

Abstract
The brain’s circuitry is established by directed migration and synaptogenesis of neurons during development. Although neurons mature and migrate in specific patterns, little is known about how neurons exit their germinal zone niche. We found that cerebellar granule neuron germinal zone exit is regulated by proteasomal degradation of Pard3A by the Seven in Absentia homolog (Siah) E3 ubiquitin ligase. Pard3A gain-of-function and Siah loss-of-function induce precocious radial migration. Time-lapse imaging using a probe to measure neuronal cell contact reveals that Pard3A promotes adhesive interactions needed for germinal zone exit by recruiting the JAM-C epithelial tight junction adhesion molecule to the neuronal cell surface. Our findings define a Siah-Pard3A signaling pathway that controls adhesion-dependent exit of neuronal progenitors or immature neurons from a germinal zone niche.
The migration of neurons from a germinal zone (GZ) to their final laminar positions is essential for morphogenesis of the developing brain (1–3); aberrations in this process are linked to profound neurodevelopmental and cognitive disorders (4). Although the substrates (5–7), guidance mechanisms (8–10), cytoskeletal elements (11–13) and post-translational modifications (14–16) required for neuronal migrations are well established, the cell-intrinsic machinery regulating when neurons gain access to permissive migration pathways to exit their GZs are unidentified (17). Developing CGNs are an excellent model to analyze the mechanisms regulating GZ exit and to elucidate migration pathway selection, as they undergo two migration phases (18–20): tangential migration near the cerebellar surface followed by radial migration away from the EGL where CGNs cross the molecular layer (ML) to eventually reside within the internal granule layer (IGL). In this study, we examined the roles of the Partitioning Defective (PAR) polarity-signaling complex and an upstream regulator in controlling CGN migration from the EGL, a GZ niche (fig. S1).
The PAR complex is an evolutionarily conserved multi-protein complex containing orthologs of Partitioning Defective-6 (Pard6), Partitioning Defective-3 (Pard3) and PKCζ, that regulates many polarized cellular processes, like cell motility, asymmetric cell division and epithelial junction formation (21). As Pard3A protein expression is low in the EGL (Fig. 1A–C), we examined whether elevated Pard3A activity induces CGN GZ exit. Expression constructs for Pard3A and the fluorescent nuclear reporter H2B-mCherry were co-electroporated into the cerebellar cortices of postnatal day 8 (P8) mice and cerebellar slices were cultured ex vivo. While control CGNs remained within the EGL after 24 hours (fig. S2A), CGNs expressing elevated Pard3A entered the ML and IGL (fig. S2B), suggesting that elevated Pard3A expression is sufficient to induce precocious GZ exit.
Figure 1. Siah is highly expressed in the EGL and ubiquitinates Pard3A protein.
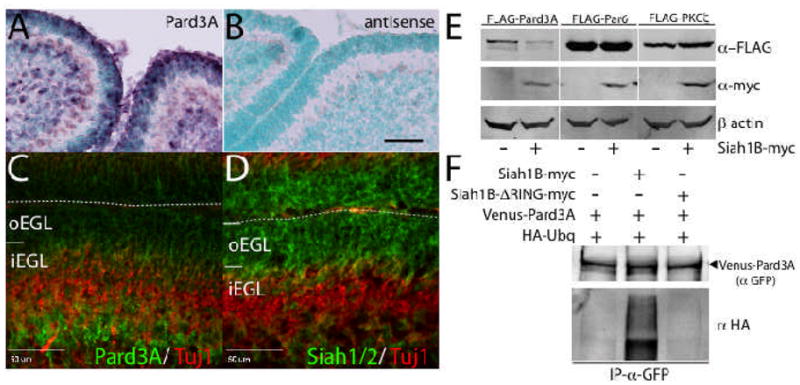
(A, B) In situ hybridization shows that Pard3A mRNA is expressed throughout the P6 EGL. Scale=80 μm (C) Immunohistochemistry of P6 mouse cerebellum for Pard3A (green) and Tuj1 (red). Pard3A expression is low in the oEGL and higher in differentiated CGNs in the iEGL. Scale=50 μm (D) Immunohistochemistry of P6 mouse cerebellum for Siah1/2 (green) and Tuj1 (red). Siah expression is high in the oEGL and absent in the iEGL. (E) Expression of Siah1B-myc reduces Pard3A but not Par6 or PKCζ protein level in HEK293 cells. (F) Siah1B-myc, but not Siah1B-ΔRING-myc, can induce ubiquitination of Venus-Pard3A in HEK293 cells.
