

Abstract
Spontaneous neurotransmitter release has been considered synaptic noise for decades. Recent work, however, suggests that synaptic events arising from action potential (AP)-independent neurotransmitter release contribute to synaptic function in a manner qualitatively different from AP-dependent release. We investigated the effect of somatic depolarization on spontaneous neurotransmitter release in neurons of the external portion (EX) of the chicken inferior colliculus. We found this manipulation affects spontaneous but not AP-dependent release onto EX neurons in a retrograde manner. This depolarization-induced suppression of spontaneous release requires activation of NMDA receptors on the presynaptic cell and postsynaptic calcium-dependent vesicle fusion. These results show a novel form of plasticity that allows the postsynaptic cell to selectively modulate spontaneous neurotransmitter release of its afferent inputs.
Introduction
Neurotransmitter release may be evoked by the invasion of the presynaptic terminal by an action potential (AP-dependent) or occur spontaneously (AP-independent). Compared with those arising from AP-dependent release, synaptic events produced by the spontaneous fusion of synaptic vesicles at the presynaptic terminal have smaller amplitude and are thus called “miniature events,” or “minis.” The idea that minis occur stochastically and presumably lack the strength and reliability of AP-dependent release led to the labeling of minis as synaptic noise of no physiological significance. However, work over the past decade has challenged this concept. Minis can modulate firing rate (Carter and Regehr, 2002) and also stabilize synaptic function at excitatory synapses (Frank et al., 2006; Sutton et al., 2006). Because of growing evidence that minis play a role in synaptic function, how their probability of occurrence is modulated is a relevant issue.
The presynaptic modulation of neurotransmission by messengers generated in the postsynaptic neuron is a widespread phenomenon (Regehr et al., 2009). Although originally restricted to gases (Brenman and Bredt, 1997; Hölscher, 1997; Boehning and Snyder, 2003), peptides (Wagner et al., 1991; Drake et al., 1994; Qian et al., 1997; Yung et al., 2001; Li et al., 2002; Acuna-Goycolea and van den Pol, 2004, 2009; Gompf et al., 2005; Kozoriz et al., 2006; Blomeley and Bracci, 2008), and molecules derived from lipid precursors (Feinmark et al., 2003; Chevaleyre et al., 2006; Sang and Chen, 2006), the list of retrograde messengers has been recently expanded to include conventional neurotransmitters such as GABA (Zilberter et al., 1999; Magnusson et al., 2008) and glutamate (Zilberter, 2000; Duguid and Smart, 2004). Here we report a novel form of synaptic plasticity induced by retrograde activation of non-postsynaptic, presumably presynaptic, NMDA receptors (NMDARs) in the external portion (EX) of the avian inferior colliculus, where endocannabinoid (eCB)-dependent long-term depression (LTD) has been described (Penzo and Peña, 2009). This novel phenomenon consists of a reduction in the frequency of minis triggered by postsynaptic depolarization, which lasts for several minutes and does not affect evoked synaptic responses.
Materials and Methods
Brain slices.
Midbrain slices were prepared from White Leghorn late chicken embryos (embryonic day 17–19) of both sexes (Charles River). Sections (350 μm thick) were made parallel to the dorsoventral axis of the optic lobe. The tissue was then incubated for 1 h at 38°C in artificial CSF (aCSF) containing the following (in mm): 139 NaCl, 2.9 KCl, 1 MgSO4, 3.14 CaCl2, 17 NaHCO3, and 12.2 d-glucose, bubbled with 95% O2 and 5% CO2, pH 7.4.
Electrophysiology.
Whole-cell patch-clamp recordings of EX neurons were obtained using standard procedures with a MultiClamp 700B amplifier (Molecular Devices). Recordings were made in the same aCSF solution as the incubation, at 37°C (Temperature Controller; Warner Instrument), delivered to the perfusion chamber at a rate of 2.5 ml per minute. The recording pipette solution contained the following (in mm): 110 K gluconate, 10 HEPES, 10 NaCl, 10 phosphocreatine (dipotassium salt), 0.2 EGTA, 5 ATP (dimagnesium salt), 0.4 GTP (trisodium salt), and 10 glucose, pH 7.2. Pipette resistance ranged from 3 to 4 MΩ.
Neurons were recorded in voltage-clamp configuration with the membrane potential held at −60 mV. Synaptic currents were evoked using monopolar stimulation of afferent inputs with a patch-type electrode of 10–20 μm tip size filled with aCSF. The stimulating electrode was placed in the central shell portion (CSh) of the inferior colliculus. Electrical stimulation consisted of paired-pulses delivered to CSh every 5 s to measure changes in paired-pulse ratio (PPR). PPR was determined for each trial and then averaged over 12 traces. Each individual pulse had an amplitude of 50–200 μA and a duration of 0.1 ms with an interpulse interval of 40 ms. Currents were low-pass filtered at 2 kHz and acquired with a 5 kHz sampling rate using Igor Pro software (WaveMetrics). The series resistance (5–20 MΩ) and the cell's input resistance were monitored throughout recordings with 100 ms test pulses of −10 mV at the beginning of each trace.
Plasticity of spontaneous release was induced by a membrane-potential step from −60 to 0 mV for 5 s (1 and 3 s steps were used in a subset of experiments), after obtaining a stable baseline of miniature EPSC (mEPSC) frequency (4–5 min). In a subset of experiments, plasticity was induced by a 5 s train of 10 ms current pulses at 5 Hz delivered in current-clamp mode.
All experiments were performed with picrotoxin (100 μm) in the bath to block GABAA receptors. TTX (1 μm) was added to the bath to isolate mEPSCs. D-APV (50 μm) was used to block NMDAR. Intracellular dialysis with autocamtide-2-related inhibitory peptide (AIP; 1 μm) and bath application of KN-93 (1 μm) were used to block CaMKII. KN-92 was used a control for KN-93. WIN55212–2 (5 μm) and AM-251 (2 or 5 μm) were used to activate and block CB1Rs, respectively. Intracellular perfusion with MK-801 (1 mm) was used to block postsynaptic NMDARs. To sequester intracellular postsynaptic calcium, the concentration of K gluconate was lowered to 80 mm and replaced with BAPTA (10 mm) in the pipette solution. Nifedipine (10 μm) was used to block L-type voltage-gated calcium channels. Evans blue (5 μm) was used to block vesicular glutamate transporters (VGLUTs). NMDA (5–30 μm) was used to activate NMDARs. Two dynamin inhibitory peptides [p4 (20 μm) and D15 (1.5 mm)] were used to prevent dynamin-dependent vesicle endocytosis. aCSF chemicals were obtained from Sigma-Aldrich. All drugs were obtained from Tocris Bioscience except for KN-92, which was bought from EMD Chemicals.
