

Abstract
The search for the mechanism of action of improgan (a non-opioid analgesic) led to the recent discovery of CC12, a compound which blocks improgan antinociception. Since CC12 is a cytochrome P450 inhibitor, and brain P450 mechanisms were recently shown to be required in opioid analgesic signaling, pharmacological and transgenic studies were performed in rodents to test the hypothesis that improgan antinociception requires brain P450 epoxygenase activity. Intracerebroventricular (icv) administration of the P450 inhibitors miconazole and fluconazole, and the arachidonic acid (AA) epoxygenase inhibitor N-methylsulfonyl-6-(2-propargyloxyphenyl)hexanamide (MS-PPOH) potently inhibited improgan antinociception in rats at doses which were inactive alone. MW06-25, a new P450 inhibitor which combines chemical features of CC12 and miconazole, also potently blocked improgan antinociception. Although miconazole and CC12 were weakly active at opioid and histamine H3 receptors, MW06-25 showed no activity at these sites, yet retained potent P450-inhibiting properties. The P450 hypothesis was also tested in Cprlow mice, a viable knock-in model with dramatically reduced brain P450 activity. Improgan (145 nmol, icv) antinociception was reduced by 37-59% in Cprlow mice, as compared with control mice. Moreover, CC12 pretreatment (200 nmol, icv) abolished improgan action (70-91%) in control mice, but had no significant effect in Cprlow mice. Thus, improgan’s activation of bulbospinal non-opioid analgesic circuits requires brain P450 epoxygenase activity. A model is proposed in which 1) improgan activates an unknown receptor to trigger downstream P450 activity, and 2) brainstem epoxygenase activity is a point of convergence for opioid and non-opioid analgesic signaling.
Keywords: pain, analgesia, opioid, non-opioid, cytochrome P450, improgan
INTRODUCTION
Improgan, a congener of the H2 antagonist cimetidine (Fig. 1A), has the preclinical profile of an effective non-opioid analgesic following icv administration [13,21,27]. Like opioids and cannabinoids, improgan acts in the periaqueductal gray and rostral ventromedial medulla (RVM) to stimulate bulbospinal analgesic circuitry [15,27]. However, improgan antinociception is not mediated by known opioid [14] or histamine receptors [25]. Recent studies suggest a link between improgan and the brain cannabinoid system [9,26], but the drug lacks direct activity at known cannabinoid receptors. Despite screening at over 100 sites, the improgan analgesic target remains unknown [13].
Figure 1.

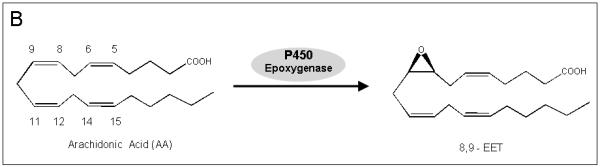
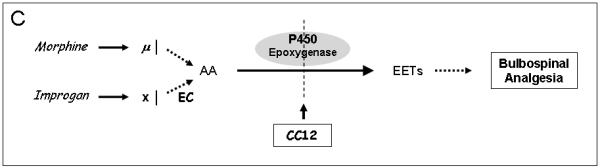
A). Chemical structures of improgan, cimetidine, four P450 inhibitors, and MS-PPOH, an epoxygenase inhibitor. Improgan, derived from the H2 antagonist cimetidine, is an imidazole-containing non-opioid analgesic. CC12, recently found to be a P450 inhibitor, blocks improgan antinociception. MW06-25, miconazole and fluconazole are also P450 inhibitors. MS-PPOH is a non-imidazole, fatty acid derivative which blocks P450-mediated epoxygenase activity. B) Synthesis of 8, 9 - EET from AA by a P450 epoxygenase pathway. Three other EET isomers (5,6-, 11,12-, and 14,15-, not shown) are also synthesized by various P450 epoxygenases [38]. C) Proposed epoxygenase transduction pathway for the analgesic actions of opioids and improgan in the brainstem. Recent results suggest that morphine stimulates bulbospinal analgesic circuits through opioid receptor activation of AA synthesis, AA epoxidation by P450s, and production of analgesic EETs [4]. CC12 and other P450/epoxygenase inhibitors (in A) block morphine antinociception by the proposed mechanism. The hypothesis that improgan acts on an unknown non-opioid receptor (X) and subsequently signals through an P450 epoxygenase mechanism is tested in the present study. Endocannabinoid (EC) release is suggested to participate in this pathway [26].
While searching for improgan’s pain-relieving mechanism, we discovered CC12 (Fig. 1A), a compound which blocks improgan antinociception [16]. Additional studies found unexpectedly that CC12 also blocked opioid and cannabinoid antinociception, suggesting an action on a common biochemical mechanism which is downstream from several analgesic receptors [16]. Recent results indicate that the CC12 target is likely to be a brain cytochrome P450 monooxygenase (P450, see below). Although P450s are best known for their roles in xenobiotic metabolism, these enzymes also participate in the synthesis and/or metabolism of many steroids, vitamins, and eicosanoids [29]. Relevant to the latter, some P450s have epoxygenase activity, i.e. they catalyze the conversion of arachidonic acid (AA) to epoxyeicosatrienoic acids (EETs, Fig. 1B, [38]). EETs have important biological functions in heart, kidney and brain [34,38,39,40].
Recently, our labs used pharmacological and transgenic approaches to show that morphine, the prototypical μ opioid analgesic, acts in the brainstem through a P450 epoxygenase signaling mechanism. In the study, Conroy et al. [4] developed a new transgenic mouse line (brain-Cpr-null) by incorporating brain neuron-specific deletion of the Cpr gene. Cpr encodes NADPH-cytochrome P450 reductase (CPR), an enzyme needed for electron transfer to microsomal P450 enzymes. As compared with control subjects, brain-Cpr-null mice showed loss of neuronal CPR, reduced brain P450 activity, and attenuated morphine antinociception [4]. Based on these and other findings, it was proposed that μ opioid receptors activate AA synthesis, AA epoxidation, and subsequent stimulation of bulbospinal analgesic circuits ([4] & Fig. 1C). Findings showing that AA can mimic opioid effects in vitro [44] and that the AA epoxidation products have antinociceptive properties in vivo [43] support this P450 mechanism. Since CC12 is a P450 inhibitor [41] which blocks morphine antinociception [16], the results of Conroy et al. [4] suggest that CC12’s anti-analgesic actions are mediated by inhibition of brain P450 activity (Fig. 1C). Although CC12 blocks improgan as well as morphine antinociception [16], improgan does not act through opioid mechanisms [14], and the mechanism by which CC12 blocks improgan’s activity has not been established. Presently, we describe two approaches to test the hypothesis that brain P450 epoxygenase activity is essential for improgan antinociception (Fig. 1C). In the first, the effects of P450 epoxygenase inhibitors were studied on improgan antinociception in rats. In the second, the effects of improgan and its antagonist CC12 were studied in Cprlow mice, a viable transgenic mouse model possessing highly attenuated P450 activity [47].
