

Abstract
Genetic information flows from DNA to macromolecular structures—the dominant force in the molecular organization of life. However, recent work suggests that metabolite availability to the hexosamine and Golgi N-glycosylation pathways exerts control over the assembly of macromolecular complexes on the cell surface, and in this capacity, acts upstream of signaling and gene expression. The structure and number of N-glycans per protein molecule cooperate to regulate lectin binding and thereby the distribution of glycoproteins at the cell surface. Congenital disorders of glycosylation provide insight as extreme hypomorphisms, whereas milder deficiencies may encompass many common chronic conditions including autoimmunity, metabolic syndrome, and aging.
Introduction
In the study of development and disease the levels of receptor ligands and their downstream signaling pathways have received considerable attention, but less clear are the determinants of receptor availability at the cell surface. A sizeable proportion of receptors and transporters are located in endomembrane compartments that exchange with the cell surface pools (Wiley and Burke, 2001). Certainly, the number of receptors at the cell surface is an important determinant of ligand-receptor complex formation that affects signal strength (Dennis et al., 2009; Gurdon and Bourillot, 2001). Many cytokine receptors and transporters are co-translationally modified with Asn (N)- and Ser/Thr (O)- linked oligosaccharides (glycans), and together with proteoglycans and glycolipids, form the glycocalyx, a ∼100 nM wide macromolecular structure that dominates the cell surface. The remodeling of N-glycans in the Golgi is sensitive to metabolism and when combined with gene-encoded differences in N-glycan number per protein, glycoprotein concentrations at the cell surface can be differentially regulated according to their affinities for endogenous lectins (Lau et al., 2007). Herein, we posit that increasing metabolic regulation of Golgi N-glycan biosynthesis in metazoan plays a role in adaptive cellular systems in postnatal life. Applying this model to disease states, we review data from mice and human studies linking genetic and metabolic dysregulation of protein N-glycosylation in congenital disorders and complex traits such as autoimmunity, cancer, and metabolic syndrome.
Glycans and cell surface regulation
Cell surface transmembrane glycoproteins are localized to and exchanged between microdomains, including clathrin coated-pits, cholesterol-rich lipid rafts, adhesion junctions between cells, substratum focal adhesion, and as discussed here, the galectin lattice. The galectins are N-acetyllactosamine-binding proteins, and their major ligands are Golgi-remodeled N-glycans common to many glycoproteins at the cell surface (Patnaik et al., 2006). Galectins are redox-sensitive proteins produced in the cytosol, and released by a non-classical pathway where they first encounter glycoproteins in cargo vesicles (Delacour et al., 2007) and at the cell surface (Hughes, 1999). Galectins, as well as the siglecs and selectins lectin families are largely absent in unicellular eukaryotes (Drickamer and Fadden, 2002). The 14 human galectins have either 1 or 2 carbohydrate recognition domains and mediate glycoprotein cross-linking (Lee and Lee, 2000). The extended C-terminus of galectin-3 forms multimeric structures (up to pentamers) driven by increasing concentrations of multivalent glycoprotein ligands, resulting in a molecular “lattice” or microdomain with irregular geometry at the cell surface (Ahmad et al., 2003; Demetriou et al., 2001). However, galectins with different lattice-forming geometries may bind glycoprotein partners selectively based on the N-glycan orientation (Brewer et al., 2002).
Members of the epidermal growth factor receptor (EGFR) family have been used extensively to study trafficking between microdomains, and sensitivity to ligand. The extracellular domain of EGFR is restrained from self-association and activation, but modeling and experimental data implicates additional extrinsic mechanism of restraint (Klein et al., 2004). One of these is galectin binding to the N-glycans on EGFR, which slows lateral diffusion and loss to coated-pit endocytosis in mammary carcinoma cells (Lajoie et al., 2007; Partridge et al., 2004). In human glioblastomas, an activating mutation in EGFR deletes part of the extracellular domain (EGFRvIII), removing 4 of 12 N-glycan sites and increasing ligand-independent dimerization (Fernandes et al., 2001). Domain II–IV normally confers intramolecular interactions that maintain the autoinhibited state, where the N-glycans in domain III contribute by blocking receptor dimerization and activation (Takahashi et al., 2008) (Tsuda et al., 2000). Therefore, in addition to opposing loss to endocytosis, N-glycans appear to “insulate” against ligand-independent EGFR dimerization. N-glycans extend from the protein surface and may restrict the approaches between monomers for EGFR dimerization. Galectin cross-linking is expected to separate glycoproteins by ∼100Å, possibly imposing a physical barrier to spontaneous dimerization (Seetharaman et al., 1998) (Figure 1). However, the lattice is not likely a barrier to ligand-mediated receptor dimerization, as various cyokine affinities for cognate receptors are ∼108-1012 M, whereas galectins affinities for glycans are lower ∼10-5-10-7 M. Galectin-3 and -8 bind to β1 integrin and regulate focal adhesion turnover and cells motility (Lagana et al., 2006; Levy et al., 2001). As expected for a multivalent system, addition of recombinant galectin to cells displays an optimum for stimulation of focal adhesion turnover. Galectin gene expression, export and redox sensitivity are likely important aspects of lattice regulation and warrant further investigation.
Figure 1. N-glycan branching pathways and the galectin lattice.
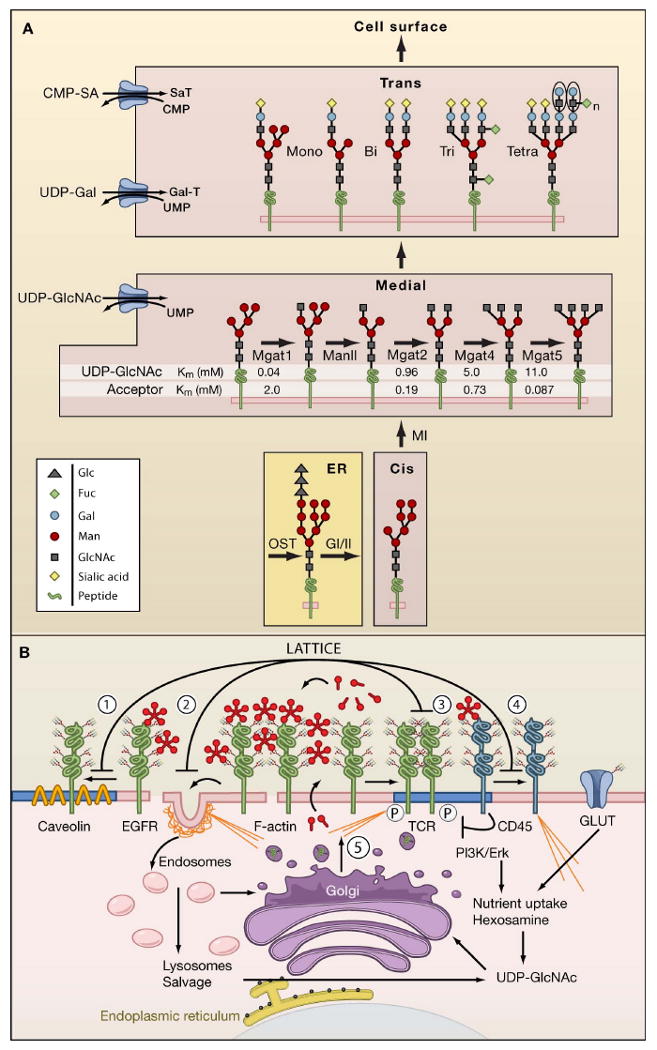
(A) N-glycan branching pathway. Oligosaccharyltransterase (OT) utilizes the pre-assembled donor Glc3Man9GlcNAc2-pp-dolichol to transfer the glycan to N-X-S/T motifs on glycoproteins in the endoplasmic reticulum (ER). Glycoproteins transit from the ER to cis, medial, and trans Golgi en route to the cell surface. The N-acetylglucosaminyltransferases enzymes, designated by their gene names (Mgat1, Mgat2, Mgat4, Mgat5) generate branched N-glycans that display a range of affinities for galectins. The Km values for Mgat1, Mgat2, Mgat4 and Mgat5 are indicated as measured in vitro for UDP-GlcNAc and acceptor glycoproteins.
(B) Dynamics of the galectin lattice. The glycocalyx is the thick carbohydrate layer surrounding the cell. Glycan structures generated in the Golgi differ in affinities for galectins. Galectins cross-link glycoprotein receptors and oppose (1) loss of epidermal growth factor receptors (EGFR) to Caveolin 1-positive microdomains, (2) coated-pit endocytosis, (3) precocious clustering of receptors, and (4) F-actin-mediated entry of T cell receptor (TCR) into and exit of CD45 from ganglioside GM1-positive microdomains (blue). (5) Nutrient supply and growth signaling increase membrane remodeling, regulate metabolite flux through the hexosamine pathway to UDP-GlcNAc and N-glycan branching (Golgi) on receptors to promote surface retention by the galectin lattice.
The α and β subunits of the T cell receptor (TCR) are N-glycosylated on at least 7 Asn-X-Ser/Thr (NXS/T) sites and the complex is assembled in the endoplasmic reticulum (ER) prior to N-glycan processing in the Golgi (Rudd et al., 1999). On naive T cells, galectin-3 binds to the T cell receptor (TCR) and opposes microfilament-dependent partitioning of TCR into lipid microdomains (ganglioside GM1-enriched), while blocking microfilament-dependent movement of the tyrosine phosphatase CD45 out of the lipid microdomains (Chen et al., 2007; Demetriou et al., 2001) (Figure 1). CD45 in lipid microdomains dampens TCR-Lck signaling. Galectin binding to TCR impedes CD8 binding, another example of insulating against complex formation that increases the threshold to TCR agonists (Demotte et al., 2008). Mice deficient in the branching enzyme β1,6-N-acetylglucosaminyltransferaseV (Mgat5) or β1,3-N-acetylglucosaminyltransferase (β3GnT2), an enzyme that extends the β1,6 branches with poly-N-acetyllactosamine, drops the barrier to TCR clustering and autoimmune disease in vivo (Demetriou et al., 2001; Togayachi et al., 2007). Selective removal of specific NXS/T sites from TCR also enhances receptor diffusion, multimerization, and activation (Kuball et al., 2009). T cell activation results in adaptive changes in glycoprotein distribution at the surface downstream of increased nutrient uptake, membrane turnover, and N-glycan branching.
Similarly, oncogenesis stimulates membrane remodeling, N-glycan branching and galectin-3 expression (Dennis et al., 1987; Lanzetti et al., 2004; Takenaka et al., 2004). Therefore tension appears to increase between membrane turnover and lattice-mediated protection of receptors from coated-pits and loss to endocytosis (Figure 1). Cancer mutations that disrupt the endocytic machinery also protect growth receptors either at the cell surface or by maintaining their activity in endosomes (Mosesson et al., 2008). For example, Avalanche and rab5 mutants in the Drosophila endocytic pathway inhibit internalization of surface receptors to the early endosome; thus an excess of surface growth receptors promotes growth signaling (Bilder, 2004). However, the scrib-dlg-lgl tumor suppressor pathway also disrupts trafficking, and thereby signaling in endosomes by tumor necrosis factor (TNF)/JNK to mediate cell death (Igaki et al., 2009). Protein phosphatases are important constitutive suppressers of growth signaling in early endosomes and lipid microdomains (Reynolds et al., 2003). The ligands for receptor tyrosine phosphatase have been elusive, and regulation may involve galectins as has been suggested for CD45 (Chen et al., 2007) (Figure 1).
