involved in both cell–cell adhesion and the regulation of gene expression (Behrens et al., 1996; Molenaar et al., 1996), was not as profoundly affected by matrix stiffness (supplementary material Fig. S7). Taken together, our findings provide insight on the effect of matrix stiffness on cell–cell contacts and EGF signalling, and the emergent patterns of proliferation in multicellular epithelial structures.
Materials and Methods
Preparation and characterization of ligand-coated polyacrylamide substrates
Polyacrylamide substrates were prepared using techniques described by Wang and colleagues (Pelham and Wang, 1997). Substrate stiffness was manipulated by varying bis-acrylamide concentrations while keeping the acrylamide concentration constant (10%). Type I collagen (Sigma) and fibronectin (Sigma) were covalently bound to the substrates using a heterobifunctional cross-linker, sulfo-SANPAH (Pierce). The surface density of adhesion ligands on the substrates was quantified and confirmed to be equivalent among gels of varying stiffness as described in supplementary material Fig. S2. Finally, the Young's modulus of polyacrylamide substrates was measured by performing compression testing as described previously (Franck et al., 2007).
Cell culture and reagents
MDCK cells were cultured in DME medium containing HEPES and L-glutamine (Invitrogen) supplemented with 10% (v/v) fetal bovine serum (Invitrogen). MCF-10A cells were cultured in DME medium/Ham's F-12 (Invitrogen) containing HEPES and L-glutamine supplemented with 5% (v/v) horse serum (Invitrogen), 20 ng/ml EGF (Peprotech), 0.5 μg/ml hydrocortisone, 0.1 μg/ml cholera toxin, 10 μg/ml insulin (Sigma), and 1% penicillin-streptomycin (Invitrogen). For experiments, adhesion-ligand-coated polyacrylamide gels bound to 25 mm circular glass coverslips (VWR) were placed in 35 mm Petri dishes (Corning), and equilibrated in growth medium for 30 minutes at 37°C. Then, cells were plated in growth medium for 24 hours and serum starved for additional 24 hours. The following antibodies were used: anti-actin (Santa Cruz), anti-BrdU (Roche Applied Science), anti-E-cadherin (BD Transduction Laboratory), anti-EGFR, anti-phospho-Thr202/Tyr204-ERK 1/2, anti-phospho-serine 473-Akt (Cell Signaling Technology), anti-ZO1 (Zymed), DECMA-1 (Sigma), and Alexa dye-labeled secondary antibodies (Invitrogen). Nuclei were stained with DAPI (Sigma) and Hoechst 33342 (Invitrogen). The pharmacological inhibitors, PD98059 and LY294002, were purchased from Calbiochem.
RNAi
siRNA (50 nM) targeting E-cadherin and control siRNA were obtained from Ambion and transfected using Lipofectamine RNAiMax (Invitrogen). This siRNA sequence has been shown to reduce specifically the expression level of E-cadherin, but not other types of cadherins, in MDCK cells (den Elzen et al., 2009).
Immunofluorescence and image acquisition
For BrdU staining, cells were fixed in 70% ethanol supplemented with 15 mM glycine (pH 2) at −20°C, blocked with 10% goat serum and 0.1% BSA in PBS, and sequentially incubated with primary and secondary antibodies. For DECMA-1, E-cadherin, EGFR and ZO-1 staining, cells were fixed in freshly prepared, ice-cold 4% paraformaldehyde (pH 7.4), permeabilized with 0.2% Triton X-100 in PBS, and blocked with Image-iT FX Signal Enhancer (Invitrogen) and 10% goat serum and 0.1% BSA in PBS in series. Cells were then sequentially incubated with corresponding primary and secondary antibodies. To stain phosphorylated ERK and Akt, cells were fixed in freshly prepared, ice-cold 2% paraformaldehyde (pH 7.4), permeabilized with 0.5% NP-40 in PBS, and dehydrated with pure methanol at −20°C. Cells were blocked with Image-iT FX Signal Enhancer and a buffer solution containing 130 mM NaCl, 7 mM Na2HPO4, 3.5 mM NaH2PO4, 7.7 mM NaN3, 0.1% bovine serum albumin, 0.2% Triton X-100, 0.05% Tween-20 (all from Sigma) in series. Cells were then sequentially incubated with corresponding primary and secondary antibodies. For the staining of phospho-proteins, phosphatase inhibitors were added at fixation and permeabilization steps at 1 mM sodium orthovanadate, 10 mM sodium fluoride, and 10 mM β-glycerophosphate (all from Sigma). Finally, cells were co-stained with either DAPI or Hoechst 33342, and mounted using ProLong Gold Antifade (Invitrogen). Fluorescence and confocal images were acquired using the Zeiss Axiovert 200M microscope and the Zeiss LSM 510 upright confocal microscope, respectively. Identical laser power and gain were used for each fluorophore. The exposure time was chosen empirically for each field such that the highest pixel intensity in a given field is close to the saturation level of the detectors. This approach ensures that the data in each image uses the full dynamic range of the detectors with the caveat that intensity values may be compared within an image but not between images.