MATERIALS AND METHODS
Materials
Unless specified otherwise, all reagents and drugs were purchased from Sigma-Aldrich (St. Louis, MO). Improgan base [14] was dissolved in dilute HCl, neutralized to pH 5.5 to 6, and diluted with either buffer or saline. CC12 hydrochloride [16] and naltrexone hydrochloride were dissolved in saline. In the double-injection studies of Figs. 3-4, MW06-25 [4] was dissolved in 100% DMSO and diluted with saline to 67% DMSO. In all other cases, MW06-25, miconazole (MP Biochemicals; Solon, OH), ketoconazole, sulconazole, clotrimazole (MP Biomedicals, Santa Ana, CA), fluconazole (LKT Laboratories; St. Paul, MN) and MS-PPOH (Cayman Chemical; Ann Arbor, MI) were dissolved in 100% DMSO. Several labs (including ours) have used these concentrations of DMSO as a diluent for icv studies without adverse effects. Dibenzylfluorescein and cDNA-expressed human CYP2C19 were purchased from BD Biosciences (Woburn, MA). Acetonitrile (HPLC grade) and magnesium chloride hexahydrate were purchased from Fisher Scientific (Pittsburgh, PA).
Figure 3.

Time course for inhibition of improgan antinociception by P450 and epoxygenase blockers. Rats were baseline tested (BL), received icv inhibitor (dose in nmol in brackets) or DMSO vehicle, and were re-tested six min later (POST). A second icv injection of improgan (Imp, 250 nmol) or saline (Sal) was then given; the interval between the end of the first and the end of the second icv injection was 15 min. Subjects were re-tested at the times after the end of the second injection (abscissa, min). Ordinate shows TF response latencies (sec, mean ± SEM) for the number of subjects in parentheses. The same DMSO / Sal and DMSO/Imp groups are shown in both A and B. . **P< 0.01 vs DMSO/Sal; #, +P < 0.05, 0.01, respectively vs DMSO/Imp.
Figure 4.

Dose-response curves for inhibition of improgan antinociception by P450 and epoxygenase blockers. Rats received two icv injections exactly as described in Fig. 3. The first contained the specified dose (abscissa, nmol) of the designated inhibitor (A-E) or vehicle (Veh), followed by improgan (Imp, 250 nmol). The Veh/Imp group consisted of either DMSO/Imp or Saline/Imp treatments, which were found to not differ from each other, and were pooled. TF latencies (mean ± SEM for n values shown) are 10 min after the end of the second injection . #Data pooled from 0.1 and 0.3 nmol doses (B); &pooled from 1 and 3 nmol doses (D); *, ** P < 0.05, 0.01, respectively vs. Veh. Inhibitory dose-response curves were fitted for MW06-25, MS-PPOH and CC12 as shown. Portions of each of the inhibitory curves are re-drawn in F. IC50 values (see legends in F) were calculated as the icv dose (nmol) achieving a latency of 6.75 sec (dashed horizontal line), based on the Imp/Veh and baseline scores (9.5 and 4.0 secs, respectively).
Animals
Male, Sprague-Dawley rats (200 - 366 g at the time of testing; Taconic Farms, Germantown, NY) were maintained on a 12-h light/ dark cycle (lights on from 0700 to 1900) and provided with food and water ad lib. Rats were housed (3-4 / group) until the time of surgery and individually thereafter. Protocols for generating wild-type (C57BL/6J) and homozygous Cprlow/low (designated here as Cprlow) mice were reported previously [47]. Briefly, Cprlow mice were generated from breeding pairs of heterozygous (Cprlow/+) mice. Wild-type and Cprlow (males only for the present study, 20 – 36 g, at least 11 weeks old) were housed as described above in groups of 5 – 8 until surgery and individually thereafter. Genotyping was performed by PCR analysis of tail DNA [47]. All animal use protocols were approved the Institutional Animal Care and Use Committee of Albany Medical College.
Rodent surgery
Following anesthesia with pentobarbital sodium (supplemented with isofluorane), icv injection guide cannulas were stereotaxically implanted into the left lateral ventricle and anchored to the skull with three stainless steel screws and cranioplast cement [5]. For rats, the coordinates for the cannula (in mm from bregma, [33]) were: AP −0.8, ML +1.5, DV −3.3. For mice, the stereotaxic location of the ventricular cannula was AP −0.5, ML +1.0, DV −2.0 mm relative to bregma [32]. After surgery, subjects were individually housed with food and water available and were allowed to recover for at least 5 to 7 days before testing. Each animal was used for a single experiment.
Nociceptive testing
Rats were tested on the hot plate (HP) and tail flick (TF) tests. For the HP test [8], animals were placed on a 52° C surface and the latency to hind paw lift or lick was recorded with a maximal exposure of 60 s. Baseline latencies were between 10 and 15 s. For the TF test [7], the ventral surface of the tail (at a location randomly selected 2-5 cm from the tip) was exposed to radiant heat, and the latency for tail movement was recorded. The heat source was set so that baseline latencies were between 3 and 4 s with a 15-s cutoff. The heat source was not adjusted for individual animals. Mouse testing was performed with the hot water tail immersion test [21]. Subjects were restrained in a conical polypropylene tube. The tail (2–3 cm) was immersed into a 55°C water bath and the latency to sudden tail movement or removal of the tail from the water was recorded. Unless specified otherwise, cutoff latencies were 10 s.
Icv injections and testing
Rats were baseline tested with a single HP test, followed by three TF tests performed at one-min intervals, with the third test used as the baseline score. As indicated in figure legends, this was sometimes followed by a systemic drug injection. Subjects were then gently secured by wrapping with a laboratory pad, the stylet was removed, and the injection cannula inserted. This cannula extends 1 mm beyond the guide to penetrate the lateral ventricle. Unless noted otherwise, icv injections were performed manually over a five-min period in a volume of 5 μl. Due to limited solubility, the highest dose of ketoconazole only (500 nmol) was administered in 7.5 μl. One min after the end of the infusion, the injection cannula was clipped and sealed approximately 2 mm above the juncture with the guide cannula. Pharmacological antagonism studies were performed with two icv injections, consisting of baseline measurements and an icv injection (as above), followed by single HP and TF tests at specified intervals, followed by a second icv injection. The second injection cannula was clipped as described above. Single HP and TF latencies were subsequently recorded at specified intervals. Successful icv injections were assured by following the movement of an air bubble in the tubing between the syringe and the cannula and by the absence of leakage. The icv procedure in mice was nearly identical to the rat procedure, except that all drug solutions were injected over a one-min period in a total volume of 2 μl/injection in all cases. After testing, rats or mice received a large dose pentobarbital sodium and India Ink (5 or 2 μl, icv, respectively). Proper distribution of the ink in the cerebroventricular system indicated successful icv injections. Data from animals with poor placements or unsuccessful injections were excluded. The identities of the drug solutions, and the specific experimental group to which each subject had been assigned were always blinded to the investigators.