We next examined the role of Siah, a PAR complex-interacting E3 ubiquitin ligase (fig. S3) expressed in the EGL (Fig. 1D), in regulating Pard3A protein level and PAR complex–dependent GZ exit. The role of Siah ligases in the morphogenesis of the vertebrate nervous system has not previously been examined (22). Epitope-tagged Siah1B immunoprecipitated Pard3A when co-expressed in HEK293 cells (fig. S3B), an interaction that required an intact Siah substrate-binding domain (fig. S3D). Furthermore, Siah1B expression reduced expression of Pard3A, but not Pard6 or PKCζ, protein (Fig. 1E) and induced Pard3A ubiquitination (Fig. 1F). Pard3A protein levels were also reduced by Siah2 but not by a dominant negative mutant ligase lacking the catalytic RING domain (Siah1B-ΔRING, fig. S3E). The Pard3A protein sequence contains two Siah degron recognition sequences Px(A,T,R)xVxP (fig. S3C) (23). Mutation of the VxP core of both Pard3A degrons to NxN or treatment with MG132 attenuated Siah-mediated reduction of Pard3A protein (fig. S3E) and blocked Siah-induced ubiquitination (fig. S3F, see fig. S2B, C and fig. S4 for additional functional differences between Pard3A and the NxN mutant in CGNs). Finally, Venus-Pard3A fluorescence signal in purified CGNs was diminished by Siah1B co-expression but not by Siah1B-ΔRING (fig. S3G). Therefore, Siah regulates Pard3A protein level through VxP degron-dependent ubiquitination.
Siah1/2 immunoreactivity in the EGL is elevated at P6, the peak time of CGN neurogenesis (Fig. 1D) and declines at P15 (see fig. S5 for comparison of Siah1 vs Siah2 expression). To detect sites of Siah activity in the developing cerebellum, we fused the Siah degron motif to the Venus fluorescent protein (fig. S6) and electroporated expression vectors for this Siah sensor in conjunction with H2B-mCherry into P8 EGL. Siah sensor fluorescence was scant in the EGL but visible in the molecular layer and IGL (fig. S7), while a negative control sensor containing the NxN mutations showed fluorescence in all cerebellar layers. These results suggest that Siah activity is high in CGNs within the EGL and decreases during GZ exit.
To assess Siah function in GZ exit, we electroporated shRNAs silencing Siah1B and Siah2 (fig. S8A) or an expression construct for dominant-negative Siah1B-ΔRING into P8 EGL. After 1 day of ex vivo culture, CGNs transfected with a control shRNA remained in the EGL. Silencing of Siah2, but not of Siah1B, and expression of Siah1B-ΔRING increased CGN migration toward the IGL (Fig. 2A, B and see fig. S9A for additional analysis). We next evaluated whether Siah activity regulates CGN GZ exit using a gain of function approach. Expression vectors for mouse Siah1B or Siah2 were electroporated into P8 EGL. After 2 days of ex vivo culture, control CGNs entered the molecular layer and IGL (Fig. 2C, D and see fig. S9B for additional analysis), while Siah1B- or Siah2-expressing CGNs remained within the EGL. EGL exit was similarly blocked by Pard3A silencing (fig. S8B, C). Elevated Pard3A expression in Siah-expressing cells rescued the Siah phenotype, confirming that Siah-mediated blockade of EGL exit is not only reversible but also Pard3A-dependent (Fig. 2C, D). Finally, we assessed proliferation status of CGNs in our experiments using EdU incorporation. While elevated Siah2 activity has no effect on EdU incorporation, Siah2 silencing and Pard3A gain of function, conditions that induce early GZ exit, significantly reduced EdU incorporation, suggesting a linkage between migration initiation and cell cycle exit (fig. S10).
Figure 2. Siah activity attenuates GZ exit by negatively regulating Pard3A.