In addition to coated-pit endocytosis and the galectin lattice, receptors exchange with other specialized membrane microdomains. For example, EGFR is tethered by epithelial cell junctions where it binds to the tumor suppressor neurofibromatosis (NF2/Merlin), which reduces receptor sensitivity to EGF (Curto et al., 2007). Adhesion junctions are lost with cancer progression, in a process known as the epithelial to mesenchymal transistion (EMT). EMT promotes actin remodeling, which stimulates increased trafficking of EGFR between endosomes and the cell surface. As a consequence, the ruffling edges of cells recruit and activate EGFR with lower ligand requirements (Moro et al., 2002).
Caveolin1 (Cav1) clusters in cholesterol-rich rafts and binds to conserved motifs found in EGFR and other signaling proteins resulting in loss of responsiveness to growth factors (Okamoto et al., 1998). CAV1 maps to a tumor suppressor locus (D7S522; 7q31.1) (Lee et al., 2002), but up-regulation of N-glycan branching in mouse mammary tumor cells acts dominantly to protect EGFR from Cav1 suppression (Lajoie et al., 2007). The galectin lattice protects EGFR from loss to Cav1 microdomains (Lajoie et al., 2007). Cav-1 binds cholesterol in rafts and stabilizes the structure by reducing diffusion, and interestingly, galectin-4 and -9 binding to glycoproteins in lipid rafts has a stabilizing effect (Braccia et al., 2003; Tanikawa et al., 2008). As outlined below, both N-glycan number (sequence-encoded NXS/T) and Golgi N-glycan branching (context-dependent) determine glycoprotein affinities for the lattice (Lau et al., 2007).
N-glycoform diversity
N-glycosylation of proteins begins with the co-translational transfer of Glc3Man9GlcNAc2 to ∼ 70% of lumenal N-X-S/T (where X≠Pro) motifs by oligosaccharyltransferase (Apweiler et al., 1999). N-glycosylation is enhanced by neighboring aromatic amino acids and turns that expose the motifs at the convex surface of the peptide, but less efficient in the proximity of disulfide bonds, the transmembrane domain, and at the termini of protein (Jones et al., 2005). After transfer and trimming of 2 glucose (Glc) units, the protein chaperones calnexin and calreticulin bind to Glc1Man9GlcNAc2 and the remaining Glc residue is recycled by α-glucosidase II and ER α-glucosyltransferase until folding is complete, allowing transit to the Golgi (Helenius and Aebi, 2004). However, most NXS/T sites are not required for protein folding, and site densities in most animals glycoproteins is higher than expected based on sequence composition, consistent with a post-Golgi function at the cell surface.
Chaperone-assisted protein folding employs an ancient and relatively homogenous N-glycan structure required prior to the Golgi. In contrast, an explosion in structural diversity has occurred with metazoan evolution of the Golgi pathways. Mannose residues are removed in the cis Golgi, then the N-acetylglucosaminyltransferases I, II, III IV, V (encoded by Mgat1, Mgat2, Mgat3, Mgat4a/b, and Mgat5) initiate the branches in a sequential pathway (Schachter, 1986) (Figure 1). Differing from the other Mgat enzymes, the N-acetylglucosamine (GlcNAc) added to the β-linked mannose residue by Mgat3 is not sequential nor elongated with galactose, and can block further branching by Mgat2, 4 and 5. In the trans Golgi, β1,4 galactosyltransferase catalyzes the addition of galactose to GlcNAc residues to generate the N-acetyllactosamine units, the epitope for galectin binding. The β1,6GlcNAc-branched product of Mgat5 is the preferred acceptor for extension with additional N-acetyllactosamine units, which increases affinity for galectins-1, -3, -8, and -9 (Hirabayashi et al., 2002). For example, galectin-3 binds to mono-, bi-, tri-, tetra-antennary N-glycans, and tetra-antennary N-glycans with polyN-acetyllactosamine with Kd's of 4.0, 1.4, 0.78, 0.69 and >0.19 μM, respectively, a synergistic increase with respect to N-acetyllactosamine units (Galβ1,4GlcNAcβ1,3). Importantly, structural diversity is magnified by partial saturation of branching at each N-glycan, as well as variable termination with sialic acid (SA), fucose (Fuc) and/or N-acetylgalactosamine (GalNAc). Terminal α2,6SA reduces N-glycan affinity for galectins, whereas GalNAc addition may increase affinities (Amano et al., 2003; Stowell et al., 2008).
In theory the diversity of the polymerization of monosaccharides into branched chains can increase in a factorial manner. However, the potential for N-glycan structural diversity in animal tissues is bounded by the specificities of Golgi enzymes encoded in animal genomes and their tissue-specific patterns of gene expression. Nonetheless, more than 40 structures (variations on branching and extension) have been identified on EGFR (Stroop et al., 2000). The protein environment of NXS/T sites can limit the accessibility to branching enzymes (Do et al., 1994), but even with some site-specific bias considerable numbers of glycoforms are possible (Figure 2). For example, 8 NXS/T sites in EGFR are occupied with N-glycans, and if we use a conservative estimate of 14 possible structures at each site, the potential number of glycoforms is 203,490, ∼10 times the number of surface EGFR molecules per cell (Lau et al., 2007). Glycoproteins differ in encoded NXS/T number, and this should influence the requirements of Golgi remodeling as a determinant of lattice avidity and surface retention.
Figure 2. Congenital Disorders of Glycosylation and glycoforms.
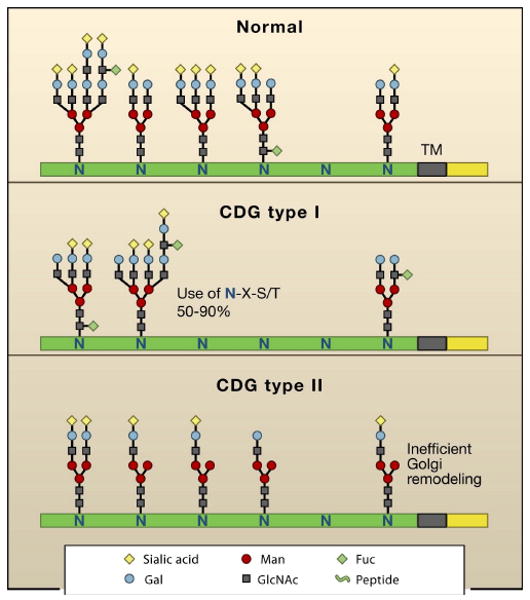
(A) Glycoprotein isoforms or glycofoms differ based on N-glycan structures present at each N-X-S/T site (not all sites are occupied). CDG type I alters glycoform distribution due to deficiencies in Glc3Man9GlcNAc2-pp-dolichol biosynthesis leading to incomplete N-X-S/T site occupancy. CDG type II are deficiencies in Golgi remodeling or glycoprotein trafficking.
NXS/T multiplicity encodes lattice affinity
The incidence of canonical N-glycosylation sequons (NXS/T, where X≠P) varies considerably between different glycoproteins. Receptor kinases that stimulate growth and proliferation such as insulin receptor, EGFR, and platelet derived growth factor receptor (PDGFR)] have ∼5 times more NXS/T sites, longer extracellular domains and more sites per 100 amino acids than receptors that mediate organogenesis, differentiation and arrest (such as Tie1, Musk, Ltk, ROR1/2, DDR1, TβR, and EphR) (Lau et al., 2007). These observations suggest that N-glycan numbers per peptide (multiplicity) is a conserved and functionally significant feature of receptor kinases. NXS/T sites are the primary determinant in N-glycan multiplicity, and together with N-glycan branching determine glycoform structures and affinity for the galectin lattice. through. Membrane glycoproteins with only one or two N-glycans per peptide are markedly dependent on branching for stable association with the galectin lattice (Lau et al., 2007). Examples of low multiplicity glycoproteins shown to be dependent on branching are discussed below and include glucose transporter (GLUT4, GLUT2), cytotoxic T lymphocyte antigen–4 (CTLA-4), and TGF-β receptors (TβR).
Interaction of metabolism with N-glycan number and diversity
Fructore-6-P, glutamine and acetyl-CoA are central to carbohydrate, nitrogen, and fatty acid metabolism, and also required for de novo uridine diphosphate-N-acetylglucosamine (UDP-GlcNAc) biosynthesis by hexosamine pathway (Grigorian et al., 2007). The Golgi UDP-GlcNAc antiporter exchanges uridine monophosphate (UMP) for UDP-GlcNAc and establishes the steady state amounts of UDP-GlcNAc inside the Golgi. Although difficult to measure, estimates suggest that UDP-GlcNAc concentration is 15-fold higher in the Golgi (∼1.5mM) than in the cytoplasm (Waldman and Rudnick, 1990). UDP-GlcNAc levels are well above the Km for Mgat1 at 0.04 mM, but Mgat1 has a relatively low affinity for acceptor N-glycan at ∼2 mM (Figure 1). This relationship is reversed for Mgat4 and Mgat5 enzymes, where UDP-GlcNAc concentrations become limiting (Lau et al., 2007; Sasai et al., 2002). In other words, Mgat1 and Mgat2 activities are suboptimal and limited by affinity for acceptors, whereas Mgat4 and Mgat5 activities are limited by UDP-GlcNAc concentration. Thus, the degree of N-glycan branching is dependent on rates of bulk protein synthesis, notably 5′cap-dependent mRNA translation stimulated by mTor/S6 kinase signaling, as well as the availability of key metabolites to UDP-GlcNAc biosynthesis. As a result, the high affinity galectin ligands, the tri- and tetra- antennary N-glycans, increase in an ultrasensitive manner as a function of UDP-GlcNAc availability (Lau et al., 2007). Ultrasensitivity describes a stimulus/response relationship in which the response rises sharply over a narrow range of stimulus (a sigmoid curve) with a Hill coefficient (nH) ≫1 indicating the deviation from a graded (hyperbolic) response curve (Ferrell, 1996). These pathway dynamics result from decreasing Mgat1, 2, 4, 5 activities moving down the pathway, decreasing affinities for the common substrate (Km for UDP-GlcNAc), sequential order, and limited reaction times due to transit of glycoprotein substrates through the Golgi (Lau et al., 2007).
TCR and growth-promoting receptor tyrosine kinases (such EGFR, insulin receptor, PDGFR) where N-glycan multiplicity is relatively high, a significant fraction of receptors are associated with the lattice, and only a slow graded increase occurs with rising UDP-GlcNAc (1.5 to 6 mM). In contrast, glycoproteins with 1 or 2 N-glycans (such as TβR, CTLA-4, and GLUT4) are largely below the threshold for stable association with the galectin lattice, but increase in a switch-like (sigmoidal) response to increasing UDP-GlcNAc concentration, mirroring the ultrasensitive output of tri- and tetra-antennary N-glycans by the branching pathway. Experimental data and computational modeling are consistent with this model for the retention of glycoproteins on the cell surface. Their half-lives are proportional to avidities for the lattice and cell surface retention is countered by constitutive endocytosis (Lau et al., 2007). To summarize, ultrasensitivity of the N-glycan branching pathway to UDP-GlcNAc allows differential regulation of surface glycoproteins based on N-glycan number and metabolite flux. Thus, metabolic supply to the hexosamine pathway titrates glycoprotein expression at the cell surface with response dynamics characteristic of N-glycan number.