Opioid receptor assays
Opioid receptor binding assays were performed as previously described with minor modifications [31]. Membrane fractions (containing 50, 20, or 100 μg protein) were prepared from CHO cells that stably expressed either the human μ, δ or κ opioid receptor, respectively. Membranes were incubated in 1 ml of 50 mM Tris-HCl, pH 7.5, with 12 different concentrations of inhibitors, along with either [3H]DAMGO (0.25 nM, a μ-selective agonist), [3H]naltrindole (0.2 nM, a δ-selective antagonist), or [3H]U69,593, (1 nM, a κ-selective agonist). The final concentration of DMSO was less than 1%, which had negligible effects when tested alone. Incubation times of 60 min, 60 min, and 3 hr were used for measuring μ, κ, and δ binding, respectively. Samples were filtered through glass fiber filters, which had been soaked in 0.25% polyethylenimine for δ and κ binding. Filters were washed three times with 3-ml of cold Tris buffer and counted in 2 ml of Ecoscint A scintillation fluid. Data are presented as mean % inhibition (± SEM) of binding from three experiments performed in triplicate.
Histamine H3 receptor assay
An SK-N-MC cell line stably expressing the human H3 receptor was cultured and used for radioligand binding as described [22]. Cell homogenates were incubated for 40 min at 25°C with approximately 1 nM [3H]N-methylhistamine in 25 mM KPO4 buffer and 140 mM NaCl (pH 7.4 at 25°C), with or without competing ligands. Bound radioligands were collected on 0.3% polyethyleneimine-pretreated Whatman GF/C filters, washed three times with 3 ml of ice-cold washing buffer (4°C) containing 25mMTris-HCl and 140 mM NaCl (pH 7.4 at 4°C), and counted. Data are mean ± SEM from three experiments each performed in triplicate.
Enzyme Activity
Inhibition of CYP2C19 activity was determined as described [6]. Reaction mixtures containing NADPH-regenerating system (1.3 mM NADP+, 3.3 mM glucose-6-phosphate, 0.4 U/mL glucose-6-phosphate dehydrogenase, and 3.3 mM magnesium chloride) in 50 mM potassium phosphate buffer (pH 7.4) and inhibitors were preincubated at 37 °C for 10 min, followed by addition of 1 pmol of CYP2C19 and dibenzylfluorescein (0.5 μM final concentration), and subsequent incubation at 37 °C for 30 min (total volume = 200 μl in 1.0 % acetonitrile). The reaction was terminated by the addition of 75 μL of 2N sodium hydroxide, followed by a 2-h 37°C incubation. Fluorescence was measured with a Victor3 140 Multilabel Counter (Perkin Elmer, Waltham, MA; excitation: 485 nm [bandwidth= 14 nm]; emission:535 nm [bandwidth = 25 nm]). Leukocyte AA 12-lipoxygenase activity was assayed as recently described [4].
Data analysis and curve fitting
Nociceptive latencies were subjected to the specified analysis of variance followed by Fisher’s least signficant differences post-hoc testing (Statistica Software, Statsoft, Inc., Tulsa, OK,). All curve-fitting was performed with Prism 5.0 (GraphPad Software Inc., San Diego, CA). For binding studies, Ki values of unlabeled compounds were calculated from the equation Ki = (IC50)/1 + S, where S = (concentration of radioligand)/(Kd of radioligand) as described [2]. The Kd values for [3H]DAMGO, [3 H]U69,593, and [3H]naltrindole were 0.56, 0.34, and 0.10 nM, respectively. Ki values for CYP2C19 in Table 1 were calculated from IC50 values and used a Km of 0.54 μM. Inhibition of improgan antinociception was quantified from in vivo dose-response curves by non-linear regression of the dose-related portions of the data using Prism’s sigmoidal dose-response (variable slope) function. Dose-response curves for three compounds demonstrated both potentiation and inhibition of improgan antinociception. For these drugs, fits were performed by constraining Prism’s ‘TOP’ variable to 12.5, 13.0 and 13.5 sec for MS-PPOH, MW06-25, and CC12, respectively. Converged solutions yielded slopes in the range of 2 to 3, ‘BOTTOM’ values between 3 and 4 sec (i.e. baseline latencies), and ED50 values with 95% confidence intervals. Dose-response curves for the other two inhibitors (miconazole and fluconazole) did not follow the above pattern, did not provide an appropriate estimate for the “TOP” variable, and were therefore unsuitable for curve-fitting. Because of these results, IC50 values for all of the inhibitors were defined as the dose achieving a 50% reduction in improgan antinociception. Based on improgan-activated (control) and baseline latencies (i.e. 9.5 and 4.0 sec, respectively), IC50 values were thus calculated as the icv dose (nmol) achieving a mean latency of 6.75 sec in the presence of improgan. For the three drugs with fitted dose-response curves, IC50 values and confidence intervals were calculated from the fitted functions. IC50 values for the other two inhibitors were approximated by logarithmic interpolation.
Table 1.
Pharmacological activities of P450 and epoxygenase inhibitors
| Compound | Ki (nM ± SEM) | IC50 (95% C.I.) icv nmol |
|||||
|---|---|---|---|---|---|---|---|
| μ opioid a | δ opioid b | κ opioid c | H3d | CYP2C19 | Leukocyte 12-LOX e |
Est. Anti- Analgesicf |
|
| CC12 g | 7.1 × 103 | > 104 | ~ 104 | 22 | 19.9 | > 104 | 66.6 (43.8-89.4) |
| Miconazole | 1,400 ± 65 | 590 ± 29 | 760 ± 120 | 4,467 ± 447 | 23.7 g | > 104 | 0.2 |
| Sulconazole | 1,100 ± 36 | 690 ± 44 | 960 ± 51 | 31.0 | |||
| Clotrimazole | 96 ± 6.4 | 770 ± 25 | 260 ± 8.7 | 316 | |||
| Fluconazole | > 104 | > 104 | > 104 | > 104 | 1,050 g | 0.02 | |
| Ketoconazole | > 104 | > 104 | > 104 | 1,800 | |||
| MW06-25 g | > 104 | > 104 | > 104 | > 104 | 19.1 | > 104 h | 5.4 (2.1-8.7) |
| MS-PPOH | > 104 | > 104 | > 104 | 22.2 × 103 | > 104 g | 0.007 (0.002 – 0.012) |
|
Ki values for receptors were calculated from percent inhibition of binding from three experiments performed in triplicate. The solvent used for all receptor studies (1% DMSO) had no effect when tested alone.