P8 EGL was co-electroporated with the indicated expression constructs and H2B-mCherry. After 24 (A) or 48 (C) hours of ex vivo culture, the migration distance of H2B-labeled CGN from the pial layer (outer dashed line) was analyzed in 3 imaging experiments. (A) Most control and Siah1B-silenced cells remain within the EGL (dashed lines) at 24 h, while Siah2-silenced and Siah1B-ΔRING over-expressing cells pre-maturely entered the ML and IGL. (B) Migration distance versus frequency plot of control (n=872, black), Siah1B-silenced (n=960, red), Siah2-slienced (n=926, blue) or Siah1B-ΔRING over-expressing (n=927, green) CGNs. (C) While control cells entered the ML and IGL after 48 h, Siah1B and Siah2 over-expressing cells remained in the EGL. Addition of Pard3A to Siah-expressing CGNs restored migration. (D) Migration distance versus frequency plot of control (n=909, black), Siah1B-expressing (n=970, red), Siah2-expressing (n=921, blue) or Siah1B+Pard3A-expressing (n=919, green) CGNs.
Having found that Siah activity controls CGN GZ exit, we next examined whether Siah regulates CGN migration mode. We introduced expression vectors for mouse Siah1B or Siah1B plus Pard3A into purified CGNs via nucleofection and examined migration by time-lapse microscopy of microcultures. Control CGNs exhibited radial-like migration that persisted in the direction of leading process extension (Fig. 3A, Movie 1, figs. S11 and S12). Siah-expressing cells were motile but did not elaborate long leading processes or neurites and were less directionally persistent (Fig. 3A, Movie 2). Ubiquitin ligase activity was required for the Siah phenotype, as Siah1B-ΔRING expression did not alter migration (Movie 3). Finally, increased Pard3A expression restored normal leading process extension (fig. S11C) and directional persistence to Siah-expressing neurons (Fig. 3A, Movie 4). Therefore, the antagonistic relationship between Siah activity and Pard3A regulates the directional persistence of migrating CGNs but not cell motility.
Figure 3. Siah activity and Pard3A regulate transition from tangential to radial migration.

(A) Purified CGNs were nucleofected with Centrin2-Venus, H2B-mCherry and the indicated constructs. After 18 h in culture, frame-to-frame migration angle of H2B-mCherry–labeled nuclei was analyzed in three separate experiments. Pseudocolored nuclei show representative migration paths. Control cells (nangle=3140) and Siah1B+Pard3A (nangle=4743) expressing cells migrated predominantly forward (315°–45°), while the migration pattern of Siah1B-expressing cells (nangle=4480) was randomized. The quadrant (315°–45°, 45°–135°, 135°–225°, 225°–315°) distribution of Siah1B expressing cells differed significantly from that of controls (p=0.001, χ2 test), while that of Siah1B+Pard3A did not (p=0.670, χ2 test). (B) P8 EGL was electroporated with H2B-mCherry and the indicated constructs. Cerebellar slices were incubated for 28 h and then imaged for 20 h. Migration endpoint angles of H2B-mCherry–labeled nuclei were tracked, binned and plotted in three separate experiments. Colored lines indicate migration paths; arrowheads indicate direction of migration. Control cells (n=431) display tangential migration parallel to the EGL (255°–285°/75°–105°) and radial migration perpendicular to the EGL (105°–255°), while Siah1B-expressing cells (n=454) display predominantly tangential and pial-directed migration (285°–75°). Migration of cells expressing Siah1B differed significantly from that of controls (p=2.056×10−8, χ2 test), while that of cells expressing Siah1B+Pard3A did not (n= 452, p=0.301, χ2 test).
To examine CGN migration pathway selection, we electroporated P8 EGL with Siah1B or Siah1B plus Pard3A expression constructs and examined the migration of H2B-mCherry–labeled CGNs by long-term time-lapse microscopy. Control CGNs migrated extensively parallel to the cerebellar slice surface before migrating radially toward the IGL (Fig. 3B, Movie 5, fig. S12E). Elevated Siah activity reduced radial migration and increased the percentage of neurons migrating toward the pial surface while the percentage of CGNs migrating tangentially within the EGL was unaffected; this phenotype was rescued by increased Pard3A expression (Fig. 3B; Movies 6 and 8). Expression of Siah1B-ΔRING, which appeared to drive GZ exit (Fig. 2A), did not adversely affect movement to the IGL, in vitro directional persistence or migration pathway selection (fig. S12 and S13). These results show that Siah activity intrinsically regulates CGN migration mode and suggest that Siah regulation of Pard3A activity constitutes a switch controlling tangential versus radial migration as CGNs exit the EGL.