Consequences of N-glycan number
Receptor kinases and cancer
In non-transformed epithelial cells, surface EGFR, PDGF, IGFR first increase in a shallow graded manner with increasing UDP-GlcNAc, followed by TβR in a switch-like response. These dynamics of receptors regulation by hexosamine/Golgi/ lattice have features of an oscillating system that may support tissue homeostasis (Lau et al., 2007). The same stimulus (UDP-GlcNAc) promotes a Michaelis-Menten response and a sigmoidal response for high and low multiplicity receptors, respectively. The intervening delay allows growth signaling and anabolic metabolism to prevail before the onset of negative regulation as UDP-GlcNAc rises to levels that retain the low multiplicity receptor. The TβR and receptor tyrosine kinase pathways often oppose each other in proliferation and differentiation. For example, self-renewal and pluripotency of cultured human embryonic stem cells is maintained by a precise ratio of fibroblast growth factor (bFGF) to Noggin, a bone morphogenic protein (BMP)/TGF-β antagonist (Xu et al., 2005). Similarly, BMP4 promotes the differentiation of tumor-initiating stem cells and inhibits growth of human glioblastoma cells in mice (Piccirillo et al., 2006). Canonical TβR/SMAD2/3 signaling also slows cell cycle progression by suppressing c-myc expression (Matsuura et al., 2004; Seoane et al., 2004). c-Myc activity in tumors stimulates the uptake of glucose and glutamine, the major carbon sources for energy in cancer cells (DeBerardinis et al., 2008), and stimulates anabolic metabolism including UDP-GlcNAc supply (DeBerardinis et al., 2008; Morrish et al., 2009). Low multiplicity of TβR and membrane remodeling in cancer cells reduces its surface levels. Although Raf/Mak/Ets activation increases Mgat4 and Mgat5 gene expression (Buckhaults et al., 1997; Ishida et al., 2005; Takamatsu et al., 1999), this appears to be insufficient to protect surface TβR, and conditions in cancer cells favor high multiplicity growth receptors over TβR (Lau et al., 2007). Metabolic supplements to UDP-GlcNAc can restore TβR signaling and slow growth suggesting that with a better understanding of N-glycosylation and metabolism, it may be possible to restore a “non-transformed” distribution of surface glycoproteins (Lau and Dennis, 2008; Mendelsohn et al., 2007).
T cell activation
T cell activation and subsequent arrest/apoptosis are regulated by gene expression, but are also temporally affected by metabolism and environment cues (Alegre et al., 2001; Frauwirth et al., 2002). Activation of naïve T cells requires TCR clustering above a threshold number, which induces Erk/PI3K signaling, glucose uptake and multiple rounds of cell division. The interval until growth arrest 4-5 days later is determined in part by stimulation of metabolite flux into UDP-GlcNAc supply (Lau et al., 2007). Similar to TβR, CTLA-4 has 2 NXS/T sites and displays an ultrasensitive response to UDP-GlcNAc to attain the branching required for surface retention and growth arrest (Lau et al., 2007). The levels of CTLA-4 transcript and intracellular protein increase markedly with TCR signaling during the early growth phase (Chikuma and Bluestone, 2002), but endosomal trafficking limits expression of CTLA-4 at the cell surface, keeping it below the threshold for the induction of growth arrest until the avidity of CTLA-4 for the galectin lattice increases. Surface CTLA-4 promotes T cell motility, which dampens TCR-CD28 signaling in blast cells leading to quiescent memory cell status or apoptosis (Schneider et al., 2006). T cells deficient in Mgat5 are hypomorphic for CTLA-4 at the cell surface but not inside the cells, and surface levels can be rescued by supplementation to UDP-GlcNAc as well as via maximal CD28 co-stimulation which drives metabolism. Compensation for the absence of tetra-antennary glycans in Mgat5 deficient cells occurs by supplementation to early reactions leading to more tri-antennary N-glycans. Galectin-1-deficient mice are also susceptibility to autoimmune disease, and both galectins-1 and -3 play a role in T cells apoptosis (Stillman et al., 2006; Toscano et al., 2007).
Glucose transporters
The glucose transporters GLUT2 and GLUT4, each with a single N-glycan, are dependent on branching and galectin binding for optimal surface retention (Lau et al., 2007; Ohtsubo et al., 2005). Insulin receptor (INSR) has 18 potential sites of N-glycan attachment, thus a large disparity with the GLUTs, and conducive to differential regulation by UDP-GlcNAc. INSR stimulates glucose uptake and surface GLUT4 in muscle with a response described as “quantum” (ultrasensitive), and this effectively shuttles excess serum glucose into muscle (Coster et al., 2004). Supplementation to the hexosamine pathway increases surface GLUT4 in a sharp sigmoidal response, and mutation of the GLUT4 NXS/T site blocks the response to UDP-GlcNAc. GLUT4 transgenic mice display increased UDP-GlcNAc levels in muscle, indicating positive feedback to the hexosamine pathway (Buse et al., 1996). Metabolites can change very rapidly in cells regulated largely by allosteric mechanisms (Bailey, 1998), but the hexosamine/Golgi/lattice may adapt the cell surface to reflect conditions averaged over time. In this regard, diabetic hyperglycemia leads to TGF-β-dependent tissue fibrosis, possibly due to a hexosamine-dependent increase in surface retention of TβR (Kolm-Litty et al., 1998).
The hexosamine pathway
UDP-GlcNAc is required in the biosynthesis of most classes of extracellular glycopolymers, including the N- and O-glycans, glycolipids, chitin, hyluronic acid, and heparin sulfate proteoglycans. UDP-GlcNAc is essential in all three phylogenetic kingdoms, and the de novo synthesis pathway may have originated with passage of life through the “RNA world”. As a vestige of this heritage, RNA transcripts for glutamine:fructose-6P aminotransferase (GFAT) in gram-negative bacteria have a 5′ aptomer that binds GlcNAc for allosteric control of its ribozyme activity. GlcNAc binding induces cleavage of the message and downregulates GFAT, which is a rate-limiting enzyme of the pathway (Winkler et al., 2004). GFAT catalyzes isomerase and amination reactions using glutamine as the amine donor, and the enzyme itself is allosterically inhibited by its product glucosamine-6-phosphate (GlcN-6P) and by UDP-GlcNAc in mammalian cells (Broschat et al., 2002). The substrates of the de novo pathway, fructose-6P, glutamine and acetyl-CoA are highly regulated intermediates of carbon and nitrogen metabolism (Figure 3).
Figure 3. Biosynthesis of Sugar-nucleotides.
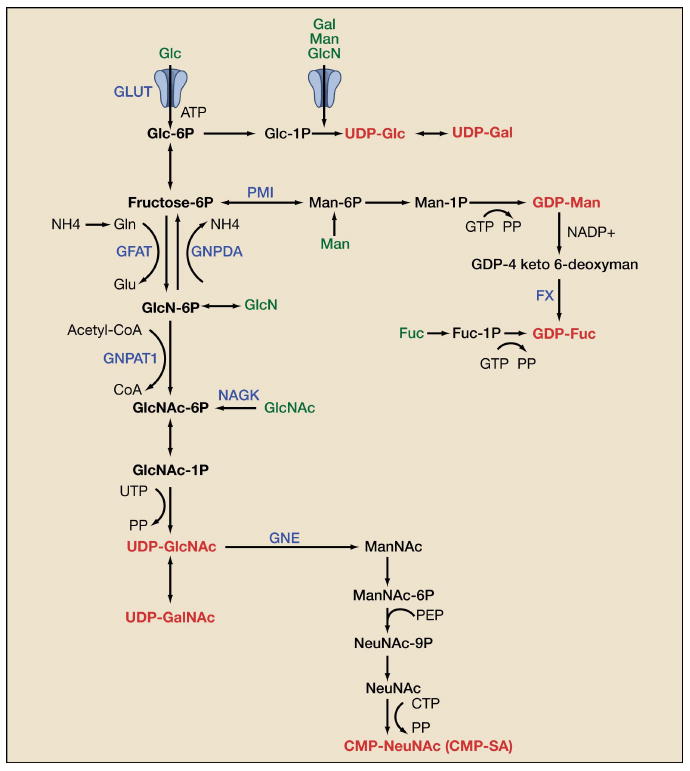
De novo biosynthesis of UDP-GlcNAc is dependent on glucose, glutamine, acetyl-CoA and uridine. UDP-GlcNAc is also the precursor for UDP-GalNAc and CMP-SA biosynthesis. Gal, Man, and GlcN salvage/import (green) are facilitated by the hexose transporters (GLUTs), whereas GlcNAc and Fuc are taken up by bulk endocytosis. The sugar nucleotides are in red. Genes in blue are discussed in the text. Metabolite abbreviations: glucose (Glc), galactose (Gal), mannose (Man), glucosamine (GlcN), fucose (Fuc), N-acetylgalactosamine (GalNAc), N-acetylglucosamine (GlcNAc), N-acetylneuraminic acid (NeuNAc), glutamine (Gln).
In mammalian cells, N-acetylglucosamine (GlcNAc) and glucosamine (GlcN) are salvaged into the hexosamine pathway from glycoconjugate turnover by hydrolysis of glycosidic linkages in lysosomes and transported into the cytoplasm by uncharacterized transporter(s), where they are phosphorylated and re-enter the UDP-GlcNAc pool. There are two routes for salvage of GlcN-6P, either acetylation to GlcNAc-6P by glucosamine-6P N-acetyltransferase GNPAT1/Emeg32 (Boehmelt et al., 2000) and UDP-GlcNAc production, or deamination and conversion to fructose-6P by glucosamine-6-phosphate isomerase/oscillin (GNPDA) and entry into glycolysis (Zhang et al., 2003) (Figure 3). Feedback inhibition of GFAT by GlcN-6P with possible activation of GNPDA may convert most GlcN to fructose-6P. In contrast, the product of GlcNAc-6-kinase (NAGK), is transferred directly into UDP-GlcNAc as there appears to be little feedback inhibition beyond GNPAT1 (Grigorian et al., 2007). Thus, GlcNAc is spared from catabolism, efficiently salvaged, and contributes to anabolic pathways of glycoprotein modification in cultured mammalian cells.
UDP-GlcNAc is a precursor to cytidine monophosphate-sialic acid (CMP-SA), the sugar-nucleotide donor utilized by siayltransferases to cap N- and O-glycans (Figure 3). Loss-of-function mutations in the gene encoding UDP-GlcNAc 2-epimerase/ManNAc kinase (GNE/MNK) results in an autosomal recessive neuromuscular disorder in humans and early death in mice (Hinderlich et al., 1997). Supplementation of pregnant mice with ManNAc prolongs the survival of the Gne mutant pups (Galeano et al., 2007), further supporting a role for amino-sugars as therapeutics.
O-GlcNAc transferase (OGT) adds O-GlcNAc to Ser/Thr residues on many cytosolic proteins, and removal by O-GlcNAcase results in rapid turnover (Kreppel and Hart, 1999). OGT overexpression in liver or fat promotes insulin resistance and hyperleptinemia in mice (McClain et al., 2002). O-GlcNAc addition to hepatic FoxO1 promotes the expression of genes for gluconeogenesis (Housley et al., 2008). Similarly, OGT competes at AMPK phosphorylation sites in CRTC2 allowing nuclear translocation and transcription of gluconeogenesis genes (Dentin et al., 2008). Glucose starvation activates AMPK and paradoxically, also up-regulates OGT gene expression in cultured HepG2 hepatocellular carcinoma cells (Taylor et al., 2008). Recently, Drosophila super sex combs (Sxc/OGT) has been shown to be a member of the Polycomb group (PcG), and mutation of Sxc provide strong evidence that O-GlcNAc is required for transcriptional repression of homeobox (HOX) proteins (Gambetta et al., 2009). N-glycan branching regulates glucose homeostasis in whole animal studies (Cheung et al., 2007; Ohtsubo et al., 2005), and further work is required to determine how OGT, N-glycan branching and the hexosamine pathway interact.