[3H]DAMGO binding
[3H]Naltrindole binding
[3H]U69,593 binding
[3H]-Nα-methylhistamine binding
AA 12-lipoxygenase
In vivo IC50 values estimated by regression from Fig. 4 as described in Methods
Data from [4]
MW06-25 also had no effect (Ki > 10 μM) on cyclooxygenase-1, cyclooxygenase-2, and platelet 12-lipoxygenase [4].
RESULTS
Effects of inhibitors of P450 and epoxygenase in rats
In rats, large icv doses (250-500 nmol) of the anti-fungal P450 inhibitors miconazole, sulconazole and ketoconazole produced notable antinociception 7 – 11 min after administration on the TF (Fig. 2A) and HP (not shown) tests. These effects were reversible, as shown by a return to baseline latencies at the 31 min time point. ANOVA of Fig. 2A (between groups: drug; within groups [repeated measures]:time) found a significant (P<0.001) main effect of time and a significant (P<0.05) drug by time interaction term. Dose-response studies confirmed the antinociception produced by these three compounds (Fig. 2B). In studies with additional P450 inhibitors, clotrimazole (50 [n=3], 250 [n=4], and 500 nmol [n=8]), MW06-25 (50, 250, and 500 nmol, all n=3) and fluconazole (250 and 500 nmol, both n=3) had no significant antinociceptive effects on the TF or HP tests (data not shown). The lowest icv dose of any of the compounds tested (50 nmol, Fig. 2B) had no effects on nociceptive thresholds. Surprisingly, systemic pre-treatment with the opioid antagonist naltrexone completely blocked the antinociception produced by sulconazole, but not that produced by ketoconazole (TF: Fig. 2C, HP data not shown). The antagonism of miconazole antinociception by naltrexone seemed similar in magnitude to that seen with sulconazole, but the former effect did not achieve robust statistical significance (P = 0.058, Fig. 2C). ANOVA of data from Fig. 2C (between groups #1: icv drug; between groups #2: naltrexone) found significant main effects of icv drug (P<0.05) and naltrexone (P<0.01), and a significant (P<0.05) interaction term.
Figure 2.



Antinociceptive effects of imidazole-containing (“azole”) anti-fungal P450 inhibitors. Subjects were baseline (BL) tested, received an icv injection of drug (dose given in nmol) or vehicle, and were re-tested at the post-icv times shown (A, abscissa). Ordinate shows TF response latencies (sec, mean ± SEM) for the number of subjects given in parentheses. B) Dose-response curves are shown for TF latencies taken 11 min after injection. Abscissa is dose of drug (nmol, icv). Data from vehicle and 11 min groups in B are reproduced from A. C) Effects of naltrexone on azole-induced antinociception. Subjects were BL tested as in A, received naltrexone (5 mg/kg, i.p.) or saline, and were re-tested 15 min later. Nineteen min after i.p. injection, subjects received the icv treatment shown (abscissa, dose in nmol). Ordinate shows TF response latencies 11 min post-icv azole treatment for the number of subjects indicated in each group. Rats receiving icv DMSO (some of which also received ip saline) served as the Vehicle group in A-C. The azole treatment groups in A and B included data from respective ip saline-treated subjects shown in C. . *, **P< 0.05, 0.01 vs vehicle; ++P<0.01 vs. saline-sulconazole.
Because miconazole and sulconazole produced naltrexone-sensitive antinociception, several of the azole anti-fungal compounds were screened in vitro for activity at opioid and other sites (Table 1). Surprisingly, miconazole and sulconazole showed weak to moderate activity on the μ, δ, and κ opioid receptors (Ki values of 0.6 – 1.4 μM). Clotrimazole was even more potent than these drugs at the μ receptor, with a Ki value of 96 nM (Table 1).
Since it was presently hypothesized that these anti-fungal P450 inhibitors should block antinociception (instead of produce antinociception, as in Fig. 2), doses of these drugs much lower than those shown in Fig. 2 were tested as inhibitors of improgan antinociception (Fig. 3). Icv miconazole (10 nmol), MW06-25 (10 nmol), and fluconazole (100 nmol) all caused a nearly-complete antagonism of improgan antinociception (TF: Fig. 3, HP data not shown). The epoxygenase inhibitor MS-PPOH (a non-imidazole fatty acid derivative, Fig. 1A) was also tested as a modulator of improgan antinociception. At the 30 nmol dose (icv), this drug produced virtually complete attenuation of improgan antinociception (TF: Fig. 3B, HP data not shown). ANOVA of all of the improgan-treated groups of Fig. 3A and 3B (between groups: inhibitors; within groups [repeated measures]: time) found a significant main effect of time (P<0.001) and a significant (P<0.02) drug by time interaction term. None of these inhibitors had significant effects on either nociceptive test when administered alone at the doses specified in Fig. 3 (n=3, data not shown). In addition, no behavioral or motor abnormalities were observed in subjects receiving these doses of drugs.
Detailed dose-response studies with these P450 blockers (Fig. 4) confirmed potent inhibition of improgan antinociception in all cases, but additional complexities were observed. Although CC12 and its congener MW06-25 produced dose-dependent inhibition of improgan, both compounds significantly enhanced antinociception at slightly lower doses. MW06-25 was approximately ten-fold more potent than CC12 on both the inhibitory and stimulatory effects (Fig. 4). The MS-PPOH dose-response curve showed a similar pattern, but revealed an extraordinary potency, with doses from 0.01 to 250 nmol producing virtually complete inhibition (Fig. 4). The lowest doses of MS-PPOH tended to enhance improgan antinociception, but the trend did not reach statistical significance. The miconazole curve likewise revealed a range of inhibitory doses (0.3 – 10 nmol). Unlike these other compounds, however, no miconazole-induced potentiation of improgan was observed (Fig. 4). Fluconazole gave the most complex dose-response curve. Doses between 10 and 500 nmol resembled the stimulatory and inhibitory actions seen by CC12, yet much lower doses of this drug (0.1 to 3 nmol) produced consistent, complete inhibition of improgan responses (Fig. 4). Separate ANOVAs of the effects of each inhibitor in Fig. 4 (between groups: dose of drug; within groups [repeated measures]:time) found highly significant (P<0.01) dose by time interaction terms for all five inhibitors. Estimates of inhibitory IC50 values for these compounds ranged from 7 pmol to 67 nmol (Fig. 4F). The rank order of anti-analgesic potencies of these drugs was MS-PPOH > fluconazole > miconazole > MW06-25 > CC12.