Although the PAR complex activates cytoskeletal elements that propel migration (13), we hypothesized that the Siah-Pard3A module controls adhesion during GZ exit as Pard3A is essential for junction formation in epithelial cells. We therefore focused our approach on the analysis of JAM-C, a tight-junction component that directly interacts with Pard3A and is required for epithelial adhesion (24). Immunostaining revealed that JAM-C is not only expressed in differentiating CGNs but also labels sites of CGN contact (fig. S14A, B). We examined JAM-C function in GZ exit by electroporating P8 EGL with a shRNA silencing JAM-C (fig. S15A) or with a dominant-negative fragment of the JAM-C cytoplasmic domain (JAM-C-DN) previously shown to competitively inhibit Pard3A binding to endogenous JAM-C and block epithelial junction formation (24) (fig. S15B). After 2 days of ex vivo culture, both JAM-C–silenced and JAM-C-DN–expressing CGNs remained predominantly within the EGL, but migration was unaffected by expression of JAM-C-DNΔ9, a JAM-C cytoplasmic domain lacking the Pard3A binding motif (fig. S15C). JAM-C-DN expression inhibited migration induced by Siah2 silencing, indicating that interaction between JAM-C and Pard3A is required for normal and Siah2 loss of function induced GZ exit (fig. S15C).
Having found that JAM-C is required for GZ exit, we next asked whether JAM-C mediated cell contacts are regulated by the Siah-Pard3A pathway in MDCK polarized epithelial cells, a well-established system to assay PAR complex–dependent adhesion. Elevated Siah1B expression dissolved ZO-1, junctional adhesion molecule C (JAM-C) and Pard3A-labeled MDCK junctions, and these effects were reversed by Pard3A expression (fig. S16A). Elevated Siah1B expression also altered the localization of endogenous JAM-C in cultured CGNs (fig. S16B). To directly measure JAM-C contact in live CGNs we developed a novel JAM-C-based fluorescent probe by fusing super ecliptic pHluorin, a pH-sensitive conditional fluorophore (25), with the extracellular domain of JAM-C (fig. S17A). High JAM-C-pHluorin signal revealed sites of neuron-neuron or neuron-glial contact (as indicated by f-actin and Pard3A accumulation), validating the JAM-C probe (Fig. 4A, fig. S17B; Movies 9, 10). We expressed JAM-C-pHluorin and Siah1B, Siah1B-ΔRING, Siah1B plus Pard3A or Siah2 shRNA into purified CGNs via nucleofection and assayed cell contacts by time-lapse microscopy of microcultures. Control CGNs displayed robust contacts with neighboring cells, while Siah1B gain of function inhibited JAM-C contacts (Fig. 4B; Movies 11,12). Increased Pard3A expression restored contact formation to near wild-type levels (Movie 14). Finally, Siah2 silencing increased JAM-C contact nearly 2-fold (Movie 15). Therefore, Siah activity inhibits Pard3A-dependent JAM-C adhesion.
Figure 4. Siah activity regulates GZ exit by modulating the formation of Pard3A-dependent JAM-C adhesions.

(A) Time lapse imaging of CGNs nucleofected to express JAM-C-pHluorin. Fluorescence is low before cell contact. Upon establishment of stable contacts, JAM-C-pHluorin signal fluorescence intensifies. (B) Purified CGNs were electroporated to co-express JAM-C-pHluorin and the indicated constructs. After 18 h, the abundance of JAM-C-pHluorin contacts were analyzed. Control cells (n=64 cells, 16045 puncta) displayed robust JAM-C contacts. Siah expression (n=37 cells, 2579 puncta) significantly reduced JAM-C-pHluorin–positive puncta. Siah1B-ΔRING (n=30 cells, 7589 puncta) and Siah1B+Pard3A (n=32 cells, 8069 puncta) cells were similar to controls, while Siah2 silencing (n=34 cells, 10709 puncta) increased JAM-C-pHluorin contact. (C, D) CGNs in P8 EGL were co-electroporated with the indicated constructs and H2B-mCherry. After 24 or 48 h of culture, the distance of H2B-labeled cells from the pial layer (outer dashed line) was analyzed in three separate experiments. Grey shading shows percentage of cells found in the EGL; red overlay indicates the average migration distribution of control cells (error bar, SD). (C) JAM-C over-expression (n=891) did not induce migration from the EGL, but the JAM-C-Nectin3 fusion molecule (n=874), a Pard3A independent JAM-C variant, induced CGN migration from the EGL at 24 h. Control vs JAM-C, p=0.58; vs JAM-C-Nectin3, p=5.50×10−26 (χ2 test). (D) JAM-C-Nectin3 (n=971) expression rescues migration of Siah1B-expressing cells. Control vs Siah1B+JAM-C-Nectin3, p=0.95 (χ2 test).