Quantification of immunofluorescence signals of phospho-proteins
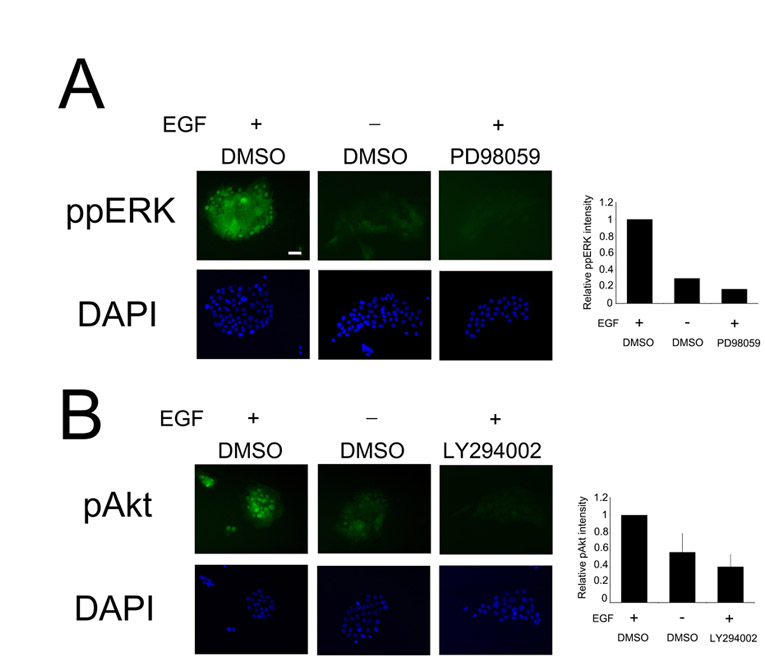
Nuclear phosphorylated ERK and Akt signal intensities were quantified by first tracing the perimeter of each nucleus, which were identified by the DAPI co-stain. The area and the total FITC intensity of each nucleus were determined using MATLAB. The average background level was multiplied by the area of the nucleus and was subtracted from the total nuclear FITC intensity to determine the final phosphorylated ERK and Akt levels for each nucleus. The stimulation of nuclear phosphorylated ERK and Akt signals by EGF and sensitivity to pharmacological inhibition of the MEK and PI3K pathways are described in supplementary material Fig. S8.
Cell lysis and western blot analysis
Cells were washed in PBS and lysed in modified RIPA buffer: 50 mM Tris-HCl (pH 7.5), 150 mM NaCl, 1% Triton X-100, 0.5% Nonidet P-40, 0.25% sodium deoxycholate, 50 mM β-glycerophosphate (pH 7.3), 10 mM NaPP, 30 mM NaF, 1 mM benzamidine, 2 mM EGTA, 1 mM sodium orthovanadate, 1 mM dithiothreitol, 5 μg/ml aprotinin, 5 μg/ml leupeptin, 1 μg/ml pepstatin and 1 mM phenylmethylsulfonyl fluoride. Cell lysate was centrifuged and the supernatant was collected as whole cell lysate. Whole cell lysates were resolved by SDS-PAGE on 7.5–10% gels and blotted onto nitrocellulose membranes (BioRad). The membranes were blocked in 3% (w/v) milk in TBST, and sequentially incubated with primary and HRP-conjugated secondary antibodies. Blots were treated with Supersignal West Femto substrate (Pierce) and images were obtained using the Versa-Doc 3000 imager (BioRad).
Statistical analyses
Student's t-test and analysis of variance (ANOVA), post-hoc tests were conducted using Microsoft Excel software and SPSS (PASW statistics 18) software, respectively.