The inhibitory interfering peptide of SNARE-dependent exocytosis made from the C-terminal domain of SNAP-25 with a point mutation of Ser 187 to Ala (SNAP-25, 10 μm) and its scrambled version were from Anaspec and kindly provided by Dr. Alberto Pereda (Albert Einstein College of Medicine, Bronx, NY).
Statistical analysis.
The statistical significance of changes was assessed with the paired t test (Systat Software). Plasticity of synaptic responses was quantified by comparing the last three data points of the baseline recording with the last three data points of each recording, except for testing the effect of NMDA, in which comparisons were made between the three points preceding and following the start of NMDA application. Cumulative probabilities were compared with the Kolmogorov–Smirnov test.
Results
Depolarization-induced suppression of spontaneous release
We performed whole-cell voltage-clamp recordings of EX neurons in the presence of 100 μm picrotoxin. TTX (1 μm) was added to the aCSF to isolate mEPSCs (minis). Evoked EPSCs (eEPSCs) were recorded by stimulating afferent inputs to EX neurons in CSh. For studying evoked responses, electrical stimulation was delivered every 5 s and 12 traces were recorded per minute (see Materials and Methods). For mEPSC recordings, 5 s of continuous recording were acquired every 6 s. To induce plasticity, we stepped the membrane potential of recorded neurons from −60 to 0 mV for 5 s. Although this manipulation had no effect on either the amplitude (n = 8; 102.0 ± 4.9% of control; p > 0.1) (Fig. 1A) of eEPSCs or the PPR [n = 8; 0.87 ± 0.03 and 0.87 ± 0.02 (before and after, respectively); p > 0.1] (Fig. 1B), we observed a slow reduction in the frequency (n = 5; 59.5 ± 4.4% of control; p < 0.001) (Fig. 1C,D) but not amplitude (n = 5; 95.0 ± 2.8% of control; p > 0.1) (Fig. 1C,D), of mEPSCs. Cumulative plots for mEPSC amplitude and frequency before and after the depolarization-induced suppression of spontaneous release (DSSR) induction are also shown (Fig. 1E–H). Shorter depolarizations also elicited DSSR, although the resulting suppression recovered faster (Fig. 2A) (n = 6 for 1 s depolarization and n = 5 for 3 s depolarization; 100.7 ± 8.2% and 77.4 ± 5.2% of control, respectively; p > 0.1 and p < 0.01, respectively). DSSR was monitored for 8 min in the majority of the experiments. However, longer recordings were conducted in a subset of experiments, which showed that DSSR could still be observed after 15 min (n = 5; 52.8 ± 4.3% of control; p < 0.001) (Fig. 2B). In addition, the postsynaptic depolarization that induced DSSR also triggered a reduction in the frequency of spontaneous excitatory synaptic currents (sEPSCs) recorded in the absence of TTX (n = 6; 65. 4 ± 3.8% of control; p < 0.001) (Fig. 2C). To test whether more physiological forms of postsynaptic depolarization also modulate spontaneous release, we measured the effect of spike trains. Spiking of the postsynaptic cell evoked by 10 ms current steps recorded in current-clamp mode (5 Hz for 5 s) also reduced the frequency of sEPSCs, in a manner similar to postsynaptic depolarization (n = 6; 54.8. 4 ± 3.0% of control; p < 0.001) (Fig. 2C). These findings indicate that suppression of spontaneous release can be triggered by physiologically relevant stimuli.
Figure 1.
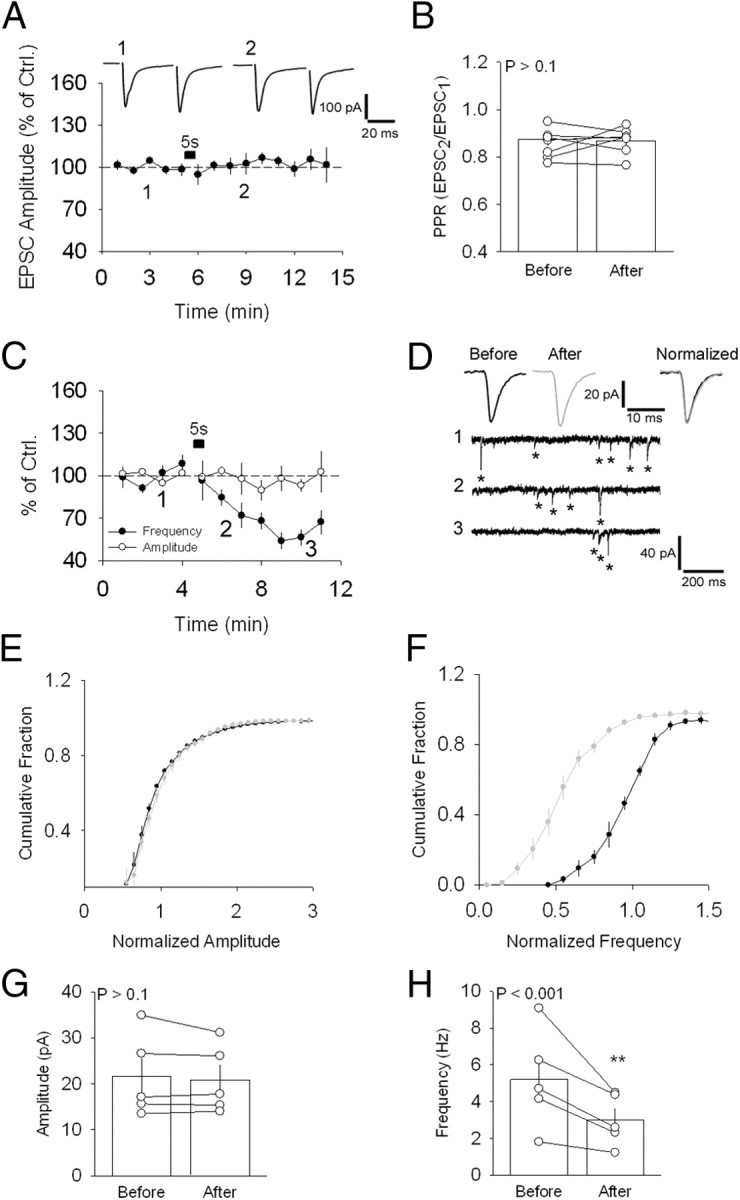
Depolarization-induced suppression of spontaneous release. A, Absence of effect of a 5 s depolarizing step (−60–0 mV) on evoked synaptic responses at the CSh–EX synapse. Sample traces are shown above. B, PPR values for the neurons shown in A. C, Average effect of the same depolarizing step on mEPSC frequency and amplitude in the presence of TTX. D, Sample traces showing a reduction in frequency of mEPSCs before (1) and following (2, 3) the current step. Sample average mEPSCs are shown above, separately (left and middle), and normalized and superimposed (right). Asterisks indicate the mEPSCs detected by the analysis software. E, Average cumulative distribution of the amplitude of mEPSCs recorded before (black circles) and after (gray circles) DSSR induction for the neurons in C. F, Same as in E for mEPSC frequency. G, Average mEPSC amplitude for the neurons in C before and after DSSR induction. H, Same as G for mEPSC frequency. **p < 0.01. Error bars indicate SEM and example traces are averages of 30–40 singles traces. Ctrl., Control.