If Pard3A binding to the JAM-C cytoplasmic domain is essential for JAM-C–dependent GZ exit, as it is for epithelial tight junction formation (24), we hypothesized that swapping the JAM-C cytoplasmic domain with that of a Pard3-independent adhesion receptor would create a Siah-insensitive receptor. We fused extracellular domains of JAM-C to the cytoplasmic domain of Nectin-3, a non-Pard3A–binding adhesion receptor required for epithelial cell adherens junctions (see fig. S15B for schematic) and electroporated P8 EGL with expression vectors for wild-type mouse JAM-C and JAM-C-Nectin3. While CGNs expressing wild-type JAM-C remained in the EGL, expression of JAM-C-Nectin3 spurred early EGL exit (Fig. 4C). JAM-C-Nectin3 expression also fully rescued the EGL exit of CGNs expressing Siah1B, directly demonstrating that elevated JAM-C cell contact overcomes Siah inhibition of GZ exit (Fig. 4D).
Newborn neurons must exercise plasticity in order to integrate into the vertebrate brain (26–28). Our results show that post-translational ubiquitination of Pard3A by Siah controls whether cerebellar granule neurons will follow the tangential migration that keeps them within the EGL of the mouse cerebellum or the radial migration path they use to exit their germinal zone and migrate to the IGL. Migration pattern plasticity is invoked by the production of tight junction JAM-C cell contacts necessary for the integration of new neurons into the developing cerebellar cortex. These cell interactions resemble those known to regulate mesenchymal epithelial transitions as new epithelial cells integrate into developing epithelia (29, 30).
Supplementary Material
Acknowledgments
We are grateful to Drs. James Morgan, Michael Dyer, Martine Roussel and Nagi Ayad for critically reading the manuscript, Drs. Akira Sawa, Stanislav Zakharenko, and Brenda Schulman for insightful discussions, and Samuel Connell, Regan Baird and Karl Kilborn (Intelligent Imaging Innovations) for timely data analysis advice and support. We thank Dr. Gordon Chan for providing space for some of the revision experiments and Sharon Naron for excellent editorial assistance. Dr. Gero Miesenbock and MSKCC provided the pHluorin reporter, Dr. Atsushi Miyawaki provided Venus, Dr. Franck Polleux provided pCIG2, Dr. Michel Aurrand-Lions provided anti-JAM-C antiserum, Dr. Ivan Dikic provided the HA-Ubiquitin construct and Dr. Akira Sawa shared Siah reagents. Supported by American Lebanese Syrian Associated Charities (ALSAC, DJS), a National Cancer Institute Cancer Center Support Grant (DJS) and a March of Dimes Basil O’Connor Starter Scholar Research Award (DJS).
Footnotes
Supplementary Online Material contains Full Materials and Methods, Figs. S1 to S17, References and 15 Movies. A figure summarizing the main result of this paper is available in Supplementary Information.