Supplementary Material
Acknowledgments
We thank D. Shen for help with image analysis, the Caltech Biological Imaging Center for access to a confocal microscope, and J. Notbohm and G. Ravichandran for help in mechanical characterization of polyacrylamide gels. This work was supported by the NCI-USC Physical Sciences of Oncology Center (U54CA143907) and the Jacobs Foundation at Caltech. Deposited in PMC for release after 12 months.
Footnotes
Supplementary material available online at http://jcs.biologists.org/cgi/content/full/124/8/1280/DC1
References
- Assoian R. K. (1997). Anchorage-dependent cell cycle progression. J. Cell Biol. 136, 1-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balda M. S., Garrett M. D., Matter K. (2003). The ZO-1-associated Y-box factor ZONAB regulates epithelial cell proliferation and cell density. J. Cell Biol. 160, 423-432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balkovetz D. F., Pollack A. L., Mostov K. E. (1997). Hepatocyte growth factor alters the polarity of Madin-Darby canine kidney cell monolayers. J. Biol. Chem. 272, 3471-3477 [DOI] [PubMed] [Google Scholar]
- Behrens J., von Kries J. P., Kuhl M., Bruhn L., Wedlich D., Grosschedl R., Birchmeier W. (1996). Functional interaction of beta-catenin with the transcription factor LEF-1. Nature 382, 638-642 [DOI] [PubMed] [Google Scholar]
- Butcher D. T., Alliston T., Weaver V. M. (2009). A tense situation: forcing tumour progression. Nat. Rev. Cancer 9, 108-122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen R. H., Sarnecki C., Blenis J. (1992). Nuclear localization and regulation of erk- and rsk-encoded protein kinases. Mol. Cell. Biol. 12, 915-927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curto M., Cole B. K., Lallemand D., Liu C.-H., McClatchey A. I. (2007). Contact-dependent inhibition of EGFR signaling by Nf2/Merlin. J. Cell Biol. 177, 893-903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahmane A., Gil S., Brehier A., Davy J., Feger J. (1996). Kinetic analysis of epidermal growth factor endocytosis in rat hepatocytes. Effects of diabetes. Eur. J. Cell Biol. 69, 335-342 [PubMed] [Google Scholar]
- de Rooij J., Kerstens A., Danuser G., Schwartz M. A., Waterman-Storer C. M. (2005). Integrin-dependent actomyosin contraction regulates epithelial cell scattering. J. Cell Biol. 171, 153-164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- den Elzen N., Buttery C. V., Maddugoda M. P., Ren G., Yap A. S. (2009). Cadherin adhesion receptors orient the mitotic spindle during symmetric cell division in mammalian epithelia. Mol. Biol. Cell 20, 3740-3750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Discher D. E., Janmey P., Wang Y. L. (2005). Tissue cells feel and respond to the stiffness of their substrate. Science 310, 1139-1143 [DOI] [PubMed] [Google Scholar]
- Engler A. J., Sen S., Sweeney H. L., Discher D. E. (2006). Matrix elasticity directs stem cell lineage specification. Cell 126, 677-689 [DOI] [PubMed] [Google Scholar]
- Fogg V. C., Liu C. J., Margolis B. (2005). Multiple regions of Crumbs3 are required for tight junction formation in MCF10A cells. J. Cell Sci. 118, 2859-2869 [DOI] [PubMed] [Google Scholar]
- Franck C., Hong S., Maskarinec S. A., Tirrell D. A., Ravichandran G. (2007). Three-dimensional full-field measurements of large deformations in soft materials using confocal microscopy and digital volume correlation. Exp. Mech. 47, 427-438 [Google Scholar]
- French A. R., Tadaki D. K., Niyogi S. K., Lauffenburger D. A. (1995). Intracellular trafficking of epidermal growth factor family ligands is directly influenced by the pH sensitivity of the receptor/ligand interaction. J. Biol. Chem. 270, 4334-4340 [DOI] [PubMed] [Google Scholar]
- Gottardi C. J., Arpin M., Fanning A. S., Louvard D. (1996). The junction-associated protein, zonula occludens-1, localizes to the nucleus before the maturation and during the remodeling of cell-cell contacts. Proc. Natl. Acad. Sci. USA 93, 10779-10784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo W. H., Frey M. T., Burnham N. A., Wang Y. L. (2006). Substrate rigidity regulates the formation and maintenance of tissues. Biophys. J. 90, 2213-2220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamaratoglu F., Willecke M., Kango-Singh M., Nolo R., Hyun E., Tao C., Jafar-Nejad H., Halder G. (2006). The tumour-suppressor genes NF2/Merlin and Expanded act through Hippo signalling to regulate cell proliferation and apoptosis. Nat. Cell Biol. 8, 27-36 [DOI] [PubMed] [Google Scholar]