Figure 2.

Characteristic of DSSR. A, Average effect of 1 s (white circles), 3 s (gray circles), and 5 s (black circles) (reproduced from Fig. 1C) depolarizating steps on mEPSC frequency. B, Average plot showing that DSSR is still observed 15 min after induction. B, Stimulation of postsynaptic cells by either somatic depolarization (depol) or action potentials suppresses sEPSC frequency similar to DSSR. Sample spikes used to trigger suppression of sEPSC frequency are shown in C. D, Postsynaptic cell loading with the dynamin inhibitory peptides p4 (black circles) and dominant-negative peptide composed of 15 amino acids (D15; gray circles) does not prevent DSSR. E, Intracellular dialysis with BAPTA (black circles) and bath application of nifedipine (white circles) prevented DSSR. F, Averaged data plot showing the effect of AM-251 on DSSR. Error bars indicate SEM. Ctrl., Control; Stim, stimulation.
The frequency of mEPSC depends both on the probability of release (P) and on the number of active release sites (N). A reduction in the frequency of mEPSC could thus be due to a decrease in either P or N. To confirm that a change in P is responsible for DSSR, we tested whether postsynaptic silencing of synaptic sites due to removal of AMPA receptors, which would affect N, could account for the observed reduction in mEPSC frequency. A DSSR-inducing protocol was applied to neurons loaded with a dynamin inhibitory peptide p4 (20 μm), which prevents endocytosis and thus, receptor internalization. p4 did not prevent DSSR (n = 5; 67.8 ± 4.5% of control; p < 0.001) (Fig. 2D) and had no significant effect in mEPSC amplitude (n = 5; 107.8 ± 2.3% of control; p > 0.05) (Fig. 2D). In addition, DSSR was not affected by loading of EX cells with a dominant-negative peptide composed of 15 amino acids, which disrupts the interaction between dynamin and amphiphysin (n = 4; 63.4 ± 5.3 of control; p < 0.001) (Fig. 2D). Together with the absence of effect on evoked responses of postsynaptic depolarizations, these results suggest that DSSR is mediated by a reduction in release probability of spontaneous transmission and is not a consequence of synaptic silencing.
CaMKII-dependent retrograde activation of putative presynaptic NMDARs mediates DSSR
We next investigated the molecular mechanism responsible for DSSR induction. DSSR was prevented by loading the postsynaptic cell with the calcium chelator BAPTA (10 mm) and by bath application of the L-type channel blocker nifedipine (10 μm) (n = 4 for each condition; 95.8 ± 5.4% of control for BAPTA and 115.3 ± 9.3% of control for nifedipine; p > 0.1 for both) (Fig. 2E). In addition, this phenomenon was independent of CB1R activation, as it was unaffected by bath application of the CB1R antagonist AM-251 (n = 6; 68.6 ± 5.3% of control; p < 0.001) (Fig. 2F), suggesting that a retrograde messenger other than eCBs was responsible for DSSR.
To determine whether NMDAR activation was required for DSSR, voltage steps were performed in the presence of either bath-applied D-APV (50 μm) or intracellular dialysis with MK-801 (iMK-801; 1 mm). D-APV (n = 5; 105.9 ± 2.6% of control; p > 0.1) (Fig. 3A,B), but not iMK-801 (n = 6; 62.4 ± 3.9% of control; p < 0.001) (Fig. 3A,B), prevented DSSR. These results are consistent with the existence of presynaptic NMDARs in EX, as previously reported (Penzo and Pena, 2009). Activation of presynaptic NMDARs requires the presence of glutamate. We hypothesize that depolarization of the postsynaptic cell induces glutamate release that acts retrogradely on these NMDARs. To test this hypothesis, we used Evans blue, a potent inhibitor of VGLUT, dialyzed through the patch pipette. Evans blue (5 μm) prevented DSSR induction (n = 5; 100.5 ± 7.2% of control; p > 0.1) (Fig. 3C,D), suggesting that glutamate released by the postsynaptic cell is required for DSSR. We next investigated whether, consistent with the hypothesis of a retrograde glutamatergic effect, DSSR requires postsynaptic vesicle exocytosis. To test this, we used a short inhibitory peptide of SNARE-dependent exocytosis, made from the C-terminal domain of SNAP-25. In blind experiments, we found that DSSR was abolished when the SNAP-25 inhibitory peptide (n = 5; 95.9 ± 3.9% of control; p > 0.1) (Fig. 4A,B) was loaded into the postsynaptic cell and not in control experiments with a scrambled peptide (n = 4; 61.7 ± 5.4% of control; p < 0.001) (Fig. 4A,B). These results suggest that DSSR is induced by SNARE-dependent retrograde transmission through putative presynaptic NMDARs.
Figure 3.

Activation of non-postsynaptic NMDARs is required for DSSR. A, Bath application of D-APV (black circles) but not iMK-801 (white circles) prevents DSSR. B, Sample traces for A, before (1) and after (2) somatic depolarization. C, DSSR blocking by Evans blue (black circles). D, Sample traces for C. Asterisks indicate the mEPSCs detected by the analysis software. Error bars indicate SEM and example traces are averages of 30–40 singles traces. Ctrl., Control.
Figure 4.