N-glycan branching in evolution and development
Phylogenetics provides insight into the role of an ultrasensitive N-glycan branching pathway. Most mature N-glycans in Arabidopsis, C. elegans, and Drosophila are mannose-terminated structures, and compared to mammals, very little branching occurs beyond mono- and a trace of bi-antennary (Zhu et al., 2004). Curiously, β-N-acetylhexosaminidase activity in these species specifically removes most of the GlcNAc residues added by Mgat1, and this blocks further branching (Gutternigg et al., 2007). Considering enzyme affinities for UDP-GlcNAc, the levels of mono-antennary N-glycans may depend largely on competition between Mgat1 and β-hexosaminidase, whereas the supply of UDP-GlcNAc may regulate the formation of bi-anntennary N-glycans by Mgat2 (Km ∼1 mM for Mgat2 and <0.1 mM for Mgat1) (Figure 1). N-glycan branching in C. elegans and Drosophila may be more dependent on developmental regulation of Golgi enzymes and less on UDP-GlcNAc metabolism (Figure 4). Furthermore, Golgi β-N-acetylhexosaminidase activity is not found in mammalian cells. This corresponds with the evolutionary expansion of the branching pathway in vertebrates (which have more Mgat genes) generating higher affinity galectin ligands. As described above, these features are key to differential regulation of glycoprotein with high verses low numbers of N-glycan attachment sites. Most receptor kinases in C. elegans and Drosophila have high NXS/T multiplicity, and interestingly, vertebrates show an expansion of genes encoding receptors with lower multiplicity (Lau et al., 2007). Notably these are TGF-β/BMP, Eph receptors, and some classes of receptor tyrosine kinases. Evolutionary trends in N-glycan branching and NXS/T multiplicity may be due to selection pressures for conditional control of growth and the adaptive immune system in longer-lived animals.
Figure 4. Mgat deficiencies.
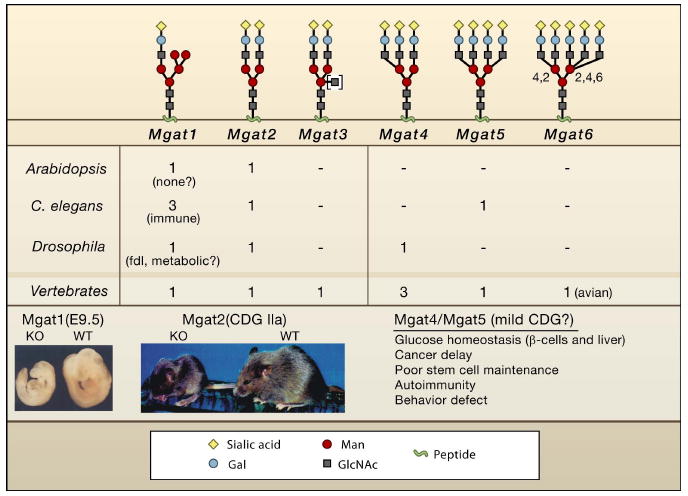
Listed are the number of genes encoding Mgat activities corresponding to the branching pattern shown above. C. elegans triple Mgat1 mutant worms develop normally, but display altered sensitivities to pathogenic bacteria (Shi et al., 2006). Mutation of Mgat1 in Drosophila leads to fused lobes in the brain, and loss of motility in adult flies, possibly reflecting both developmental and metabolic deficiencies (Sarkar et al., 2006). A fused lobes (fdl) phenotype is also observed for loss of β-hexosaminidase, the enzyme that specifically removes GlcNAc added by Mgat1 (Leonard et al., 2006). It is possible that a precise level of Mgat1 product regulates a morphogen gradient that defines the boundary between lobes. Mgat1 deficient embryos are growth impaired and display defects in neural tube closure (Ioffe and Stanley, 1994; Metzler et al., 1994). Mice lacking Mgat2 are small and display severe defects in multiple organs, which is comparable to human CDG-IIa. Reproduced by permission of Oxford University Press: Wang et al. (2001) Glycobiology, 11 (12), 1051-1070. The product of Mgat3 shown in brackets can be found in any of the structures shown, but its presence blocks further branching.
Mouse embryos lacking all N-glycan branches due to Mgat1 deficiency die by E9.5 with defects in growth and morphogenesis (Ioffe and Stanley, 1994; Metzler et al., 1994) (Figure 4). Mgat2 initiates the second antenna and displays an absolute requirement for the prior action of Mgat1 and α-Mannosidase II, consistent with sequential evolution and regulation of the pathway described above. Human Mgat2 deficiency (CDG-IIa) (Tan et al., 1996) and mice lacking Mgat2 (Wang et al., 2001) display similar developmental and postnatal defects. Mice with mutations in genes encoding enzymes further down the branching pathway are viable, but display postnatal defects in the immune system, metabolism, tissue renewal, and delays in cancer development. Mgat4a and Mgat5 both generate galectin ligands, but have opposing effects on systemic glucose homeostasis in gene knockout studies. Mgat4a is required for tri- and tetra- antennary N-glycans in pancreatic β cells, whereas Mgat5 generates tetra-antennary branches and is expressed more widely. Mgat5-deficient mice are resistant to weight gain on an enriched diet, are hypersensitive to fasting, and are mildly hypoglycemic (Cheung et al., 2007). In contrast, mice deficient in Mgat4a display hyperglycemia, obesity, and insulin insufficiency on an enriched diet (Ohtsubo et al., 2005). Mgat4a is required in the modification of the single N-glycan chain on GLUT2, which binds to galectins and promotes cell surface retention. Elevated GLUT2 increases glucose uptake in β cells, acting as a stimulus for insulin secretion at peak serum glucose. Insulin responses appear normal in Mgat5 deficient mice, but changes in GLUT4 surface retention in muscle and fat coupled with impaired glucagon responses in liver may contribute to the lean phenotype (Cheung et al., 2007). Surface expression of glucagon receptor and cAMP signaling are deficient in hepatocytes from Mgat5-/- mice, which may contribute to these phenotypes. Resistance to weight-gain in Mgat5 deficient mice is rescued by supplementing their drinking water with GlcNAc but not GlcN (A. Johswich, M. Ryczko, J. Dennis, unpublished data).
Muscle satellite cells and bone marrow cells from Mgat5 deficient mice display an imbalance favors Smad2/3 to Erk signaling, accompanied by an increased ratio of differentiation to renewal. In vivo, bone marrow osteoprogenitors and muscle satellite cells are depleted and the normal aging processes in bone and muscle of Mgat5 deficient mice is accelerated (Cheung et al., 2007). Stem cell maintenance requires trophic factors and surface levels of cognate receptors, which both decline with normal aging (Conboy et al., 2005; Shiraha et al., 2000). It is possible that intervention directed at hexosamine and other metabolic pathways might support the “youthful” distributions of surface glycoproteins that more effectively support stem cell renewal. Profiling or proteomics of the cell surface by mass spectrometry has the potential to reveal critical changes in receptor and transporters (Wollscheid et al., 2009). The Klotho gene was identified as a senescence-suppressor, and encoding a putative α2,6 sialidase that enhances N-glycan affinity for galectin-1 and protects glycoproteins from coated-pit endocytosis. Loss of Klotho results in increased retention of the surface renal Ca2+ and K+ channels TRPV5 and ROMK1, each with one available NXS/T site (Cha et al., 2009; Cha et al., 2008). Adaptation of the cell surface by metabolic flux to Golgi N-glycosylation in youth may buffer against environmental stresses and genetic variation, but decline with aging due to a shift from glycolytic to oxidative metabolism (Petersen et al., 2003). Thus life history and genetic background may interact through N-glycan-dependent regulation of glycoproteins to reveal late-onset diseases such as autoimmunity, metabolic disease, and cancer.
Congenital Disorders of Glycosylation
Congenital disorders of glycosylation (CDG) are hypomorphic defects in N-glycan biosynthesis, and provide insight into the functions of N-glycans in humans. Type I CDGs are deficiencies in various enzymes in the biosynthetic pathway for Glc3Man9GlcNAc2-pp-dolichol, the donor for N-glycan addition. As a result, in CDG usage of NXS/T sites is reduced (reviewed in (Jaeken and Matthijs, 2007) (Figure 2).
CDG-Ib is a deficiency in phosphomannose isomerase (PMI, Figure 3) that decreases the supply of guanosine diphosphate mannose (GDP-Man) for dolichol-pp-oligosaccharide synthesis. Unlike most CDGs, the CDG-Ib defect can be treated with a monosaccharide supplement that alleviates the chronic postnatal phenotypes (Freeze, 1998). Failure to thrive, coagulopathies, protein-losing enteropathy, and liver fibrosis observed in CDG-Ib are improved by supplementing the diet with mannose, which bypasses the defective step (conversion of Fru-6P to Man-6P) and increases Man-6P and GDP-Man by the action of hexokinase. Glucose deprivation in cultured cells causes reduced glycosylation of sites in EGFR and altered distribution at the plasma membrane (Konishi and Berk, 2003). Low glucose conditions impairs Glc3Man9GlcNAc2 –pp-dol biosynthesis giving rise to smaller intermediates and incomplete N-glycosylation. The resulting unfolded protein response activates PERK, which inhibits general translation by phosphorylating the translation initiation factor eIF2α, and this allows Glc3Man9GlcNAc2 –pp-dol and N-glycosylation levels to recover (Shang et al., 2007). Fibroblast from patients with type I CDG displayed similar but moderate induction of PERK kinase, suggesting that ER and metabolic stress may be a common feature of CDG-I (Lecca et al., 2005).
Subjects with defects in the Golgi and secretory pathway are classified as having type II CDG, which is characterized by reduced branching or extension to the N-glycans (Figure 2). The clinical phenotypes of type I and II CDG are often similar. In addition to those clinical features noted above, features of type II CDG include psychomotor retardation, ataxia, seizures, retinopathy, and dysmorphic features. These deficiencies arise from loss-of-function mutations in genes related to remodeling; notably Mgat2 (CDG-IIa), α-glucosidase I (CDG-IIb), GDP-Fucose Golgi transporter (CDG-IIc), and β1,4-galactosyltransferase (CDG-IId) (Jaeken and Matthijs, 2007). Leukocyte adhesion deficiency type II (LAD II or CDG-IId), is caused by a hypomorphic mutation in the Golgi GDP-Fuc transporter that reduces cell surface fucosylated glycans and patients suffer recurrent infections due to a deficiency in selectin ligands. The fucose salvage pathway has the capacity to generate very high concentrations of GDP-Fuc, sufficient to overcome a partial defect in Golgi GDP-fucose transport in patients (Marquardt et al., 1999) (Figure 3). Similarly, fucose fed to mice deficient in GDP-keto-4-keto-6-deoxymannose 3,5-epimerase-4-reductase (FX), restores fucose content in glycoproteins (Smith et al., 2002). Although the developmental defects associated with CDG are irreversible, postnatal defects can in some cases be rescued by dietary supplements to sugar-nucleotide pools. The similarities in CDG clinical phenotypes suggest that NXS/T site usage (CDG type I) and Golgi N-glycan remodeling (CDG type II) are largely convergent for glycoprotein localization at the cell surface and in matrix.