Effects of improgan and CC12 in Cprlow mice
In wild-type mice, improgan (145 nmol, icv) produced significant, near-maximal antinociception 10 - 30 min after administration (Figs. 5C-E). CC12 pretreatment (200 nmol, icv) had no effect alone (‘Post’, Fig. 5B), but significantly reduced improgan actions by 91%, 70% and 70% at the 10, 20 and 30 min test periods, respectively (Fig. 5C-E). In Cprlow mice not treated with CC12, average improgan antinociception was decreased by 59%, 37% and 41% as compared with control mice at the three respective post-test periods (Fig. 5C-E). Post-hoc testing confirmed the genotype difference in improgan responses at the ten min interval (Fig. 5C). In further contrast, CC12 pretreatment had no significant effects on improgan antinociception in Cprlow mice at any of the time points (Figs. 5C-E). ANOVA of the data of Fig. 5A-E (between groups #1: genotype; between groups #2: drug treatments; within groups [repeated measures]:time) found significant (P<0.001) main effects of drug treatment and time, and significant (P<0.05) genotype by drug, drug by time, and genotype by drug by time interaction terms. The results from this experiment were subjected to additional data analysis by computing area-under-the curve calculations (i.e. sums of 10-30 min latency scores) for each subject (Fig. 5F). This analysis confirmed: a) robust improgan antinociception and its antagonism by CC12 in control mice, b) significantly reduced improgan antinociception in Cprlow (vs. wild-type) mice, and c) no antagonism of improgan responses by CC12 in Cprlow mice (Fig. 5F). ANOVA of the AUC data of Fig. 5F (between groups #1: genotype; between groups #2: drug treatments) a found significant (P<0.001) main effect of drug treatment and a significant (P<0.05) genotype by drug interaction term.
Figure 5.

Improgan antinociception and its modulation by CC12 in wild-type (WT) and Cprlow mice. Subjects were baseline tested (A), and received CC12 (200 nmol, or pH 4.5 saline vehicle, Veh, icv). Ten min after the end of infusion, they were re-tested (Post, B), received improgan (Imp, 145 nmol, icv) or saline (Veh), and were re-tested 10 (C), 20 (D) and 30 (E) min after the end of the second injection. For each subject, the combined latency scores at 10, 20, and 30 min are shown as areas under the curve (F). Nociceptive tail immersion latencies (sec, ordinate, mean ± SEM for the number of subjects specified in bars in (F) are shown for each treatment group. *,**P<0.05, 0.01, respectively vs Veh/Veh at the same time in the same genotype; ++P<0.01, vs Veh/Imp at the same time in the same genotype; #P<0.05 vs WT Veh/Imp at the same time.
DISCUSSION
A role for P450s in antinociceptive drug action (Fig. 1C) requires that P450 inhibitors behave as antagonists of analgesic drugs. However, the presently-observed antinociceptive effects of large doses of the P450 blockers miconazole, sulconazole and ketoconazole (Fig. 2) are the opposite of this hypothesis. Earlier studies described the antinociceptive properties of the P450 inhibitor proadifen [12,20], but the antinociceptive properties of other P450 inhibitors have not been previously reported. Although these drugs are used clinically, they do not penetrate the blood-brain barrier [10]; this barrier was circumvented presently by icv drug administration. Despite the extensive literature on miconazole-like P450 inhibitors (Suppl. Table 1), the affinities of these drugs for some receptors (Table 1) have not been previously reported. The ability of naltrexone to antagonize azole-induced antinociception (Fig. 2C, observed earlier with proadifen [12]) may be relevant to the opioid receptor affinities for some of these drugs (Table 1), but this requires further study.
In contrast to the results with high doses of P450 inhibitors (250 – 500 nmol, Fig. 2), the prediction that these drugs should inhibit improgan antinociception was verified with lower doses (, Figs. 3, 4). Miconazole is a potent, versatile inhibitor of many P450 isoforms (Suppl. Table 1), and this drug has been used to show biological roles for P450 epoxygenases [46,48]. However, since miconazole has many non-P450 actions (Suppl. Table 1), the inhibition of improgan antinociception by the more selective inhibitor fluconazole ([49] & Suppl. Table 1) considerably strengthens the P450 hypothesis, and may help to identify the analgesia-relevant P450 isoforms.
The finding that miconazole behaved as an improgan antagonist with a 300-fold greater potency than that of CC12 (Fig. 4, Table 1) prompted the design of MW06-25, a CC12 isomer more closely resembling the structure of miconazole. MW06-25 has an N-substituted side chain, a feature found in miconazole, but not in CC12 (Fig. 1). Like CC12 and miconazole, MW06-25 is a potent P450 inhibitor (Table 1). Unlike these other drugs, however, MW06-25 lacks affinity for the H3 and other receptors (Table 1), thereby demonstrating an enhanced P450 selectivity. The potent antagonism of improgan antinociception by MW06-25 (Fig. 4) also supports the P450 hypothesis.
Many P450s (e.g. CYP4A2/3 [45], CYP4X1 [42], and members of the 2B, 2C, and 2J families [18,35]) can convert AA to EETs (Fig. 1B). But P450s can also metabolize EETs [50], and can hydroxylate AA to several hydroxyeicosatetraenoic acids [50]. All of these reactions can be inhibited by the heme-binding, miconazole-like P450 inhibitors, and all should be impaired in the Cprlow mice. To further distinguish epoxygenase vs non-epoxygenase P450 mechanisms, we used MS-PPOH (Fig. 1A), a selective P450 epoxygenase blocker [1,34,36,45,48]. In vivo brain studies with MS-PPOH are limited [36], but the extraordinarily potent inhibition of improgan antinociception by this compound (Fig. 4) strongly implicates a brain epoxygenase pathway in improgan action. The weak activity of MS-PPOH on our prototype P450 (CYP2C19, Table 1) confirms that the relevant P450 epoxygenase has not been found.
Since P450s are associated with drug metabolism, the inhibition of drug action in vivo by a P450 inhibitor can mean that drug metabolism is required to produce an active metabolite. A well known example is codeine, which is activated by conversion to morphine by hepatic CYP2D [3]. Although brain P450s can metabolize drugs [23,24], the presently-observed inhibition of improgan antinociception by P450 blockers is not likely to be due to a drug-drug metabolic interaction for the following reasons: 1) All drugs were administered directly into the brain, circumventing hepatic metabolism. 2) The rapid onset and short duration of antinociceptive activity following icv improgan (onset within 5 – 10 min, rapid decline thereafter, Fig. 3) is not commensurate with the expected time course of brain metabolic activation. The activity of a brain-injected pro-drug would not be immediate, but instead should develop gradually over 30 to 60 min (e.g. see [24]). 3) The P450 blocker CC12 not only inhibits the antinociceptive activity of improgan, but also the effects of morphine (which is metabolized in the rat by glucuronidation, not by P450 oxidation [19]), and the effects of a cannabinoid [16]. Thus, the presently-observed effects of P450 inhibitors on improgan action are most likely to represent a pharmacodynamic (vs pharmacokinetic) drug – drug interaction (Fig. 1C).