Postsynaptic vesicle fusion and CaMKII are required for DSSR. A, SNAP-25 interfering peptide (black circles) and absence of this effect with a scramble peptide (white circles). B, Sample traces for A. C, Summary plots showing the effects of two CaMKII inhibitors (Inhb.) on DSSR: bath-applied KN-93 (black circles) and intracellular dialysis with AIP (white circles). D, Sample traces for C. Asterisks indicate the mEPSCs detected by the analysis software. Error bars indicate SEM and example traces are averages of 30–40 singles traces. Ctrl., Control.
CaMKII is associated to synaptic vesicles (Ouimet et al., 1984) and phosphorylates both synapsin I and SNARE proteins in vitro (Wang, 2008). Because EX neurons are rich in CaMKII (Penzo and Pena, 2009) and DSSR is SNARE-dependent, we tested whether CaMKII could be involved in DSSR. In separate experiments, bath application of the CaMKII antagonist KN-93 (1 μm; n = 6; 97.6 ± 3.8% of control; p > 0.1) (Fig. 4C,D) and intracellular dialysis with AIP (1 μm; n = 6; 110.8 ± 2.7% of control; p > 0.1) (Fig. 4C,D), which also inhibits CaMKII, prevented DSSR. KN-92, the inactive version of KN-93, was used as control (n = 5; 51.8 ± 3.7% of control; p < 0.001) (Fig. 4C,D). These results indicate that CaMKII is a postsynaptic mediator of DSSR.
NMDARs control spontaneous neurotransmitter release
An assumption made earlier is that the non-postsynaptic NMDARs required for DSSR are presynaptic. To test this assumption, we compared the frequency and amplitude of mEPSCs recorded in the presence and absence of the NMDAR antagonist D-APV. Events recorded from seven neurons had an average amplitude of 36.0 ± 7.2 pA and an average frequency of 4.0 ± 1.1 Hz (Fig. 5B). Following a stable baseline (4–6 min), bath application of D-APV significantly reduced the frequency (2.7 ± 0.6 Hz; p < 0.05) but not the amplitude (38.1 ± 8.9 pA; p > 0.1) of spontaneous events (Fig. 5B). This result is consistent with the involvement of presynaptic NMDARs. Similar effects of NMDARs in neurotransmitter release have been reported by others (for review, see Corlew et al., 2008).
Figure 5.

NMDAR-dependent control of release. A, Sample traces showing mEPSCs recordings in the presence of TTX before and after bath application of D-APV for two neurons (top). Each trace represents 2 s of a 6 s sweep recording. Bottom, Time course of D-APV-induced suppression of mEPSC frequency. B, Bar plots show that D-APV induces a significant decrease in the frequency (right) but not the amplitude (left) of mEPSCs. Sample average mEPSCs are shown above, separately (left and middle). Normalized and superimposed traces are shown on the right. White circles represent each data point and bars show averages and SEM. *p < 0.05 (statistically significant).
As described above, blocking NMDARs with D-APV prevents DSSR. However, D-APV application alone reduces the frequency of mEPSCs. Although the reduction in mEPSC frequency following D-APV application is smaller than DSSR (32% and 41%, respectively), it is possible that the absence of DSSR in slices treated with D-APV results from occlusion due to an already suppressed transmission caused by the D-APV. An alternative explanation is that NMDARs are tonically activated by ambient glutamate, thereby enhancing neurotransmitter release in basal conditions. Blocking these tonically active receptors would thus reduce spontaneous release. To determine whether this was the case, we investigated the effect of NMDAR activation on mEPSC frequency. We tested the effect of 5 and 30 μm NMDA on spontaneous release. Bath application of 5 μm NMDA induced a significant increase in the frequency of mEPSC (n = 4; 145.2 ± 11.9% of control; p < 0.01) (Fig. 6A,B). This increase in mEPSC frequency was transient and not blocked by loading the postsynaptic cell with MK-801 (1 mm; n = 4; 124.3 ± 10.2% of control; p < 0.05) (Fig. 6B). Conversely, bath perfusion with 30 μm NMDA induced a decrease in mEPSC frequency (n = 5; 69.5 ± 4.7% of control; p < 0.001) (Fig. 6C,D). This decrease in mEPSC frequency persisted after NMDA washout and was not prevented by iMK-801 (n = 4; 75.9 ± 5.8% of control; p < 0.01) (Fig. 6D). Together, these results suggest that the NMDAR exerts a dynamic control on neurotransmitter release. Whereas application of low dose of the NMDA transiently facilitates release, a higher dose produces a persistent suppression. We conclude that the reduction in mEPSC frequency following D-APV application is likely due to a suppression of the facilitating effect of NMDAR activation under basal conditions, whereas DSSR is likely to be a plasticity triggered by increased activation of NMDARs on the presynaptic cell.
Figure 6.

Bidirectional effect of NMDA on spontaneous neurotransmitter release. A, Enhancement in mEPSC frequency following bath application of 5 μm NMDA in a single cell. B, Summary plots of the effect of 5 μm NMDA on mEPSC frequency in control cells (black circles) and in cells with iMK-801 (white circles). C, Sample plot showing a decrease in mEPSC frequency following bath application of 30 μm NMDA in a single cell. D, Summary plots of the effect of 30 μm NMDA on mEPSC frequency in control cells (black circles) and in iMK-801 (white circles). Error bars indicate SEM. Ctrl., Control.
Suppression of evoked neurotransmission by coincident postsynaptic depolarization and CB1R activation
Modulation of spontaneous release is cannabinoid-independent and mediated by a retrograde signal that does not affect evoked (AP-dependent) neurotransmission. In light of previous work showing that LTD of evoked transmission at the CSh–EX synapse requires both CB1R and presynaptic NMDAR activation (Penzo and Peña, 2009), we reasoned that activation of presynaptic NMDARs alone by DSSR-inducing stimuli might not be sufficient to suppress evoked transmission. If this was the case, then coupling postsynaptic depolarization to pharmacological activation of CB1R should be sufficient to induce long-lasting suppression of evoked neurotransmission at this synapse. To test this prediction, we bath applied the CB1R agonist WIN, which reduces the amplitude of eEPSCs and is reversible by CB1R antagonists (Penzo and Peña, 2009). Pairing bath application of WIN and postsynaptic depolarization prevented recovery of eEPSC amplitude following washout of WIN with the CB1R antagonist AM-251, thus converting an otherwise transient depression into an LTD (n = 6; 80.9 ± 1.5% of control; p < 0.001) (Fig. 7A) This suppression was expressed as a long-term increase in PPR, consistent with a decrease in neurotransmitter release probability (n = 5; p < 0.01) (Fig. 5B). In control experiments, D-APV was applied 1 min prior to and following the depolarization to test whether NMDARs are required for this depolarization-induced suppression of AP-dependent release. As predicted, 50 μm D-APV prevented the effect of depolarization on the WIN-mediated reduction of the EPSC amplitude (n = 5; 96.5 ± 3.0% of control; p > 0.1) (Fig. 7A,B). These findings are consistent with coincident presynaptic NMDAR and CB1R activation being necessary to trigger LTD (Sjöström et al., 2003; Bender et al., 2006; Penzo and Peña, 2009) and demonstrate that somatic depolarization can trigger the release of a retrograde messenger that leads to the activation of presynaptic NMDAR.