References
- 1.Rakic P. J Comp Neurol. 1972 May;145:61. doi: 10.1002/cne.901450105. [DOI] [PubMed] [Google Scholar]
- 2.Hatten ME. Science. 2002 Sep 6;297:1660. doi: 10.1126/science.1074572. [DOI] [PubMed] [Google Scholar]
- 3.Metin C, Vallee RB, Rakic P, Bhide PG. J Neurosci. 2008 Nov 12;28:11746. doi: 10.1523/JNEUROSCI.3860-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kato M, Dobyns WB. Hum Mol Genet. 2003 Apr 1;12(Spec No 1):R89. doi: 10.1093/hmg/ddg086. [DOI] [PubMed] [Google Scholar]
- 5.Fishell G, Hatten ME. Development. 1991 Nov;113:755. doi: 10.1242/dev.113.3.755. [DOI] [PubMed] [Google Scholar]
- 6.Anton ES, Kreidberg JA, Rakic P. Neuron. 1999 Feb;22:277. doi: 10.1016/s0896-6273(00)81089-2. [DOI] [PubMed] [Google Scholar]
- 7.Elias LA, Wang DD, Kriegstein AR. Nature. 2007 Aug 23;448:901. doi: 10.1038/nature06063. [DOI] [PubMed] [Google Scholar]
- 8.Polleux F, Whitford KL, Dijkhuizen PA, Vitalis T, Ghosh A. Development. 2002 Jul;129:3147. doi: 10.1242/dev.129.13.3147. [DOI] [PubMed] [Google Scholar]
- 9.Zhou P, et al. Neuron. 2007 Jul 5;55:53. [Google Scholar]
- 10.Renaud J, et al. Nat Neurosci. 2008 Apr;11:440. doi: 10.1038/nn2064. [DOI] [PubMed] [Google Scholar]
- 11.Schaar BT, McConnell SK. Proc Natl Acad Sci U S A. 2005 Sep 20;102:13652. doi: 10.1073/pnas.0506008102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tsai JW, Bremner KH, Vallee RB. Nat Neurosci. 2007 Jul 8;10:970. doi: 10.1038/nn1934. [DOI] [PubMed] [Google Scholar]
- 13.Solecki DJ, et al. Neuron. 2009 Jul 16;63:63. [Google Scholar]
- 14.Patrick GN, Zhou P, Kwon YT, Howley PM, Tsai LH. J Biol Chem. 1998 Sep 11;273:24057. doi: 10.1074/jbc.273.37.24057. [DOI] [PubMed] [Google Scholar]
- 15.Suetsugu S, et al. Biochem J. 2004 Nov 15;384:1. doi: 10.1042/BJ20041103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Karakuzu O, Wang DP, Cameron S. Development. 2009 Mar;136:943. doi: 10.1242/dev.029363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rakic P. Contact Regulation of Neuronal Migration. In: Edelman GM, Thiery JP, editors. The Cell in Contact: Adhesion and Junctions of Morphogenetic Determinants. Wiley and Sons; New York: 1985. [Google Scholar]
- 18.Edmondson JC, Hatten ME. J Neurosci. 1987 Jun;7:1928. doi: 10.1523/JNEUROSCI.07-06-01928.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ryder EF, Cepko CL. Neuron. 1994 May;12:1011. doi: 10.1016/0896-6273(94)90310-7. [DOI] [PubMed] [Google Scholar]
- 20.Komuro H, Yacubova E, Rakic P. J Neurosci. 2001 Jan 15;21:527. doi: 10.1523/JNEUROSCI.21-02-00527.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Solecki DJ, Govek EE, Tomoda T, Hatten ME. Genes Dev. 2006 Oct 1;20:2639. doi: 10.1101/gad.1462506. [DOI] [PubMed] [Google Scholar]
- 22.House CM, Moller A, Bowtell DD. Cancer Res. 2009 Dec 1;69:8835. doi: 10.1158/0008-5472.CAN-09-1676. [DOI] [PubMed] [Google Scholar]
- 23.House CM, et al. Structure. 2006 Apr;14:695. [Google Scholar]
- 24.Ebnet K, et al. EMBO J. 2001 Jul 16;20:3738. doi: 10.1093/emboj/20.14.3738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Miesenbock G, De Angelis DA, Rothman JE. Nature. 1998 Jul 9;394:192. doi: 10.1038/28190. [DOI] [PubMed] [Google Scholar]
- 26.Noctor SC, Martinez-Cerdeno V, Ivic L, Kriegstein AR. Nat Neurosci. 2004 Feb;7:136. doi: 10.1038/nn1172. [DOI] [PubMed] [Google Scholar]
- 27.Bultje RS, et al. Neuron. 2009 Jul 30;63:189. doi: 10.1016/j.neuron.2009.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yi JJ, Barnes AP, Hand R, Polleux F, Ehlers MD. Cell. 2010 Jul 9;142:144. doi: 10.1016/j.cell.2010.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Thiery JP, Sleeman JP. Nat Rev Mol Cell Biol. 2006 Feb;7:131. doi: 10.1038/nrm1835. [DOI] [PubMed] [Google Scholar]
- 30.Chaffer CL, Thompson EW, Williams ED. Cells Tissues Organs. 2007;185:7. doi: 10.1159/000101298. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.