The inhibitory dose-response curves for the P450 epoxygenase blockers on improgan antinociception are complicated by biphasic (and even triphasic) actions of some of these drugs (Fig. 4). If, as suggested by these curves, different brain P450s play opposing roles in pain modulation, then a particular spectrum of action of a single inhibitor on these enzymes could produce a specfic pattern of biphasic effects. This ‘dual P450’ idea seems consistent with the hypothesis that EETs mediate improgan antinociception (Fig. 1C), since P450s can perform both EET biosynthesis and EET metabolism [50]. Alternatively, off-target actions could account for the improgan-enhancing effects of these drugs, since many P450 inhibitors have side effects on ion channels and other enzymes (Suppl. Table 1). The biphasic actions of the more selective agents MS-PPOH and fluconazole may not support this possibility. Further studies with these compounds (e.g. in P450-deficient animal models) will help to clarify these issues.
Genetic approaches to the study of P450 in mammals have been hampered by the large number of P450 genes (over 120 in the mouse [28]). In contrast, CPR, an electron provider required for microsomal P450 and heme oxygenase function, is encoded by a single gene [37]. However, germ-line, whole-body inactivation of Cpr produced 100% embryonic lethality [30,37], preventing assessment of P450 functions in adult knockouts. Wu and colleagues [47] developed a transgenic mouse model (Cprlow) which surmounts these obstacles. In these mice, embryonic insertion of the neo gene into Cpr led to widespread reductions in CPR and P450 activities, with limited embryonic lethality and a 90% reduction in brain CPR activity [47]. As predicted, improgan antinociception was significantly reduced in Cprlow as compared with wild-type mice (Fig. 5). The finding that improgan responses were reduced but not eliminated in Cprlow mice may be due to the fact that P450 activity is reduced, but not eliminated in this mouse model; a small amount of Cpr expression is retained in these animals [47]. The robust antagonism of improgan by CC12 in control mice, and the virtual elimination of this effect in Cprlow mice show that CC12’s anti-analgesic activity in normal subjects depends on brain P450 activity. The fact that Cprlow mice not only have a deficiency in P450 activity, but also in heme oxygenase activity (another CPR-requiring enzyme [47]), suggests an alternative, non-P450 explanation for the present results. However, heme oxygenase is not likely to account for changes in improgan or CC12 effects in these mice, since miconazole and fluconazole (which block improgan antinociception) do not inhibit the brain form of heme oxygenase (Suppl. Table 1).
The presently-established P450 epoxygenase requirement for improgan antinociception provides significant progress toward understanding this drug’s mechanism of action. The present model (Fig. 1C) suggests that improgan activates an unknown site that is distinct from known opioid, cannabinoid or histamine receptors [9,14,25,26]. Endocannabinoid release is proposed to follow receptor activation, yet precede a P450-mediated step. Improgan-evoked release of endocannabinoids has not been directly shown, but the model is based on 1) blockade of improgan antinociception by the CB1 antagonist rimonabant [9], 2) lack of rimonabant-sensitive improgan responses in CB1-deficient mice [17], 3) reduced improgan antinociception in cannabinoid-tolerant mice [26], 4) lack of improgan affinity for known cannabinoid receptors [9], and 5) inhibition of cannabinoid antinociception by the P450 inhibitor CC12 [16]. In addition, recent in vivo studies of RVM neuronal activity found that CC12 reverses some (but not all) of improgan’s actions [11]. These results support the hypothesis that a P450-based mechanism follows activation of the improgan receptor. The alternative hypothesis (that CC12 is an improgan receptor blocker) predicts that CC12 should block all improgan actions, but none of the effects of opioids or cannabinoids; this is not what was found. Many elements of the proposed model (Fig. 1C) remain untested.
The present findings also broaden the significance of the recently-discovered P450 epoxygenase mechanism for opioid analgesia [4]. For example, dose-response curves with P450 inhibitors showed both antinociceptive (Fig. 2) as well as anti-analgesic (Fig. 4) actions The former could lead to novel analgesic drug development. In addition, two kinds of P450-deficient mutant mice (Cprlow, Fig. 5, and brain-Cpr- null, [4] ) have now been shown to lack normal responses to analgesic drugs (improgan and morphine, respectively). The present studies are also the first to show that the anti-analgesic activity of P450 inhibitors in control subjects is not observable in P450-deficient mice (Fig. 5). Finally, taken with other recent results, the present findings show that both non-opioid (improgan) and opioid (morphine) analgesic drugs use P450 epoxygenase mechanisms, suggesting a point of convergence for these two kinds of signaling (Fig. 1C). Further studies are needed to investigate this hypothesis, and to identify the relevant P450 isoforms, their cellular localization(s), and the products of their activity.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by U.S. National Institutes of Health grants DA03816 (L.H.), ES07462 (X.D.), and DA00360 (J.M.B.). We thank Dr. Jeff Carlson for valuable discussions regarding statistical analyses, and Erik Istre for technical assistance. The authors declare no competing financial interests.
Non-standard abbreviations
- AA
arachidonic acid
- CPR
NADPH-cytochrome P450 reductase
- EET
epoxyeicosatrienoic acid
- HP
hot plate
- icv
intracerebroventricular
- MS-PPOH
N-methylsulfonyl-6-(2-propargyloxyphenyl)hexanamide
- P450
cytochrome P-450 monooxygenase
- RVM
rostral ventromedial medulla
- TF
tail flick
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- [1].Bhardwaj A, Northington FJ, Carhuapoma JR, Falck JR, Harder DR, Traystman RJ, Koehler RC. P-450 epoxygenase and NO synthase inhibitors reduce cerebral blood flow response to N-methyl-D-aspartate. Am J Physiol Heart Circ Physiol. 2000;279:H1616–H1624. doi: 10.1152/ajpheart.2000.279.4.H1616. [DOI] [PubMed] [Google Scholar]