Figure 7.

Coincident CB1R activation and postsynaptic depolarization trigger long-lasting suppression of evoked responses. A, Pooled data show the effect of bath application of the CB1R agonist WIN coupled to somatic depolarization. Following a WIN-induced 15% depression of EPSCs' amplitude, EX neurons were transiently depolarized (as in A). After 11 min of WIN application, AM-251 was applied for the remainder of the experiment (black circles). LTD triggered by coincident CB1R activation and postsynaptic depolarization was prevented by bath application of D-APV (white circles). B, PPR values for neurons in A. White circles represent each data point and bar graphs show their average and SEM. **p < 0.01. Ctrl., Control; Depol., depolarization.
Input selectivity of DSSR
The data presented so far suggest that DSSR is triggered by a mechanism that selectively affects spontaneous transmission, leaving responses evoked by electrical stimulation unchanged. Our hypothesis is that both types of release take place at the same synapses. An alternative explanation of our results, however, is that the inputs producing the spontaneous responses are different from those activated by electrical stimulation. Although this problem is very difficult to resolve in our preparation, we conducted experiments to address the question.
We have found two major inputs to EX neurons: one arising from the CSh and another from the deep layers of the optic tectum (OT). Although exact homology has not been demonstrated, this type of connectivity resembles that found in barn owls, where a feedback from OT to the auditory midbrain plays a role in experience-dependent plasticity (Knudsen, 2002). We could elicit eEPSCs by placing an additional stimulating electrode in the deep layers of OT and in the tectotegmental tract of the inferior colliculus. Electrical stimulation of these sites induced synaptic currents in EX neurons (Fig. 8A). As we observed no cross-suppression with paired stimulation (Fig. 8A), we determined that these inputs were independent from those excited by CSh stimulation.
Figure 8.

DHPG suppresses both EPSCs induced by CSh stimulation and spontaneous transmission, but not EPSCs induced by OT stimulation. A, Schematic of the midbrain-slice preparation with two stimulating electrodes (one in CSh and one in the tectotegmental tract). Test showing absence of cross-facilitation/suppression and therefore input independence. B, Average effect of DHPG on CSh–eEPSCs (black circles) and OT–eEPSCs (white circles). C, Average effect of D-APV (black circles) and iMK-801 (white circles) on DHPG-induced LTD of CSh–eEPSCs. D, Suppression of spontaneous transmission in EX neurons induced by DHPG. Error bars indicate SEM. Ctrl., Control; CCo, central core portion of the inferior colliculus; S, stimulating electrode; R, recording electrode.
To further distinguish these inputs, we investigated the effect of group I mGluR activation at OT and CSh synapses onto EX neurons. It is known that group I mGluR activation by (S)-3,5-dihydroxyphenylglycine hydrate (DHPG; 50 μm) induces LTD at CSh–EX synapses (Penzo and Peña, 2009). Here, we performed a variation of this protocol. DHPG was applied to the bath for 7–8 min following a stable baseline while simultaneously monitoring CSh–eEPSCs and OT–eEPDCs on EX neurons. Whereas DHPG induced LTD of CSh–eEPSCs (n = 4; 67.8 ± 2.5% of control; p < 0.001) (Fig. 8B), no effect was observed on OT–eEPSCs (n = 4; 99.5 ± 6.3% of control; p > 0.1) (Fig. 8B). This result suggests that group I mGluR activation selectively depresses CSh–EX synapses. Similar to DSSR, DHPG-mediated LTD of CSh–eEPSCs was prevented and partially reversed by D-APV (n = 4; 109.4 ± 5.9% of control; p < 0.05) (Fig. 8C) but not by iMK-801 (n = 5; 68.5 ± 1.9% of control; p < 0.001) (Fig. 8C). DHPG application also induced a persistent decrease in the frequency of mEPSCs onto EX neurons (n = 6; 55.1 ± 4.2% of control; p < 0.001) (Fig. 8D). These results show that the CSh input shares specific regulatory mechanisms with the inputs mediating the bulk of spontaneous events onto EX neurons. This evidence suggests that the mEPSCs and eEPSCs investigated in this study arise, largely, from the same input, i.e., the CSh–EX synapse.
Discussion
Retrograde messengers allow neurons to modulate the strength of their afferent inputs (Regehr et al., 2009). Because different forms of transmitter release are present at synapses, the selectivity of retrograde messengers to regulate these diverse forms of neurotransmission remains an open question. Here we have investigated the role of retrograde transmission in the modulation of AP-independent release. We present a novel form of plasticity induced by postsynaptic depolarization that requires activation of NMDARs on the presynaptic cell and that exclusively affects spontaneous release.
Depolarization-induced suppression of spontaneous release
Work over the past decade has challenged the concept of spontaneous neurotransmitter release as mere synaptic noise. A recent report showed that minis alone can adjust the threshold for plasticity at single synapses (Lee et al., 2010). Also, minis and synaptic events induced by action potentials may act on different subsets of postsynaptic receptors at a single terminal (Atasoy et al., 2008). This suggests that spontaneous and evoked release belong to different functional microdomains within a single synapse. Our results are consistent with this view. At the CSh–EX synapse of the avian midbrain, postsynaptic depolarization induces a persistent suppression of spontaneous release by retrograde activation of presynaptic NMDARs, which does not affect AP-dependent release. Unlike in the cerebellum, where postsynaptic depolarization of Purkinje cells triggers an increase in both the frequency and amplitude of inhibitory minis (Duguid and Smart, 2004), retrograde activation of presynaptic NMDARs in EX reduces the frequency of minis while leaving their amplitude unaltered, thus implicating glutamate as a putative retrograde messenger. DSSR blockade by selectively inhibition of VGLUTs on the postsynaptic cell is consistent with this idea.