Glycosylation defects in complex human diseases
Metabolism is embedded with multiple feedback mechanisms, but homeostatic set-points for glucose and fatty acid metabolism change with aging in manner that is poorly understood (Petersen et al., 2003). Autoimmune disease and sporadic cancers are examples of diseases with stochastic etiologies caused by one or a few cells expanding in a pathological manner (Dennis et al., 2002). Therefore, small changes in homeostasis can be clinically important in large cell populations, given sufficient time and external stresses.
CTLA-4 has low N-glycan multiplicity, is subject to high rates of constitutive endocytosis (Alegre et al., 2001), and requires the galectin lattice to promote T cell growth arrest (Lau et al., 2007). A common polymorphism in CTLA-4 (T17A) reduces N-glycan occupancy at the two NXS/T sites by ∼50%, and diminishes surface expression (Anjos et al., 2002). Although CTLA-4-deficient mice display widespread autoimmunity (Waterhouse et al., 1995), CTLA-4(Ala17) alone contributes little risk to multiple sclerosis in humans. However, the addition of fewer N-glycans can be viewed as a selective “CDG type I deficiency” with the potential to promote disease in combination with Golgi polymorphisms (“mild CDG type II deficiency”). Indeed, polymorphisms in human Mgat1 alter branching and interact with CTLA-4(Ala17), and coinheritance cooperatively suppresses CTLA-4 surface expression and synergistically increases the risk of multiple sclerosis, with neither variant promoting disease in the absence of the other (H.-L. Chen, J. Dennis, M. Demetriou unpublished data). Importantly, both TCR sensitivity and defects in CTLA-4 surface expression are conditional on UDP-GlcNAc supply to the N-glycan branching pathway and rescued by supplementation with GlcNAc. Dietary supplements of GlcNAc also suppress autoimmune diabetes in non-obese diabetic mice (Grigorian et al., 2007), and chronic inflammatory bowel disease in children (Salvatore et al., 2000).
Interestingly, several mouse strains, including PL/J mice, are prone to autoimmunity and are naturally hypomorphic for GlcNAc branching, with deficiencies in Mgat1, Mgat2 and Mgat5 enzyme activities (Lee et al., 2007). PL/J mice are also metabolic outliers among mouse strains displaying features similar to Mgat5-deficient mice on a 129 or C57B6 background, notably small size, resistance to a high fat diet, low respiratory quotient, and shorter lifespan. In addition to genetics, nutrient conditions during embryogenesis are known to affect skeletal development as well as metabolism later in life. A poor nutrient environment in utero appears to adapt the metabolic circuitry for harsh conditions in postnatal life (Gluckman and Hanson, 2006). Poor human prenatal nutrition coupled with the high-calorie diet readily available in industrialized nations may predispose to high body mass and diabetes (Bateson et al., 2004). Perhaps nutrient supply to the fetus or newborn influences the homeostatic set-point for hexosamine/Golgi/lattice and qualitative aspects of extracellular matrix, acting as a metabolic and self-organizing regulatory network in post-natal life.
Emerging evidence also links N-glycan branching to specific functions in the nervous system. Mgat5 is also highly expressed in the central nervous system (Granovsky et al., 1995), and mice deficient in Mgat5 display resistance to depression-like behavior in response to stress (Soleimani et al., 2007). In most sporadic amyotrophic lateral sclerosis (ALS) patients, EAAT2 protein and transport activity are reduced by 30-90%, which is not readily explained by mRNA levels. However, an allele of EAAT2 with a point mutation that removes one of two N-glycosylation sites suggests surface expression is dependent on N-glycans, possibly because of retention by lectins (Aoki et al., 1998; Trotti et al., 2001). Oxygen radical stress is a prominent feature of ALS that may also disrupt the lattice by oxidation of galectins.
Concluding remarks
Kuriyan and Eisenberg (2007) argue that an early driving force in natural selection was low-affinity colocalization of proteins that enhance mass action, and subsequent evolution of many variations on high affinity and allosteric control of protein networks. Structurally, the galectin lattice is a force for localization and organization of surface glycoproteins, but in an evolutionary sense, lectin-carbohydrate lattices may be tolerant to variation in structural details, and still maintain essential parameters of the regulatory network. Lectins bind with low monomeric affinities, but multivalency allows a very wide range of affinities, and provides flexibility for evolutionary insertion of new glycan structures. From a chemist's view, the N-glycan pathway might appear to be a very imprecise process, essentially imposing “entropy” to the interface of cells with their environment. However, in biological systems, variance is tolerated and often serves a feedback regulatory role at the network level (Coffey et al., 1998; Maamar et al., 2007; Radman, 2001). Defining all glycoforms in time and space is obviously an unattainable goal, and not likely to make a meaningful contribution to our understanding of cell behavior. Rather, glycoforms can be classified in terms of their affinity for the animal lectins and as part of a larger network that regulates membrane organization.
Finally, gene-gene and gene-environmental interactions underpin many complex trait diseases. As such, genome-wide screens in complex diseases such as type 2 diabetes, autoimmunity and cardiovascular disease, generally find individual single nucleotide polymorphisms that account for very modest risk (<1.5) and therefore only a small fraction of the total genetic risk for the disease in question (Manolio et al., 2009). Cell surface glycoproteins regulate highly adaptive systems in animals, and their regulation in a conditional manner should permit heritable phenotypic variation, evolutionary fitness, and opportunities for radiation. However, greater adaptive potential may also come with greater risk of polygenic diseases and proliferative disorders such as cancer and autoimmune disease. We suggest that genetic variations in hexosamine/N-glycan pathways and substrate glycoproteins interact with each other and with life-history changes in metabolism to account for a large portion of “missing” risk for many chronic human diseases.
Acknowledgments
The author's research is supported by grants from CIHR (J.W.D. and I.R.N.), from Genome Canada through the OGI (J.W.D.), and from NIH and NMSS (M.D.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ahmad N, Gabius HJ, Andre S, Kaltner H, Sabesan S, Roy R, Liu B, Macaluso F, Brewer CF. Galectin-3 precipitates as a pentamer with synthetic multivalent carbohydrates and forms heterogeneous cross-linked complexes. Journal of Biological Chemistry. 2003;279:10841–10847. doi: 10.1074/jbc.M312834200. [DOI] [PubMed] [Google Scholar]
- Alegre ML, Frauwirth KA, Thompson CB. T-cell regulation by CD28 and CTLA-4. Nat Rev Immunol. 2001;1:220–228. doi: 10.1038/35105024. [DOI] [PubMed] [Google Scholar]
- Amano M, Galvan M, He J, Baum LG. The ST6Gal I sialyltransferase selectively modifies N-glycans on CD45 to negatively regulate galectin-1-induced CD45 clustering, phosphatase modulation, and T cell death. J Biol Chem. 2003;278:7469–7475. doi: 10.1074/jbc.M209595200. [DOI] [PubMed] [Google Scholar]
- Anjos S, Nguyen A, Ounissi-Benkalha H, Tessier MC, Polychronakos C. A common autoimmunity predisposing signal peptide variant of the cytotoxic T-lymphocyte antigen 4 results in inefficient glycosylation of the susceptibility allele. J Biol Chem. 2002;277:46478–46486. doi: 10.1074/jbc.M206894200. [DOI] [PubMed] [Google Scholar]
- Aoki M, Lin CL, Rothstein JD, Geller BA, Hosler BA, Munsat TL, Horvitz HR, Brown RH., Jr Mutations in the glutamate transporter EAAT2 gene do not cause abnormal EAAT2 transcripts in amyotrophic lateral sclerosis. Ann Neurol. 1998;43:645–653. doi: 10.1002/ana.410430514. [DOI] [PubMed] [Google Scholar]
- Apweiler R, Hermjakob H, Sharon N. On the frequency of protein glycosylation, as deduced from analysis of the SWISS-PROT database. Biochimica et Biophysica Acta. 1999;1473:4–8. doi: 10.1016/s0304-4165(99)00165-8. [DOI] [PubMed] [Google Scholar]
- Bailey JE. Mathematical modeling and analysis in biochemical engineering: past accomplishments and future opportunities. Biotechnol Prog. 1998;14:8–20. doi: 10.1021/bp9701269. [DOI] [PubMed] [Google Scholar]
- Bateson P, Barker D, Clutton-Brock T, Deb D, D'Udine B, Foley RA, Gluckman P, Godfrey K, Kirkwood T, Lahr MM, et al. Developmental plasticity and human health. Nature. 2004;430:419–421. doi: 10.1038/nature02725. [DOI] [PubMed] [Google Scholar]
- Bilder D. Epithelial polarity and proliferation control: links from the Drosophila neoplastic tumor suppressors. Genes Dev. 2004;18:1909–1925. doi: 10.1101/gad.1211604. [DOI] [PubMed] [Google Scholar]
- Boehmelt G, Fialka I, Brothers G, McGinley MD, Patterson SD, Mo R, Hui CC, Chung S, Huber LA, Mak TW, Iscove NN. Cloning and characterization of the murine glucosamine-6-phosphate acetyltransferase EMeg32. J Biol Chem. 2000;275:12821–12832. doi: 10.1074/jbc.275.17.12821. [DOI] [PubMed] [Google Scholar]
- Braccia A, Villani M, Immerdal L, Niels-Christiansen LL, Nystrom BT, Hansen GH, Danielsen EM. Microvillar membrane microdomains exist at physiological temperature. Role of galectin-4 as lipid raft stabilizer revealed by “superrafts”. J Biol Chem. 2003;278:15679–15684. doi: 10.1074/jbc.M211228200. [DOI] [PubMed] [Google Scholar]
- Brewer CF, Miceli MC, Baum LG. Clusters, bundles, arrays and lattices: novel mechanisms for lectin-saccharide-mediated cellular interactions. Curr Opin Struct Biol. 2002;12:616–623. doi: 10.1016/s0959-440x(02)00364-0. [DOI] [PubMed] [Google Scholar]