- [2].Cheng Y, Prusoff WH. Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- [3].Cleary J, Mikus G, Somogyi A, Bochner F. The influence of pharmacogenetics on opioid analgesia: studies with codeine and oxycodone in the Sprague-Dawley/Dark Agouti rat model. J Pharmacol Exp Ther. 1994;271:1528–1534. [PubMed] [Google Scholar]
- [4].Conroy JL, Fang C, Gu J, Zeitlin SO, Yang W, VanAlstine MA, Nalwalk JW, Albrecht PJ, Mazurkiewicz JE, Snyder-Keller A, Shan Z, Zhang S, Wentland MP, Behr M, Knapp BI, Bidlack JM, Zuiderveld OP, Leurs R, Ding X, Hough LB. Opioids activate brain analgesic circuits through cytochrome P450/epoxygenase signaling. Nature Neuroscience. 2010;13:284–286. doi: 10.1038/nn.2497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Crane LA, Glick SD. Simple cannula for repeated intracerebral drug administration in rats. Pharmacol Biochem Behav. 1979;10:799–800. doi: 10.1016/0091-3057(79)90336-8. [DOI] [PubMed] [Google Scholar]
- [6].Crespi CL, Miller VP, Penman BW. Microtiter plate assays for inhibition of human, drug-metabolizing cytochromes P450. Anal Biochem. 1997;248:188–190. doi: 10.1006/abio.1997.2145. [DOI] [PubMed] [Google Scholar]
- [7].D’Amour FE, Smith DL. A method for determining loss of pain sensation. J Pharmacol Exp Ther. 1941;72:74–79. [Google Scholar]
- [8].Eddy NB, Leimbach D. Synthetic analgesics, II Dithienylbutenyl and dithienylbutylamines. J Pharmacol Exp Ther. 1953;107:385–393. [PubMed] [Google Scholar]
- [9].Gehani NC, Nalwalk JW, Razdan RK, Martin BR, Sun X, Wentland M, Abood ME, Hough LB. Significance of cannabinoid CB(1) receptors in improgan antinociception. J Pain. 2007;8:850–860. doi: 10.1016/j.jpain.2007.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Graybill JR, Craven PC. Antifungal agents used in systemic mycoses. Activity and therapeutic use. Drugs. 1983;25:41–62. doi: 10.2165/00003495-198325010-00003. [DOI] [PubMed] [Google Scholar]
- [11].Heinricher MM, Maire JJ, Lee D, Nalwalk JW, Hough LB. Physiological basis for inhibition of morphine and improgan antinociception by CC12, a P450 epoxygenase inhibitor. J.Neurophysiol. 2010 doi: 10.1152/jn.00681.2010. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Ho TK, LaBella FS, Pinsky C. Opiate properties of SKF 525A. Can J Physiol Pharmacol. 1978;56:550–554. doi: 10.1139/y78-088. old 21. [DOI] [PubMed] [Google Scholar]
- [13].Hough LB. Improgan-like analgesics: a family of compounds derived From histamine antagonists. Med Chem Res. 2004;13:78–87. [Google Scholar]
- [14].Hough LB, Nalwalk JW, Chen Y, Schuller A, Zhu Y, Zhang J, Menge WMPB, Leurs R, Timmerman H, Pintar JE. Improgan, a cimetidine analog, induces morphine-like antinociception in opioid receptor-knockout mice. Brain Res. 2000;880:102–108. doi: 10.1016/s0006-8993(00)02776-1. [DOI] [PubMed] [Google Scholar]
- [15].Hough LB, Nalwalk JW, Menge WM, Leurs R, Timmerman H. Significance of GABAergic systems in the action of improgan, a non-opioid analgesic. Life Sci. 2001;68:2751–2757. doi: 10.1016/s0024-3205(01)01080-3. [DOI] [PubMed] [Google Scholar]
- [16].Hough LB, Nalwalk JW, Phillips JG, Kern B, Shan Z, Wentland MP, Iwan Esch JP, Janssen E, Barr T, Stadel R. CC12, a high-affinity ligand for [3H]cimetidine binding, is an improgan antagonist. Neuropharmacology. 2007;52:1244–1255. doi: 10.1016/j.neuropharm.2007.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Hough LB, Nalwalk JW, Stadel R, Timmerman H, Leurs R, Paria BC, Wang X, Dey SK. Inhibition of improgan antinociception by the cannabinoid (CB)(1) antagonist N-(piperidin-1-yl)-5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-p yrazole-3-carboxamide (SR141716A): lack of obligatory role for endocannabinoids acting at CB(1) receptors. J Pharmacol Exp Ther. 2002;303:314–322. doi: 10.1124/jpet.102.036251. [DOI] [PubMed] [Google Scholar]
- [18].Iliff JJ, Jia J, Nelson J, Goyagi T, Klaus J, Alkayed NJ. Epoxyeicosanoid signaling in CNS function and disease. Prostaglandins Other Lipid Mediat. 2009 doi: 10.1016/j.prostaglandins.2009.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Kuo CK, Hanioka N, Hoshikawa Y, Oguri K, Yoshimura H. Species difference of site-selective glucuronidation of morphine. J Pharmacobiodyn. 1991;14:187–193. doi: 10.1248/bpb1978.14.187. [DOI] [PubMed] [Google Scholar]
- [20].Lehman T, Peterson GR. Naloxone-reversible analgesic action of SKF 525-A in mice. Psychopharmacology (Berl) 1978;59:305–308. doi: 10.1007/BF00426639. [DOI] [PubMed] [Google Scholar]
- [21].Li BY, Nalwalk JW, Finkel JM, Glick SD, Hough LB. SKF92374, a cimetidine analog, produces mechanical and thermal antinociception in the absence of motor impairment. Analgesia. 1997;3:15–20. [Google Scholar]
- [22].Lim HD, van Rijn RM, Ling P, Bakker RA, Thurmond RL, Leurs R. Evaluation of histamine H1-, H2-, and H3-receptor ligands at the human histamine H4 receptor: identification of 4-methylhistamine as the first potent and selective H4 receptor agonist. J Pharmacol Exp Ther. 2005;314:1310–1321. doi: 10.1124/jpet.105.087965. [DOI] [PubMed] [Google Scholar]
- [23].Miksys S, Tyndale RF. The unique regulation of brain cytochrome P450 2 (CYP2) family enzymes by drugs and genetics. Drug Metab Rev. 2004;36:313–333. doi: 10.1081/dmr-120034149. [DOI] [PubMed] [Google Scholar]
- [24].Miksys S, Tyndale RF. Brain drug-metabolizing cytochrome P450 enzymes are active in vivo, demonstrated by mechanism-based enzyme inhibition. Neuropsychopharmacology. 2009;34:634–640. doi: 10.1038/npp.2008.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Mobarakeh JI, Nalwalk JW, Watanabe T, Sakurada S, Hoffman M, Leurs R, Timmerman H, Silos-Santiago I, Yanai K, Hough LB. Improgan antinociception does not require neuronal histamine or histamine receptors. Brain Res. 2003;974:146–152. doi: 10.1016/s0006-8993(03)02572-1. [DOI] [PubMed] [Google Scholar]
- [26].Nalwalk JW, Svokos K, Hough LB. Cannabinoid-improgan cross-tolerance: Improgan is a cannabinomimetic analgesic lacking affinity at the cannabinoid CB(1) receptor. Eur J Pharmacol. 2006;549:79–83. doi: 10.1016/j.ejphar.2006.08.035. [DOI] [PubMed] [Google Scholar]