DSSR is mediated by CaMKII and SNARE-dependent vesicle exocytosis at the postsynaptic compartment. CaMKII can associate with synaptic vesicles (Ouimet et al., 1984) as well as phosphorylate SNARE proteins in vitro (Wang, 2008). In addition, CaMKII can induce glutamate release from both intact synapses (Llinás et al., 1985, 1991) and synaptosomes (Nichols et al., 1990). CaMKII interaction with Synapsin I (Llinás et al., 1985, 1991; Nichols et al., 1990, 1992) and SNARE proteins (Nielander et al., 1995; Hirling and Scheller, 1996) presumably mediates this function. Altogether, these findings strengthen the hypothesis that glutamate is directly released by the postsynaptic cell during DSSR.
A reduction in the frequency of minis induced by postsynaptic depolarization could be the result of a decrease in presynaptic function. Conversely, postsynaptic silencing by the removal of AMPA receptors from synapses could also account for our observations. Several aspects of our studies support the former hypothesis. First, DSSR requires the activation of NMDARs on the presynaptic cell. Second, blocking dynamin-dependent endocytosis does not prevent DSSR induction. Third, DSSR does not affect evoked responses. These findings support the idea that DSSR is presynaptically expressed, affecting only spontaneous release. Furthermore, DSSR is also induced by physiologically relevant depolarizing stimuli such as low-frequency spiking of the postsynaptic cell.
The question remains of what functional roles DSSR plays in EX. Given the amplitude (∼40 pA) and frequency (4–6 Hz) of mEPSCs, it is unlikely that spontaneous transmission would have a direct effect in the output of EX cells, as it is the case in stellate cells of the cerebellum (Carter and Regehr, 2002). However, as stated above, recent work suggests that even at low rates, mEPSCs can adjust the threshold for plasticity at single synapses (Lee et al., 2010). Determining whether the reduction in mEPSC frequency observed in DSSR has a similar effect in the threshold for LTD induction and/or in its expression is intriguing and requires further study.
Retrograde transmission in DSSR
Depolarization-induced suppression of both inhibition (Llano et al., 1991; Pitler and Alger, 1992, 1994; Vincent and Marty, 1993; Alger and Pitler, 1995) and excitation (Kreitzer and Regehr, 2001; Ohno-Shosaku et al., 2001) is a form of short-term plasticity that has been reported in several brain regions. These phenomena generally require endocannabinoid synthesis at the postsynaptic neuron (Kreitzer and Regehr, 2001; Wilson and Nicoll, 2001) and subsequent activation of presynaptic CB1Rs. A longer lasting plasticity reported at a cerebellar synapse, termed depolarization-induced potentiation of inhibition (DPI), is endocannabinoid-independent and presumably mediated by retrograde glutamatergic transmission (Duguid and Smart, 2004). Like DPI, DSSR is not prevented by blocking CB1Rs and requires both non-postsynaptic NMDAR activation and SNARE-dependent exocytosis at the postsynaptic membrane, suggesting glutamate as a plausible retrograde messenger. In fact, somatodendritic release of glutamate has been reported by others (Zilberter, 2000; Zilberter et al., 2005). Though in our opinion glutamate release from the postsynaptic neuron represents the most likely scenario, conclusive evidence in favor of this hypothesis is not presented here.
It is intriguing that D-APV not only prevents DSSR and DHPG-induced LTD but also unmasks a potentiation induced by stimulation protocols that would normally cause depression. This suggests that NMDAR activation might turn into depression a mechanism that would otherwise induce potentiation. What the mediators of this small potentiation are requires further studies.
Input-specificity of DSSR
An important issue regarding our results is the possibility that the inputs driving CSh-evoked responses are qualitatively distinct from those mediating mEPSCs. We have addressed this issue by comparing the role of group I mGluR activation on the function of two major inputs of EX neurons, one originating in CSh and another in OT. We have shown that DHPG induces NMDAR-dependent LTD at the CSh–EX synapse (Penzo and Peña, 2009). Here we report that this LTD is not prevented by selectively blocking postsynaptic NMDARs, and presumably requires NMDARs on the presynaptic cell. In addition, DHPG-mediated LTD is selectively expressed in CSh inputs but not in OT-evoked responses. Because DHPG suppresses the frequency of mEPSCs similarly to DSSR and CSh–eEPSCs, we conclude that inputs mediating CSh-evoked responses and mEPSCs are likely to be largely the same.
Distinct modulation of spontaneous and evoked transmitter release by retrograde messengers
As mentioned above, the postsynaptic depolarization responsible for DSSR does not affect evoked transmission onto EX neurons. However, this manipulation was sufficient to evoke LTD when CB1Rs were activated pharmacologically. These findings are consistent with growing evidence suggesting that various forms of neurotransmitter release represent qualitatively different processes. Spontaneous and nonspontaneous synaptic events are mediated by different vesicle-fusion mechanisms (Maximov et al., 2007; Chung et al., 2010) and target different populations of postsynaptic receptors (Atasoy et al., 2008). We have found that spontaneous and evoked synaptic release respond differently to retrograde messengers. Whereas retrograde activation of presynaptic NMDARs alone can reduce spontaneous release, coincident activation of CB1Rs by eCBs might be required for the postsynaptic neuron to exert a similar effect on evoked transmission. Thus, combining retrograde messengers could potentially allow for a differential modulation of distinct forms of neurotransmitter release.
Footnotes
This work was supported by National Institutes of Health Grants DC007690 and DC007690.S1, and by National Research Service Award Grant F31NS065698. We thank Pablo Castillo, Reed Carroll, Andrés Chávez, Donald Faber, Kamran Khodakhah, Alberto Pereda, and Thomas Younts for their comments and advice.