- Broschat KO, Gorka C, Page JD, Martin-Berger CL, Davies MS, Huang Hc HC, Gulve EA, Salsgiver WJ, Kasten TP. Kinetic characterization of human glutamine-fructose-6-phosphate amidotransferase I: potent feedback inhibition by glucosamine 6-phosphate. The Journal of biological chemistry. 2002;277:14764–14770. doi: 10.1074/jbc.M201056200. [DOI] [PubMed] [Google Scholar]
- Buckhaults P, Chen L, Fregien N, Pierce M. Transcriptional regulation of N-acetylglucosaminyltransferase V by the src Oncogene. Journal of Biological Chemistry. 1997;272:19575–19581. doi: 10.1074/jbc.272.31.19575. [DOI] [PubMed] [Google Scholar]
- Buse MG, Robinson KA, Marshall BA, Mueckler M. Differential effects of GLUT1 or GLUT4 overexpression on hexosamine biosynthesis by muscles of transgenic mice. The Journal of biological chemistry. 1996;271:23197–23202. doi: 10.1074/jbc.271.38.23197. [DOI] [PubMed] [Google Scholar]
- Cha SK, Hu MC, Kurosu H, Kuro-o M, Moe O, Huang CL. Regulation of renal outer medullary potassium channel and renal K(+) excretion by Klotho. Mol Pharmacol. 2009;76:38–46. doi: 10.1124/mol.109.055780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cha SK, Ortega B, Kurosu H, Rosenblatt KP, Kuro OM, Huang CL. Removal of sialic acid involving Klotho causes cell-surface retention of TRPV5 channel via binding to galectin-1. Proc Natl Acad Sci U S A. 2008;105:9805–9810. doi: 10.1073/pnas.0803223105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen IJ, Chen HL, Demetriou M. Lateral compartmentalization of T cell receptor versus CD45 by galectin-N-glycan binding and microfilaments coordinate basal and activation signaling. J Biol Chem. 2007;282:35361–35372. doi: 10.1074/jbc.M706923200. [DOI] [PubMed] [Google Scholar]
- Cheung P, Pawling J, Partridge EA, Sukhu B, Grynpas M, Dennis JW. Metabolic homeostasis and tissue renewal are dependent on beta1,6GlcNAc-branched N-glycans. Glycobiology. 2007;17:828–837. doi: 10.1093/glycob/cwm048. [DOI] [PubMed] [Google Scholar]
- Chikuma S, Bluestone JA. CTLA-4: Acting at the Synapse. Mol Interv. 2002;2:205–208. doi: 10.1124/mi.2.4.205. [DOI] [PubMed] [Google Scholar]
- Coffey MC, Strong JE, Forsyth PA, Lee PWK. Reovirus therapy of tumors with activated ras pathway. Science. 1998;282:1332–1334. doi: 10.1126/science.282.5392.1332. [DOI] [PubMed] [Google Scholar]
- Conboy IM, Conboy MJ, Wagers AJ, Girma ER, Weissman IL, Rando TA. Rejuvenation of aged progenitor cells by exposure to a young systemic environment. Nature. 2005;433:760–764. doi: 10.1038/nature03260. [DOI] [PubMed] [Google Scholar]
- Coster AC, Govers R, James DE. Insulin stimulates the entry of GLUT4 into the endosomal recycling pathway by a quantal mechanism. Traffic. 2004;5:763–771. doi: 10.1111/j.1600-0854.2004.00218.x. [DOI] [PubMed] [Google Scholar]
- Curto M, Cole BK, Lallemand D, Liu CH, McClatchey AI. Contact-dependent inhibition of EGFR signaling by Nf2/Merlin. J Cell Biol. 2007;177:893–903. doi: 10.1083/jcb.200703010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeBerardinis RJ, Lum JJ, Hatzivassiliou G, Thompson CB. The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 2008;7:11–20. doi: 10.1016/j.cmet.2007.10.002. [DOI] [PubMed] [Google Scholar]
- Delacour D, Greb C, Koch A, Salomonsson E, Leffler H, Le Bivic A, Jacob R. Apical sorting by galectin-3-dependent glycoprotein clustering. Traffic. 2007;8:379–388. doi: 10.1111/j.1600-0854.2007.00539.x. [DOI] [PubMed] [Google Scholar]
- Demetriou M, Granovsky M, Quaggin S, Dennis JW. Negative regulation of T-cell activation and autoimmunity by Mgat5 N-glycosylation. Nature. 2001;409:733–739. doi: 10.1038/35055582. [DOI] [PubMed] [Google Scholar]
- Demotte N, Stroobant V, Courtoy PJ, Van Der Smissen P, Colau D, Luescher IF, Hivroz C, Nicaise J, Squifflet JL, Mourad M, et al. Restoring the association of the T cell receptor with CD8 reverses anergy in human tumor-infiltrating lymphocytes. Immunity. 2008;28:414–424. doi: 10.1016/j.immuni.2008.01.011. [DOI] [PubMed] [Google Scholar]
- Dennis JW, Laferte S, Waghorne C, Breitman ML, Kerbel RS. Beta 1-6 branching of Asn-linked oligosaccharides is directly associated with metastasis. Science. 1987;236:582–585. doi: 10.1126/science.2953071. [DOI] [PubMed] [Google Scholar]
- Dennis JW, Lau KS, Demetriou M, Nabi IR. Adaptive Regulation at the Cell Surface by N-Glycosylation. Traffic. 2009;11:1569–1578. doi: 10.1111/j.1600-0854.2009.00981.x. [DOI] [PubMed] [Google Scholar]
- Dennis JW, Pawling J, Cheung P, Partridge EA, Demetriou M. UDP-N-acetylglucosamine:-6-D-mannoside 1,6 N-acetylglucosaminyltransferase V (Mgat5) deficient mice. Biochimica et Biophysica Acta. 2002;1573:414–422. doi: 10.1016/s0304-4165(02)00411-7. [DOI] [PubMed] [Google Scholar]
- Dentin R, Hedrick S, Xie J, Yates J, 3rd, Montminy M. Hepatic glucose sensing via the CREB coactivator CRTC2. Science. 2008;319:1402–1405. doi: 10.1126/science.1151363. [DOI] [PubMed] [Google Scholar]
- Do KY, Fregien N, Pierce M, Cummings RD. Modification of glycoproteins by N-acetylglucosaminyltransferase V is greatly influenced by accessibility of the enzyme to oligosacharide acceptors. Journal of Biological Chemistry. 1994;269:23456–23464. [PubMed] [Google Scholar]
- Drickamer K, Fadden AJ. Genomic analysis of C-type lectins. Biochemistry Society Symposium. 2002;69:59–72. doi: 10.1042/bss0690059. [DOI] [PubMed] [Google Scholar]
- Fernandes H, Cohen S, Bishayee S. Glycosylation-induced conformational modification positively regulates receptor-receptor association: a study with an aberrant epidermal growth factor receptor (EGFRvIII/DeltaEGFR) expressed in cancer cells. J Biol Chem. 2001;276:5375–5383. doi: 10.1074/jbc.M005599200. [DOI] [PubMed] [Google Scholar]
- Ferrell JE., Jr Tripping the switch fantastic: how a protein kinase cascade can convert graded inputs into switch-like outputs. Trends Biochem Sci. 1996;21:460–466. doi: 10.1016/s0968-0004(96)20026-x. [DOI] [PubMed] [Google Scholar]
- Frauwirth KA, Riley JL, Harris MH, Parry RV, Rathmell JC, Plas DR, Elstrom RL, June CH, Thompson CB. The CD28 signaling pathway regulates glucose metabolism. Immunity. 2002;16:769–777. doi: 10.1016/s1074-7613(02)00323-0. [DOI] [PubMed] [Google Scholar]
- Freeze HH. Disorders in protein glycosylation and potential therapy: Tip of an iceberg? The Journal of pediatrics. 1998;133:1–8. doi: 10.1016/s0022-3476(98)70096-4. [DOI] [PubMed] [Google Scholar]
- Galeano B, Klootwijk R, Manoli I, Sun M, Ciccone C, Darvish D, Starost MF, Zerfas PM, Hoffmann VJ, Hoogstraten-Miller S, et al. Mutation in the key enzyme of sialic acid biosynthesis causes severe glomerular proteinuria and is rescued by N-acetylmannosamine. J Clin Invest. 2007;117:1585–1594. doi: 10.1172/JCI30954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gambetta MC, Oktaba K, Muller J. Essential role of the glycosyltransferase sxc/Ogt in polycomb repression. Science. 2009;325:93–96. doi: 10.1126/science.1169727. [DOI] [PubMed] [Google Scholar]
- Gluckman PD, Hanson MA. The consequences of being born small - an adaptive perspective. Horm Res. 2006;65 3:5–14. doi: 10.1159/000091500. [DOI] [PubMed] [Google Scholar]
- Granovsky M, Fode C, Warren CE, Campbell RM, Marth JD, Pierce M, Fregien N, Dennis JW. GlcNAc-transferase V and core 2 GlcNAc-transferase expression in the developing mouse embryo. Glycobiology. 1995;5:797–806. doi: 10.1093/glycob/5.8.797. [DOI] [PubMed] [Google Scholar]
- Grigorian A, Lee SU, Tian W, Chen IJ, Gao G, Mendelsohn R, Dennis JW, Demetriou M. Control of T cell mediated autoimmunity by metabolite flux to N-glycan biosynthesis. J Biol Chem. 2007;282:20027–20035. doi: 10.1074/jbc.M701890200. [DOI] [PubMed] [Google Scholar]
- Gurdon JB, Bourillot PY. Morphogen gradient interpretation. Nature. 2001;413:797–803. doi: 10.1038/35101500. [DOI] [PubMed] [Google Scholar]
- Gutternigg M, Kretschmer-Lubich D, Paschinger K, Rendic D, Hader J, Geier P, Ranftl R, Jantsch V, Lochnit G, Wilson IB. Biosynthesis of truncated N-linked oligosaccharides results from non-orthologous hexosaminidase-mediated mechanisms in nematodes, plants, and insects. J Biol Chem. 2007;282:27825–27840. doi: 10.1074/jbc.M704235200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helenius A, Aebi M. Roles of N-linked glycans in the endoplasmic reticulum. Annu Rev Biochem. 2004;73:1019–1049. doi: 10.1146/annurev.biochem.73.011303.073752. [DOI] [PubMed] [Google Scholar]
- Hinderlich S, Stasche R, Zeitler R, Reutter W. A bifunctional enzyme catalyzes the first two steps in N-acetylneuraminic acid biosynthesis of rat liver. Purification and characterization of UDP-N-acetylglucosamine 2-epimerase/N-acetylmannosamine kinase. J Biol Chem. 1997;272:24313–24318. doi: 10.1074/jbc.272.39.24313. [DOI] [PubMed] [Google Scholar]
- Hirabayashi J, Hashidate T, Arata Y, Nishi N, Nakamura T, Hirashima M, Urashima T, Oka T, Futai M, Muller WE, et al. Oligosaccharide specificity of galectins: a search by frontal affinity chromatography. Biochimica et Biophysica Acta. 2002;1572:232–254. doi: 10.1016/s0304-4165(02)00311-2. [DOI] [PubMed] [Google Scholar]
- Housley MP, Rodgers JT, Udeshi ND, Kelly TJ, Shabanowitz J, Hunt DF, Puigserver P, Hart GW. O-GlcNAc regulates FoxO activation in response to glucose. J Biol Chem. 2008;283:16283–16292. doi: 10.1074/jbc.M802240200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes RC. Secretion of the galectin family of mammalian carbohydrate-binding proteins. Biochimica et Biophysica Acta. 1999;1473:172–185. doi: 10.1016/s0304-4165(99)00177-4. [DOI] [PubMed] [Google Scholar]