- [27].Nalwalk JW, Svokos K, Taraschenko O, Leurs R, Timmerman H, Hough LB. Activation of brain stem nuclei by improgan, a non-opioid analgesic. Brain Res. 2004;1021:248–255. doi: 10.1016/j.brainres.2004.06.066. [DOI] [PubMed] [Google Scholar]
- [28].Nelson DR, Zeldin DC, Hoffman SM, Maltais LJ, Wain HM, Nebert DW. Comparison of cytochrome P450 (CYP) genes from the mouse and human genomes, including nomenclature recommendations for genes, pseudogenes and alternative-splice variants. Pharmacogenetics. 2004;14:1–18. doi: 10.1097/00008571-200401000-00001. [DOI] [PubMed] [Google Scholar]
- [29].Ortiz de Montellano PR. Cytochrome P450: Structure, Mechanism, and Biochemistry. Kluwer Academic/Plenum Publishers; New York: 2005. [Google Scholar]
- [30].Otto DM, Henderson CJ, Carrie D, Davey M, Gundersen TE, Blomhoff R, Adams RH, Tickle C, Wolf CR. Identification of novel roles of the cytochrome P450 system in early embryogenesis: effects on vasculogenesis and retinoic acid homeostasis. Mol Cell Biol. 2003;23:6103–6116. doi: 10.1128/MCB.23.17.6103-6116.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Parkhill AL, Bidlack JM. Several delta-opioid receptor ligands display no subtype selectivity to the human delta-opioid receptor. Eur J Pharmacol. 2002;451:257–264. doi: 10.1016/s0014-2999(02)02241-0. [DOI] [PubMed] [Google Scholar]
- [32].Paxinos G, Franklin KBJ. The Mouse Brain in Stereotaxic Coordinates. Academic Press; San Diego: 2001. [Google Scholar]
- [33].Paxinos G, Watson C. The Rat Brain in Stereotaxic Coordinates. Academic Press; Sydney: 1998. [Google Scholar]
- [34].Peng X, Carhuapoma JR, Bhardwaj A, Alkayed NJ, Falck JR, Harder DR, Traystman RJ, Koehler RC. Suppression of cortical functional hyperemia to vibrissal stimulation in the rat by epoxygenase inhibitors. Am J Physiol Heart Circ Physiol. 2002;283:H2029–H2037. doi: 10.1152/ajpheart.01130.2000. [DOI] [PubMed] [Google Scholar]
- [35].Roman RJ. P-450 metabolites of arachidonic acid in the control of cardiovascular function. Physiol Rev. 2002;82:131–185. doi: 10.1152/physrev.00021.2001. [DOI] [PubMed] [Google Scholar]
- [36].Sellers KW, Sun C, Diez-Freire C, Waki H, Morisseau C, Falck JR, Hammock BD, Paton JF, Raizada MK. Novel mechanism of brain soluble epoxide hydrolase-mediated blood pressure regulation in the spontaneously hypertensive rat. FASEB J. 2005;19:626–628. doi: 10.1096/fj.04-3128fje. [DOI] [PubMed] [Google Scholar]
- [37].Shen AL, O’Leary KA, Kasper CB. Association of multiple developmental defects and embryonic lethality with loss of microsomal NADPH-cytochrome P450 oxidoreductase. J Biol Chem. 2002;277:6536–6541. doi: 10.1074/jbc.M111408200. [DOI] [PubMed] [Google Scholar]
- [38].Spector AA. Arachidonic acid cytochrome P450 epoxygenase pathway. J Lipid Res. 2009;50(Suppl):S52–S56. doi: 10.1194/jlr.R800038-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Spector AA, Norris AW. Action of epoxyeicosatrienoic acids on cellular function. Am J Physiol Cell Physiol. 2007;292:C996–1012. doi: 10.1152/ajpcell.00402.2006. [DOI] [PubMed] [Google Scholar]
- [40].Spiecker M, Liao JK. Vascular protective effects of cytochrome p450 epoxygenase-derived eicosanoids. Arch Biochem Biophys. 2005;433:413–420. doi: 10.1016/j.abb.2004.10.009. [DOI] [PubMed] [Google Scholar]
- [41].Stadel R, Yang J, Nalwalk JW, Phillips JG, Hough LB. High affinity binding of 3[H]-cimetidine to a heme-containing protein in rat brain. Drug Metab Dispos. 2008 Jan 31;36:614–621. doi: 10.1124/dmd.107.017889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Stark K, Dostalek M, Guengerich FP. Expression and purification of orphan cytochrome P450 4X1 and oxidation of anandamide. FEBS J. 2008;275:3706–3717. doi: 10.1111/j.1742-4658.2008.06518.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Terashvili M, Tseng LF, Wu HE, Narayanan J, Hart LM, Falck JR, Pratt PF, Harder DR. Antinociception produced by 14,15-epoxyeicosatrienoic acid is mediated by the activation of {beta}-endorphin and Met-enkephalin in the rat ventrolateral periaqueductal gray. J.Pharmacol.Exp.Ther. 2008;326:614–619. doi: 10.1124/jpet.108.136739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Vaughan CW, Ingram SL, Connor MA, Christie MJ. How opioids inhibit GABA-mediated neurotransmission. Nature. 1997;390:611–614. doi: 10.1038/37610. [DOI] [PubMed] [Google Scholar]
- [45].Wang MH, Brand-Schieber E, Zand BA, Nguyen X, Falck JR, Balu N, Schwartzman ML. Cytochrome P450-derived arachidonic acid metabolism in the rat kidney: characterization of selective inhibitors. J Pharmacol Exp Ther. 1998;284:966–973. [PubMed] [Google Scholar]
- [46].Watanabe H, Vriens J, Prenen J, Droogmans G, Voets T, Nilius B. Anandamide and arachidonic acid use epoxyeicosatrienoic acids to activate TRPV4 channels. Nature. 2003;424:434–438. doi: 10.1038/nature01807. [DOI] [PubMed] [Google Scholar]
- [47].Wu L, Gu J, Cui H, Zhang QY, Behr M, Fang C, Weng Y, Kluetzman K, Swiatek PJ, Yang W, Kaminsky L, Ding X. Transgenic mice with a hypomorphic NADPH-cytochrome P450 reductase gene: effects on development, reproduction, and microsomal cytochrome P450. J Pharmacol Exp Ther. 2005;312:35–43. doi: 10.1124/jpet.104.073353. [DOI] [PubMed] [Google Scholar]
- [48].Yamaura K, Gebremedhin D, Zhang C, Narayanan J, Hoefert K, Jacobs ER, Koehler RC, Harder DR. Contribution of epoxyeicosatrienoic acids to the hypoxia-induced activation of Ca2+ -activated K+ channel current in cultured rat hippocampal astrocytes. Neurosci. 2006;143:703–716. doi: 10.1016/j.neuroscience.2006.08.021. [DOI] [PubMed] [Google Scholar]
- [49].Yokoyama C, Shinjo F, Yoshimoto T, Yamamoto S, Oates JA, Brash AR. Arachidonate 12-lipoxygenase purified from porcine leukocytes by immunoaffinity chromatography and its reactivity with hydroperoxyeicosatetraenoic acids. J Biol Chem. 1986;261:16714–16721. [PubMed] [Google Scholar]
- [50].Zeldin DC. Epoxygenase pathways of arachidonic acid metabolism. J Biol Chem. 2001;276:36059–36062. doi: 10.1074/jbc.R100030200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.