References
- Acuna-Goycolea C, van den Pol A. Glucagon-like peptide 1 excites hypocretin/orexin neurons by direct and indirect mechanisms: implications for viscera-mediated arousal. J Neurosci. 2004;24:8141–8152. doi: 10.1523/JNEUROSCI.1607-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Acuna-Goycolea C, van den Pol AN. Neuroendocrine proopiomelanocortin neurons are excited by hypocretin/orexin. J Neurosci. 2009;29:1503–1513. doi: 10.1523/JNEUROSCI.5147-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alger BE, Pitler TA. Retrograde signaling at GABAA-receptor synapses in the mammalian CNS. Trends Neurosci. 1995;18:333–340. doi: 10.1016/0166-2236(95)93923-l. [DOI] [PubMed] [Google Scholar]
- Atasoy D, Ertunc M, Moulder KL, Blackwell J, Chung C, Su J, Kavalali ET. Spontaneous and evoked glutamate release activates two populations of NMDA receptors with limited overlap. J Neurosci. 2008;28:10151–10166. doi: 10.1523/JNEUROSCI.2432-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bender VA, Bender KJ, Brasier DJ, Feldman DE. Two coincidence detectors for spike timing-dependent plasticity in somatosensory cortex. J Neurosci. 2006;26:4166–4177. doi: 10.1523/JNEUROSCI.0176-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blomeley C, Bracci E. Substance P depolarizes striatal projection neurons and facilitates their glutamatergic inputs. J Physiol. 2008;586:2143–2155. doi: 10.1113/jphysiol.2007.148965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehning D, Snyder SH. Novel neural modulators. Annu Rev Neurosci. 2003;26:105–131. doi: 10.1146/annurev.neuro.26.041002.131047. [DOI] [PubMed] [Google Scholar]
- Brenman JE, Bredt DS. Synaptic signaling by nitric oxide. Curr Opin Neurobiol. 1997;7:374–378. doi: 10.1016/s0959-4388(97)80065-7. [DOI] [PubMed] [Google Scholar]
- Carter AG, Regehr WG. Quantal events shape cerebellar interneuron firing. Nat Neurosci. 2002;5:1309–1318. doi: 10.1038/nn970. [DOI] [PubMed] [Google Scholar]
- Chevaleyre V, Takahashi KA, Castillo PE. Endocannabinoid-mediated synaptic plasticity in the CNS. Annu Rev Neurosci. 2006;29:37–76. doi: 10.1146/annurev.neuro.29.051605.112834. [DOI] [PubMed] [Google Scholar]
- Chung C, Barylko B, Leitz J, Liu X, Kavalali ET. Acute dynamin inhibition dissects synaptic vesicle recycling pathways that drive spontaneous and evoked neurotransmission. J Neurosci. 2010;30:1363–1376. doi: 10.1523/JNEUROSCI.3427-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corlew R, Brasier DJ, Feldman DE, Philpot BD. Presynaptic NMDA receptors: newly appreciated roles in cortical synaptic function and plasticity. Neuroscientist. 2008;14:609–625. doi: 10.1177/1073858408322675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drake CT, Terman GW, Simmons ML, Milner TA, Kunkel DD, Schwartzkroin PA, Chavkin C. Dynorphin opioids present in dentate granule cells may function as retrograde inhibitory neurotransmitters. J Neurosci. 1994;14:3736–3750. doi: 10.1523/JNEUROSCI.14-06-03736.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duguid IC, Smart TG. Retrograde activation of presynaptic NMDA receptors enhances GABA release at cerebellar interneuron-Purkinje cell synapses. Nat Neurosci. 2004;7:525–533. doi: 10.1038/nn1227. [DOI] [PubMed] [Google Scholar]
- Feinmark SJ, Begum R, Tsvetkov E, Goussakov I, Funk CD, Siegelbaum SA, Bolshakov VY. 12-Lipoxygenase metabolites of arachidonic acid mediate metabotropic glutamate receptor-dependent long-term depression at hippocampal CA3-CA1 synapses. J Neurosci. 2003;23:11427–11435. doi: 10.1523/JNEUROSCI.23-36-11427.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank CA, Kennedy MJ, Goold CP, Marek KW, Davis GW. Mechanisms underlying the rapid induction and sustained expression of synaptic homeostasis. Neuron. 2006;52:663–677. doi: 10.1016/j.neuron.2006.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gompf HS, Moldavan MG, Irwin RP, Allen CN. Nociceptin/orphanin FQ (N/OFQ) inhibits excitatory and inhibitory synaptic signaling in the suprachiasmatic nucleus (SCN) Neuroscience. 2005;132:955–965. doi: 10.1016/j.neuroscience.2004.11.057. [DOI] [PubMed] [Google Scholar]
- Hirling H, Scheller RH. Phosphorylation of synaptic vesicle proteins: modulation of the alpha SNAP interaction with the core complex. Proc Natl Acad Sci U S A. 1996;93:11945–11949. doi: 10.1073/pnas.93.21.11945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hölscher C. Nitric oxide, the enigmatic neuronal messenger: its role in synaptic plasticity. Trends Neurosci. 1997;20:298–303. doi: 10.1016/s0166-2236(97)01065-5. [DOI] [PubMed] [Google Scholar]
- Knudsen EI. Instructed learning in the auditory localization pathway of the barn owl. Nature. 2002;417:322–328. doi: 10.1038/417322a. [DOI] [PubMed] [Google Scholar]
- Kozoriz MG, Kuzmiski JB, Hirasawa M, Pittman QJ. Galanin modulates neuronal and synaptic properties in the rat supraoptic nucleus in a use and state dependent manner. J Neurophysiol. 2006;96:154–164. doi: 10.1152/jn.01028.2005. [DOI] [PubMed] [Google Scholar]
- Kreitzer AC, Regehr WG. Cerebellar depolarization-induced suppression of inhibition is mediated by endogenous cannabinoids. J Neurosci. 2001;21:RC174. doi: 10.1523/JNEUROSCI.21-20-j0005.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MC, Yasuda R, Ehlers MD. Metaplasticity at single glutamatergic synapses. Neuron. 2010;66:859–870. doi: 10.1016/j.neuron.2010.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Gao XB, Sakurai T, van den Pol AN. Hypocretin/Orexin excites hypocretin neurons via a local glutamate neuron-A potential mechanism for orchestrating the hypothalamic arousal system. Neuron. 2002;36:1169–1181. doi: 10.1016/s0896-6273(02)01132-7. [DOI] [PubMed] [Google Scholar]
- Llano I, Leresche N, Marty A. Calcium entry increases the sensitivity of cerebellar Purkinje cells to applied GABA and decreases inhibitory synaptic currents. Neuron. 1991;6:565–574. doi: 10.1016/0896-6273(91)90059-9. [DOI] [PubMed] [Google Scholar]