- Igaki T, Pastor-Pareja JC, Aonuma H, Miura M, Xu T. Intrinsic tumor suppression and epithelial maintenance by endocytic activation of Eiger/TNF signaling in Drosophila. Dev Cell. 2009;16:458–465. doi: 10.1016/j.devcel.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ioffe E, Stanley P. Mice lacking N-acetylglucosaminyltransferase I activity die at mid-gestation, revealing an essential role for complex or hybrid N-linked carbohydrates. Proceedings of the National Academy of Sciences of the United States of America. 1994;91:728–732. doi: 10.1073/pnas.91.2.728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishida H, Togayachi A, Sakai T, Iwai T, Hiruma T, Sato T, Okubo R, Inaba N, Kudo T, Gotoh M, et al. A novel beta1,3-N-acetylglucosaminyltransferase (beta3Gn-T8), which synthesizes poly-N-acetyllactosamine, is dramatically upregulated in colon cancer. FEBS Lett. 2005;579:71–78. doi: 10.1016/j.febslet.2004.11.037. [DOI] [PubMed] [Google Scholar]
- Jaeken J, Matthijs G. Congenital disorders of glycosylation: a rapidly expanding disease family. Annu Rev Genomics Hum Genet. 2007;8:261–278. doi: 10.1146/annurev.genom.8.080706.092327. [DOI] [PubMed] [Google Scholar]
- Jones J, Krag SS, Betenbaugh MJ. Controlling N-linked glycan site occupancy. Biochim Biophys Acta. 2005;1726:121–137. doi: 10.1016/j.bbagen.2005.07.003. [DOI] [PubMed] [Google Scholar]
- Klein P, Mattoon D, Lemmon MA, Schlessinger J. A structure-based model for ligand binding and dimerization of EGF receptors. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:929–934. doi: 10.1073/pnas.0307285101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolm-Litty V, Sauer U, Nerlich A, Lehmann R, Schleicher ED. High glucose-induced transforming growth factor beta1 production is mediated by the hexosamine pathway in porcine glomerular mesangial cells. The Journal of clinical investigation. 1998;101:160–169. doi: 10.1172/JCI119875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konishi A, Berk BC. Epidermal growth factor receptor transactivation is regulated by glucose in vascular smooth muscle cells. J Biol Chem. 2003;278:35049–35056. doi: 10.1074/jbc.M304913200. [DOI] [PubMed] [Google Scholar]
- Kreppel LK, Hart GW. Regulation of a cytosolic and nuclear O-GlcNAc transferase. Role of the tetratricopeptide repeats. J Biol Chem. 1999;274:32015–32022. doi: 10.1074/jbc.274.45.32015. [DOI] [PubMed] [Google Scholar]
- Kuball J, Hauptrock B, Malina V, Antunes E, Voss RH, Wolfl M, Strong R, Theobald M, Greenberg PD. Increasing functional avidity of TCR-redirected T cells by removing defined N-glycosylation sites in the TCR constant domain. J Exp Med. 2009;206:463–475. doi: 10.1084/jem.20082487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuriyan J, Eisenberg D. The origin of protein interactions and allostery in colocalization. Nature. 2007;450:983–990. doi: 10.1038/nature06524. [DOI] [PubMed] [Google Scholar]
- Lagana A, Goetz JG, Cheung P, Raz A, Dennis JW, Nabi IR. Galectin binding to Mgat5-modified N-glycans regulates fibronectin matrix remodeling in tumor cells. Mol Cell Biol. 2006;26:3181–3193. doi: 10.1128/MCB.26.8.3181-3193.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lajoie P, Partridge EA, Guay GSN, Goetz JG, Pawling J, Lagana A, Dennis JW, Nabi IR. Plasma membrane domain organization regulates EGFR signaling in tumor cells. J Cell Biol. 2007;179:341–356. doi: 10.1083/jcb.200611106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanzetti L, Palamidessi A, Areces L, Scita G, Di Fiore PP. Rab5 is a signalling GTPase involved in actin remodelling by receptor tyrosine kinases. Nature. 2004;429:309–314. doi: 10.1038/nature02542. [DOI] [PubMed] [Google Scholar]
- Lau K, Partridge EA, Silvescu CI, Grigorian A, Pawling J, Reinhold VN, Demetriou M, Dennis JW. Complex N-glycan number and degree of branching cooperate to regulate cell proliferation and differentiation. Cell. 2007;129:123–124. doi: 10.1016/j.cell.2007.01.049. [DOI] [PubMed] [Google Scholar]
- Lau KS, Dennis JW. N-Glycans in cancer progression. Glycobiology. 2008;18:750–760. doi: 10.1093/glycob/cwn071. [DOI] [PubMed] [Google Scholar]
- Lecca MR, Wagner U, Patrignani A, Berger EG, Hennet T. Genome-wide analysis of the unfolded protein response in fibroblasts from congenital disorders of glycosylation type-I patients. Faseb J. 2005;19:240–242. doi: 10.1096/fj.04-2397fje. [DOI] [PubMed] [Google Scholar]
- Lee H, Park DS, Razani B, Russell RG, Pestell RG, Lisanti MP. Caveolin-1 mutations (P132L and null) and the pathogenesis of breast cancer: caveolin-1 (P132L) behaves in a dominant-negative manner and caveolin-1 (-/-) null mice show mammary epithelial cell hyperplasia. Am J Pathol. 2002;161:1357–1369. doi: 10.1016/S0002-9440(10)64412-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee RT, Lee YC. Affinity enhancement by multivalent lectin-carbohydrate interaction. Glycoconj J. 2000;17:543–551. doi: 10.1023/a:1011070425430. [DOI] [PubMed] [Google Scholar]
- Lee SU, Grigorian A, Pawling J, Chen IJ, Gao G, Mozaffar T, McKerlie C, Demetriou M. N-glycan processing deficiency promotes spontaneous inflammatory demyelination and neurodegeneration. J Biol Chem. 2007;282:33725–33734. doi: 10.1074/jbc.M704839200. [DOI] [PubMed] [Google Scholar]
- Leonard R, Rendic D, Rabouille C, Wilson IB, Preat T, Altmann F. The Drosophila fused lobes gene encodes an N-acetylglucosaminidase involved in N-glycan processing. J Biol Chem. 2006;281:4867–4875. doi: 10.1074/jbc.M511023200. [DOI] [PubMed] [Google Scholar]
- Levy Y, Arbel-Goren R, Hadari YR, Eshhar S, Ronen D, Elhanany E, Geiger B, Zick Y. Galectin-8 functions as a matricellular modulator of cell adhesion. Journal of Biological Chemistry. 2001;276:31285–31295. doi: 10.1074/jbc.M100340200. [DOI] [PubMed] [Google Scholar]
- Maamar H, Raj A, Dubnau D. Noise in gene expression determines cell fate in Bacillus subtilis. Science. 2007;317:526–529. doi: 10.1126/science.1140818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manolio TA, Collins FS, Cox NJ, Goldstein DB, Hindorff LA, Hunter DJ, McCarthy MI, Ramos EM, Cardon LR, Chakravarti A, et al. Finding the missing heritability of complex diseases. Nature. 2009;461:747–753. doi: 10.1038/nature08494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marquardt T, Luhn K, Srikrishna G, Freeze HH, Harms E, Vestweber D. Correction of leukocyte adhesion deficiency type II with oral fucose. Blood. 1999;94:3976–3985. [PubMed] [Google Scholar]
- Matsuura I, Denissova NG, Wang G, He D, Long J, Liu F. Cyclin-dependent kinases regulate the antiproliferative function of Smads. Nature. 2004;430:226–231. doi: 10.1038/nature02650. [DOI] [PubMed] [Google Scholar]
- McClain DA, Lubas WA, Cooksey RC, Hazel M, Parker GJ, Love DC, Hanover JA. Altered glycan-dependent signaling induces insulin resistance and hyperleptinemia. Proc Natl Acad Sci U S A. 2002;99:10695–10699. doi: 10.1073/pnas.152346899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendelsohn R, Cheung P, Berger L, Partridge EA, Lau K, Pawling J, Dennis JW. Control of tumor metabolism and growth by N-glycan processing. Cancer Res. 2007;67:9771–9780. doi: 10.1158/0008-5472.CAN-06-4580. [DOI] [PubMed] [Google Scholar]
- Metzler M, Gertz A, Sarkar M, Schachter H, Schrader JW, Marth JD. Complex asparagine-linked oligosaccharides are required for morphogenic events during post-implantation development. The EMBO Journal. 1994;13:2056–2065. doi: 10.1002/j.1460-2075.1994.tb06480.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moro L, Dolce L, Cabodi S, Bergatto E, Erba EB, Smeriglio M, Turco E, Retta SF, Giuffrida MG, Venturino M, et al. Integrin-induced epidermal growth factor (EGF) receptor activation requires c-Src and p130Cas and leads to phosphorylation of specific EGF receptor tyrosines. Journal of Biological Chemistry. 2002;277:9405–9414. doi: 10.1074/jbc.M109101200. [DOI] [PubMed] [Google Scholar]
- Morrish F, Isern N, Sadilek M, Jeffrey M, Hockenbery DM. c-Myc activates multiple metabolic networks to generate substrates for cell-cycle entry. Oncogene. 2009;28:2485–2491. doi: 10.1038/onc.2009.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosesson Y, Mills GB, Yarden Y. Derailed endocytosis: an emerging feature of cancer. Nat Rev Cancer. 2008;8:835–850. doi: 10.1038/nrc2521. [DOI] [PubMed] [Google Scholar]
- Ohtsubo K, Takamatsu S, Minowa MT, Yoshida A, Takeuchi M, Marth JD. Dietary and genetic control of glucose transporter 2 glycosylation promotes insulin secretion in suppressing diabetes. Cell. 2005;123:1307–1321. doi: 10.1016/j.cell.2005.09.041. [DOI] [PubMed] [Google Scholar]
- Okamoto T, Schlegel A, Scherer PE, Lisanti MP. Caveolins, a family of scaffolding proteins for organizing “preassembled signaling complexes” at the plasma membrane. J Biol Chem. 1998;273:5419–5422. doi: 10.1074/jbc.273.10.5419. [DOI] [PubMed] [Google Scholar]
- Partridge EA, Le Roy C, Di Guglielmo GM, Pawling J, Cheung P, Granovsky M, Nabi IR, Wrana JL, Dennis JW. Regulation of cytokine receptors by Golgi N-glycan processing and endocytosis. Science. 2004;306:120–124. doi: 10.1126/science.1102109. [DOI] [PubMed] [Google Scholar]
- Patnaik SK, Potvin B, Carlsson S, Sturm D, Leffler H, Stanley P. Complex N-glycans are the major ligands for galectin-1, -3, and -8 on Chinese hamster ovary cells. Glycobiology. 2006;16:305–317. doi: 10.1093/glycob/cwj063. [DOI] [PubMed] [Google Scholar]
- Petersen KF, Befroy D, Dufour S, Dziura J, Ariyan C, Rothman DL, DiPietro L, Cline GW, Shulman GI. Mitochondrial dysfunction in the elderly: possible role in insulin resistance. Science. 2003;300:1140–1142. doi: 10.1126/science.1082889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piccirillo SG, Reynolds BA, Zanetti N, Lamorte G, Binda E, Broggi G, Brem H, Olivi A, Dimeco F, Vescovi AL. Bone morphogenetic proteins inhibit the tumorigenic potential of human brain tumour-initiating cells. Nature. 2006;444:761–765. doi: 10.1038/nature05349. [DOI] [PubMed] [Google Scholar]
- Radman M. Fidelity and infidelity. Nature. 2001;413:115. doi: 10.1038/35093178. [DOI] [PubMed] [Google Scholar]
- Reynolds AR, Tischer C, Verveer PJ, Rocks O, Bastiaens PI. EGFR activation coupled to inhibition of tyrosine phosphatases causes lateral signal propagation. Nat Cell Biol. 2003;5:447–453. doi: 10.1038/ncb981. [DOI] [PubMed] [Google Scholar]