- Hanahan D., Weinberg R. A. (2000). The hallmarks of cancer. Cell 100, 57-70 [DOI] [PubMed] [Google Scholar]
- Kim J. H., Kushiro K., Graham N. A., Asthagiri A. R. (2009). Tunable interplay between epidermal growth factor and cell-cell contact governs the spatial dynamics of epithelial growth. Proc. Natl. Acad. Sci. USA 106, 11149-11153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein E. A., Yin L., Kothapalli D., Castagnino P., Byfield F. J., Xu T., Levental I., Hawthorne E., Janmey P. A., Assoian R. K. (2009). Cell-cycle control by physiological matrix elasticity and in vivo tissue stiffening. Curr. Biol. 19, 1511-1518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lampugnani M., Zanetti A., Corada M., Takahashi T., Balconi G., Breviario F., Orsenigo F., Cattelino A., Kemler R., Daniel T. O., et al. (2003). Contact inhibition of VEGF-induced proliferation requires vascular endothelial cadherin, beta-catenin, and the phosphatase DEP-1/CD148. J. Cell Biol. 161, 793-804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lampugnani M. G., Orsenigo F., Gagliani M. C., Tacchetti C., Dejana E. (2006). Vascular endothelial cadherin controls VEGFR-2 internalization and signaling from intracellular compartments. J. Cell Biol. 174, 593-604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J. W., Juliano R. (2004). Mitogenic signal transduction by integrin- and growth factor receptor-mediated pathways. Mol. Cells 17, 188-202 [PubMed] [Google Scholar]
- Lee S. B., Xuan Nguyen T. L., Choi J. W., Lee K. H., Cho S. W., Liu Z., Ye K., Bae S. S., Ahn J. Y. (2008). Nuclear Akt interacts with B23/NPM and protects it from proteolytic cleavage, enhancing cell survival. Proc. Natl. Acad. Sci. USA 105, 16584-16589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levental K. R., Yu H., Kass L., Lakins J. N., Egeblad M., Erler J. T., Fong S. F., Csiszar K., Giaccia A., Weninger W., et al. (2009). Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell 139, 891-906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meier R., Alessi D. R., Cron P., Andjelkovic M., Hemmings B. A. (1997). Mitogenic activation, phosphorylation, and nuclear translocation of protein kinase Bbeta. J. Biol. Chem. 272, 30491-30497 [DOI] [PubMed] [Google Scholar]
- Molenaar M., van de Wetering M., Oosterwegel M., Peterson-Maduro J., Godsave S., Korinek V., Roose J., Destree O., Clevers H. (1996). XTcf-3 transcription factor mediates beta-catenin-induced axis formation in Xenopus embryos. Cell 86, 391-399 [DOI] [PubMed] [Google Scholar]
- Nelson C. M., Jean R. P., Tan J. L., Liu W. F., Sniadecki N. J., Spector A. A., Chen C. S. (2005). Emergent patterns of growth controlled by multicellular form and mechanics. Proc. Natl. Acad. Sci. USA 102, 11594-11599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paszek M. J., Zahir N., Johnson K. R., Lakins J. N., Rozenberg G. I., Gefen A., Reinhart-King C. A., Margulies S. S., Dembo M., Boettiger D., et al. (2005). Tensional homeostasis and the malignant phenotype. Cancer Cell 8, 241-254 [DOI] [PubMed] [Google Scholar]
- Pelham R. J., Jr, Wang Y. (1997). Cell locomotion and focal adhesions are regulated by substrate flexibility. Proc. Natl. Acad. Sci. USA 94, 13661-13665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsapara A., Matter K., Balda M. S. (2006). The heat-shock protein Apg-2 binds to the tight junction protein ZO-1 and regulates transcriptional activity of ZONAB. Mol. Biol. Cell 17, 1322-1330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiseman B. S., Werb Z. (2002). Stromal effects on mammary gland development and breast cancer. Science 296, 1046-1049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin F., Pan D. (2007). Fat flies expanded the hippo pathway: a matter of size control. Sci. STKE 2007, pe12 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}