- Llinás R, McGuinness TL, Leonard CS, Sugimori M, Greengard P. Intraterminal injection of synapsin I or calcium/calmodulin-dependent protein kinase II alters neurotransmitter release at the squid giant synapse. Proc Natl Acad Sci U S A. 1985;82:3035–3039. doi: 10.1073/pnas.82.9.3035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llinás R, Gruner JA, Sugimori M, McGuinness TL, Greengard P. Regulation by synapsin I and Ca(2+)-calmodulin-dependent protein kinase II of the transmitter release in squid giant synapse. J Physiol. 1991;436:257–282. doi: 10.1113/jphysiol.1991.sp018549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magnusson AK, Park TJ, Pecka M, Grothe B, Koch U. Retrograde GABA signaling adjusts sound localization by balancing excitation and inhibition in the brainstem. Neuron. 2008;59:125–137. doi: 10.1016/j.neuron.2008.05.011. [DOI] [PubMed] [Google Scholar]
- Maximov A, Shin OH, Liu X, Südhof TC. Synaptotagmin-12, a synaptic vesicle phosphoprotein that modulates spontaneous neurotransmitter release. J Cell Biol. 2007;176:113–124. doi: 10.1083/jcb.200607021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols RA, Sihra TS, Czernik AJ, Nairn AC, Greengard P. Calcium/calmodulin-dependent protein kinase II increases glutamate and noradrenaline release from synaptosomes. Nature. 1990;343:647–651. doi: 10.1038/343647a0. [DOI] [PubMed] [Google Scholar]
- Nichols RA, Chilcote TJ, Czernik AJ, Greengard P. Synapsin I regulates glutamate release from rat brain synaptosomes. J Neurochem. 1992;58:783–785. doi: 10.1111/j.1471-4159.1992.tb09788.x. [DOI] [PubMed] [Google Scholar]
- Nielander HB, Onofri F, Valtorta F, Schiavo G, Montecucco C, Greengard P, Benfenati F. Phosphorylation of VAMP/synaptobrevin in synaptic vesicles by endogenous protein kinases. J Neurochem. 1995;65:1712–1720. doi: 10.1046/j.1471-4159.1995.65041712.x. [DOI] [PubMed] [Google Scholar]
- Ohno-Shosaku T, Maejima T, Kano M. Endogenous cannabinoids mediate retrograde signals from depolarized postsynaptic neurons to presynaptic terminals. Neuron. 2001;29:729–738. doi: 10.1016/s0896-6273(01)00247-1. [DOI] [PubMed] [Google Scholar]
- Ouimet CC, McGuinness TL, Greengard P. Immunocytochemical localization of calcium/calmodulin-dependent protein kinase II in rat brain. Proc Natl Acad Sci U S A. 1984;81:5604–5608. doi: 10.1073/pnas.81.17.5604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penzo MA, Peña JL. Endocannabinoid-mediated long-term depression in the avian midbrain expressed presynaptically and postsynaptically. J Neurosci. 2009;29:4131–4139. doi: 10.1523/JNEUROSCI.5466-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitler TA, Alger BE. Postsynaptic spike firing reduces synaptic GABAA responses in hippocampal pyramidal cells. J Neurosci. 1992;12:4122–4132. doi: 10.1523/JNEUROSCI.12-10-04122.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitler TA, Alger BE. Depolarization-induced suppression of GABAergic inhibition in rat hippocampal pyramidal cells: G protein involvement in a presynaptic mechanism. Neuron. 1994;13:1447–1455. doi: 10.1016/0896-6273(94)90430-8. [DOI] [PubMed] [Google Scholar]
- Qian J, Colmers WF, Saggau P. Inhibition of synaptic transmission by neuropeptide Y in rat hippocampal area CA1: modulation of presynaptic Ca2+ entry. J Neurosci. 1997;17:8169–8177. doi: 10.1523/JNEUROSCI.17-21-08169.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regehr WG, Carey MR, Best AR. Activity-dependent regulation of synapses by retrograde messengers. Neuron. 2009;63:154–170. doi: 10.1016/j.neuron.2009.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sang N, Chen C. Lipid signaling and synaptic plasticity. Neuroscientist. 2006;12:425–434. doi: 10.1177/1073858406290794. [DOI] [PubMed] [Google Scholar]
- Sjöström PJ, Turrigiano GG, Nelson SB. Neocortical LTD via coincident activation of presynaptic NMDA and cannabinoid receptors. Neuron. 2003;39:641–654. doi: 10.1016/s0896-6273(03)00476-8. [DOI] [PubMed] [Google Scholar]
- Sutton MA, Ito HT, Cressy P, Kempf C, Woo JC, Schuman EM. Miniature neurotransmission stabilizes synaptic function via tonic suppression of local dendritic protein synthesis. Cell. 2006;125:785–799. doi: 10.1016/j.cell.2006.03.040. [DOI] [PubMed] [Google Scholar]
- Vincent P, Marty A. Neighboring cerebellar Purkinje cells communicate via retrograde inhibition of common presynaptic interneurons. Neuron. 1993;11:885–893. doi: 10.1016/0896-6273(93)90118-b. [DOI] [PubMed] [Google Scholar]
- Wagner JJ, Evans CJ, Chavkin C. Focal stimulation of the mossy fibers releases endogenous dynorphins that bind kappa 1-opioid receptors in guinea pig hippocampus. J Neurochem. 1991;57:333–343. doi: 10.1111/j.1471-4159.1991.tb02132.x. [DOI] [PubMed] [Google Scholar]
- Wang ZW. Regulation of synaptic transmission by presynaptic CaMKII and BK channels. Mol Neurobiol. 2008;38:153–166. doi: 10.1007/s12035-008-8039-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson RI, Nicoll RA. Endogenous cannabinoids mediate retrograde signalling at hippocampal synapses. Nature. 2001;410:588–592. doi: 10.1038/35069076. [DOI] [PubMed] [Google Scholar]
- Yung WH, Leung PS, Ng SS, Zhang J, Chan SC, Chow BK. Secretin facilitates GABA transmission in the cerebellum. J Neurosci. 2001;21:7063–7068. doi: 10.1523/JNEUROSCI.21-18-07063.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zilberter Y. Dendritic release of glutamate suppresses synaptic inhibition of pyramidal neurons in rat neocortex. J Physiol. 2000;528:489–496. doi: 10.1111/j.1469-7793.2000.00489.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zilberter Y, Kaiser KM, Sakmann B. Dendritic GABA release depresses excitatory transmission between layer 2/3 pyramidal and bitufted neurons in rat neocortex. Neuron. 1999;24:979–988. doi: 10.1016/s0896-6273(00)81044-2. [DOI] [PubMed] [Google Scholar]
- Zilberter Y, Harkany T, Holmgren CD. Dendritic release of retrograde messengers controls synaptic transmission in local neocortical networks. Neuroscientist. 2005;11:334–344. doi: 10.1177/1073858405275827. [DOI] [PubMed] [Google Scholar]