- Rudd PM, Wormald MR, Stanfield RL, Huang M, Mattsson N, Speir JA, DiGennaro JA, Fetrow JS, Dwek RA, Wilson IA. Roles for glycosylation of cell surface receptors involved in cellular immune recognition. Journal of Molecular Biology. 1999;293:351–366. doi: 10.1006/jmbi.1999.3104. [DOI] [PubMed] [Google Scholar]
- Salvatore S, Heuschkel R, Tomlin S, Davies SE, Edwards S, Walker-Smith JA, French I, Murch SH. A pilot study of N-acetyl glucosamine, a nutritional substrate for glycosaminoglycan synthesis, in paediatric chronic inflammatory bowel disease. Aliment Pharmacol Ther. 2000;14:1567–1579. doi: 10.1046/j.1365-2036.2000.00883.x. [DOI] [PubMed] [Google Scholar]
- Sarkar M, Leventis PA, Silvescu CI, Reinhold VN, Schachter H, Boulianne GL. Null mutations in Drosophila N-acetylglucosaminyltransferase I produce defects in locomotion and a reduced life span. J Biol Chem. 2006;281:12776–12785. doi: 10.1074/jbc.M512769200. [DOI] [PubMed] [Google Scholar]
- Sasai K, Ikeda Y, Fujii T, Tsuda T, Taniguchi N. UDP-GlcNAc concentration is an important factor in the biosynthesis of beta1,6-branched oligosaccharides: regulation based on the kinetic properties of N-acetylglucosaminyltransferase V. Glycobiology. 2002;12:119–127. doi: 10.1093/glycob/12.2.119. [DOI] [PubMed] [Google Scholar]
- Schachter H. Biosynthetic controls that determine the branching and microheterogeneity of protein-bound oligosaccharides. Biochemistry and Cell Biology. 1986;64:163–181. doi: 10.1139/o86-026. [DOI] [PubMed] [Google Scholar]
- Schneider H, Downey J, Smith A, Zinselmeyer BH, Rush C, Brewer JM, Wei B, Hogg N, Garside P, Rudd CE. Reversal of the TCR stop signal by CTLA-4. Science. 2006;313:1972–1975. doi: 10.1126/science.1131078. [DOI] [PubMed] [Google Scholar]
- Seetharaman J, Kanigsberg A, Slaaby R, Leffler H, Barondes SH, Rini JM. X-ray crystal structure of the human galectin-3 carbohydrate recognition domain at 2.1-A resolution. J Biol Chem. 1998;273:13047–13052. doi: 10.1074/jbc.273.21.13047. [DOI] [PubMed] [Google Scholar]
- Seoane J, Le HV, Shen L, Anderson SA, Massague J. Integration of Smad and forkhead pathways in the control of neuroepithelial and glioblastoma cell proliferation. Cell. 2004;117:211–223. doi: 10.1016/s0092-8674(04)00298-3. [DOI] [PubMed] [Google Scholar]
- Shang J, Gao N, Kaufman RJ, Ron D, Harding HP, Lehrman MA. Translation attenuation by PERK balances ER glycoprotein synthesis with lipid-linked oligosaccharide flux. J Cell Biol. 2007;176:605–616. doi: 10.1083/jcb.200607007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi H, Tan J, Schachter H. N-glycans are involved in the response of Caenorhabditis elegans to bacterial pathogens. Methods Enzymol. 2006;417:359–389. doi: 10.1016/S0076-6879(06)17022-6. [DOI] [PubMed] [Google Scholar]
- Shiraha H, Gupta K, Drabik K, Wells A. Aging fibroblasts present reduced epidermal growth factor (EGF) responsiveness due to preferential loss of EGF receptors. The Journal of biological chemistry. 2000;275:19343–19351. doi: 10.1074/jbc.M000008200. [DOI] [PubMed] [Google Scholar]
- Smith PL, Myers JT, Rogers CE, Zhou L, Petryniak B, Becker DJ, Homeister JW, Lowe JB. Conditional control of selectin ligand expression and global fucosylation events in mice with a targeted mutation at the FX locus. J Cell Biol. 2002;158:801–815. doi: 10.1083/jcb.200203125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soleimani L, Roder JC, Dennis JW, Lipina T. Beta N-acetylglucosaminyltransferase V (Mgat5) deficiency reduces the depression-like phenotype in mice. Genes Brain Behav. 2007 doi: 10.1111/j.1601-183X.2007.00358.x. [DOI] [PubMed] [Google Scholar]
- Stillman BN, Hsu DK, Pang M, Brewer CF, Johnson P, Liu FT, Baum LG. Galectin-3 and galectin-1 bind distinct cell surface glycoprotein receptors to induce T cell death. J Immunol. 2006;176:778–789. doi: 10.4049/jimmunol.176.2.778. [DOI] [PubMed] [Google Scholar]
- Stowell SR, Arthur CM, Mehta P, Slanina KA, Blixt O, Leffler H, Smith DF, Cummings RD. Galectin-1, -2, and -3 exhibit differential recognition of sialylated glycans and blood group antigens. J Biol Chem. 2008;283:10109–10123. doi: 10.1074/jbc.M709545200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stroop CJ, Weber W, Gerwig GJ, Nimtz M, Kamerling JP, Vliegenthart JFG. Characterization of the carbohydrate chains of the secreted form of the human epidermal growth factor receptor. Glycobiology. 2000;10:901–917. doi: 10.1093/glycob/10.9.901. [DOI] [PubMed] [Google Scholar]
- Takahashi M, Yokoe S, Asahi M, Lee SH, Li W, Osumi D, Miyoshi E, Taniguchi N. N-glycan of ErbB family plays a crucial role in dimer formation and tumor promotion. Biochim Biophys Acta. 2008;1780:520–524. doi: 10.1016/j.bbagen.2007.10.019. [DOI] [PubMed] [Google Scholar]
- Takamatsu S, Oguri S, Minowa MT, Yoshida A, Nakamura K, Takeuchi M, Kobata A. Unusually high expression of N-acetylglucosaminyltransferase-IV a in human choriocarcinoma cell lines: a possible enzymatic basis of the formaiton of abnormal biantennary sugar chain. Cancer Research. 1999;59:3949–3953. [PubMed] [Google Scholar]
- Takenaka Y, Fukumori T, Raz A. Galectin-3 and metastasis. Glycoconj J. 2004;19:543–549. doi: 10.1023/B:GLYC.0000014084.01324.15. [DOI] [PubMed] [Google Scholar]
- Tan J, Dunn J, Jaeken J, Schachter H. Mutations in the MGAT2 gene controlling complex N-glycan synthesis cause carbohydrate-deficient glycoprotein syndrome type II, an autosomal recessive disease with defective brain development. American journal of human genetics. 1996;59:810–817. [PMC free article] [PubMed] [Google Scholar]
- Tanikawa R, Tanikawa T, Okada Y, Nakano K, Hirashima M, Yamauchi A, Hosokawa R, Tanaka Y. Interaction of galectin-9 with lipid rafts induces osteoblast proliferation through the c-Src/ERK signaling pathway. J Bone Miner Res. 2008;23:278–286. doi: 10.1359/jbmr.071008. [DOI] [PubMed] [Google Scholar]
- Taylor RP, Parker GJ, Hazel MW, Soesanto Y, Fuller W, Yazzie MJ, McClain DA. Glucose Deprivation Stimulates O-GlcNAc Modification of Proteins through Up-regulation of O-Linked N-Acetylglucosaminyltransferase. J Biol Chem. 2008;283:6050–6057. doi: 10.1074/jbc.M707328200. [DOI] [PubMed] [Google Scholar]
- Togayachi A, Kozono Y, Ishida H, Abe S, Suzuki N, Tsunoda Y, Hagiwara K, Kuno A, Ohkura T, Sato N, et al. Polylactosamine on glycoproteins influences basal levels of lymphocyte and macrophage activation. Proc Natl Acad Sci U S A. 2007;104:15829–15834. doi: 10.1073/pnas.0707426104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toscano MA, Bianco GA, Ilarregui JM, Croci DO, Correale J, Hernandez JD, Zwirner NW, Poirier F, Riley EM, Baum LG, Rabinovich GA. Differential glycosylation of TH1, TH2 and TH-17 effector cells selectively regulates susceptibility to cell death. Nat Immunol. 2007;8:825–834. doi: 10.1038/ni1482. [DOI] [PubMed] [Google Scholar]
- Trotti D, Aoki M, Pasinelli P, Berger UV, Danbolt NC, Brown RH, Jr, Hediger MA. Amyotrophic lateral sclerosis-linked glutamate transporter mutant has impaired glutamate clearance capacity. J Biol Chem. 2001;276:576–582. doi: 10.1074/jbc.M003779200. [DOI] [PubMed] [Google Scholar]
- Tsuda T, Ikeda Y, Taniguchi N. The Asn-420-linked sugar chain in human epidermal growth factor receptor suppresses ligand-independent spontaneous oligomerization. Possible role of a specific sugar chain in controllable receptor activation. J Biol Chem. 2000;275:21988–21994. doi: 10.1074/jbc.M003400200. [DOI] [PubMed] [Google Scholar]
- Waldman BC, Rudnick G. UDP-GlcNAc transport across the Golgi membrane: electroneutral exchange for dianionic UMP. Biochemistry. 1990;29:44–52. doi: 10.1021/bi00453a006. [DOI] [PubMed] [Google Scholar]
- Wang Y, Tan J, Sutton-Smith M, Ditto D, Panico M, Campbell RM, Varki NM, Long JM, Jaeken J, Levinson SR, et al. Modeling human congenital disorder of glycosylation type IIa in the mouse: conservation of asparagine-linked glycan-dependent functions in mammalian physiology and insights into disease pathogenesis. Glycobiology. 2001;11:1051–1070. doi: 10.1093/glycob/11.12.1051. [DOI] [PubMed] [Google Scholar]
- Waterhouse P, Penninger JM, Timms E, Wakeham A, Shahinian A, Lee KP, Thompson CB, Griesser H, Mak TW. Lymphoproliferative disorders with early lethality in mice deficient in Ctla-4. Science. 1995;270:985–988. doi: 10.1126/science.270.5238.985. [DOI] [PubMed] [Google Scholar]
- Wiley HS, Burke PM. Regulation of receptor tyrosine kinase signaling by endocytic trafficking. Traffic. 2001;2:12–18. doi: 10.1034/j.1600-0854.2001.020103.x. [DOI] [PubMed] [Google Scholar]
- Winkler WC, Nahvi A, Roth A, Collins JA, Breaker RR. Control of gene expression by a natural metabolite-responsive ribozyme. Nature. 2004;428:281–286. doi: 10.1038/nature02362. [DOI] [PubMed] [Google Scholar]
- Wollscheid B, Bausch-Fluck D, Henderson C, O'Brien R, Bibel M, Schiess R, Aebersold R, Watts JD. Mass-spectrometric identification and relative quantification of N-linked cell surface glycoproteins. Nat Biotechnol. 2009;27:378–386. doi: 10.1038/nbt.1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu RH, Peck RM, Li DS, Feng X, Ludwig T, Thomson JA. Basic FGF and suppression of BMP signaling sustain undifferentiated proliferation of human ES cells. Nat Methods. 2005;2:185–190. doi: 10.1038/nmeth744. [DOI] [PubMed] [Google Scholar]
- Zhang J, Zhang W, Zou D, Chen G, Wan T, Li N, Cao X. Cloning and functional characterization of GNPI2, a novel human homolog of glucosamine-6-phosphate isomerase/oscillin. J Cell Biochem. 2003;88:932–940. doi: 10.1002/jcb.10444. [DOI] [PubMed] [Google Scholar]
- Zhu S, Hanneman A, Reinhold VN, Spence AM, Schachter H. Caenorhabditis elegans triple null mutant lacking UDP-N-acetyl-D-glucosamine:alpha-3-D-mannoside beta1,2-N-acetylglucosaminyltransferase I. Biochem J. 2004;382:995–1001. doi: 10.1042/BJ20040793. [DOI] [PMC free article] [PubMed] [Google Scholar]