

Abstract
Bright (B cell regulator of IgH transcription) transactivates the immunoglobulin heavy chain (IgH) intronic enhancer, Eµ, by binding to matrix attachment regions (MARs), sites necessary for DNA attachment to the nuclear matrix. Here we report that Bright interacts with the ubiquitous autoantigen Sp100, a component of promyelocytic leukemia nuclear bodies (PML NBs), and with LYSp100B/Sp140, the lymphoid-restricted homolog of Sp100. Both in intact cells and in nuclear matrix preparations, the majority of Bright and Sp100 colocalize within PML NBs. In contrast, Bright colocalizes with only a small fraction of LYSp100B while inducing a redistribution of the majority of LYSp100B from its associated nuclear domains (LANDs) into nucleoplasm and cytoplasm. Sp100 represses the MAR-binding and transactivation activity of Bright. LYSp100B interacts more weakly with Bright but requires significantly higher levels than Sp100 to inhibit MAR binding. However, it strongly stimulates Bright transactivation through Eµ. We suggest that Sp100 and LYSp100B interactions with Bright have different consequences for IgH transcription, potentially through differential association of Eµ MARs with nuclear matrix- associated PML NBs and LANDs.
Keywords: autoantigen/nuclear matrix attachment/PML nuclear bodies/transcription regulation
Introduction
The eukaryotic nucleus is highly organized into distinct domains, which spatially separate different biochemical processes (Spector, 1993; Lamond and Earnshaw, 1998). A growing body of evidence indicates that DNA replication, RNA transcription and pre-mRNA splicing take place in defined nuclear domains. However, the precise structure and functional role for these domains are poorly understood. The systematic characterization of antibodies produced in autoimmune gastritis (Szostecki et al., 1990) led to the identification of one such nuclear structure, termed the nuclear body (NB; Ascoli and Maul, 1991; reviewed in Doucas and Evans, 1996). Promyelocytic leukemia (PML) NBs (also termed PML oncogenic domains, Kr or ND10 bodies) are best defined by electron microscopic studies of acute promyleocytic leukemia (APL) cells; they contain the PML transcription factor, the Sp100 antoantigen and at least four other proteins in a macromolecular multiprotein complex (Ascoli et al., 1991; Maul et al., 1993, 1995; Dyck et al., 1994; Koken et al., 1994; Zhu et al., 1994; Korioth et al., 1995; Boddy et al., 1996; Ishov et al., 1996). PML NBs are present in all cultured cell lines. They are dense, spherical particles, 0.3–0.5 µm in diameter (reviewed in Sternsdorf et al., 1997). Several studies suggest that PML NBs play a role in the regulation of cell growth and proliferation (Koken et al., 1994; Mu et al., 1994; Weis et al., 1994; Chang et al., 1995; Grignani et al., 1996).
A second PML NB-like subnuclear structure was defined by the unique nuclear localization pattern of the Sp100 homolog, Sp140/LYSp100B (Bloch et al., 1996; Dent et al., 1996). LYSP100-associated nuclear dots (LANDs) appear to be distinct in location and morphology from PML NBs in B-lymphocytic cell lines (Dent et al., 1996). Since LYSp100B is only expressed in lymphocytes, it has been postulated that LANDs function in a tissue-restricted manner. Sp100 and LYSp100B share high identity (49%) in their N-termini, whereas the extended C-terminus of LYSp100 contains bromodomains and plant homeobox domains (PHDs). As suggested by the presence of these domains, coactivator activity was inferred for Sp140 by virtue of its Gal4 DNA-binding domain fusion activity in transfected COS cells (Bloch et al., 1999). Bloch et al. (1999), using ectopic expression of Sp140 in non-B cells, argue against the existence of separate LAND substructures, opting to refer to them as a subset of PML NDs. Taking these findings together, it is clear that nuclear bodies are heterogeneous with respect to composition, cell type and function.
PML NBs appear to be associated with the nuclear matrix, as evidenced by fractionation experiments and DNase and RNase resistance (Dyck et al., 1994). Although originally thought of as a structural component of the nucleus, the nuclear matrix may play an important role in transcriptional regulation of gene expression (reviewed in Bell et al., 1998; Stein et al., 1998). Nuclear matrix attachment regions (MARs) often flank promoters or enhancers, and have been implicated in this regulatory mechanism (Stief et al., 1989; McKnight et al., 1992; Forrester et al., 1994; Zong and Scheuermann, 1995; Oancea et al., 1997). For example, the Bright transcriptional activator (Herrscher et al., 1995) stimulates transcription driven by the immunoglobulin heavy chain (IgH) enhancer, Eµ, by binding ATC motifs within its flanking MARs. Highly specific binding within the minor groove is achieved by virtue of two newly described domains (reviewed in Webb et al., 1999): A self association/tetramerization domain, termed REKLES for a heptapeptide conserved within this region among Bright orthologs, and a DNA-binding region, termed ARID (AT-rich interaction domain). The Bright ARID defined a new family of DNA-binding proteins, including SWI1, a component of the yeast SWI–SNF complex, which has been shown to remodel chromatin (Peterson and Lamkun, 1995). Like SWI1, all ARID proteins bind A/T-rich DNA, but only family members that contain both ARID and REKLES (e.g. Bright, Dri and Dril; Webb et al., 1999) bind specifically to A/T-rich MAR motifs.
Bright was originally identified as a B-cell-specific, gel-shifted complex (Webb et al., 1991). Its DNA-binding activity can be significantly increased by induction via a number of cell-surface receptors (e.g. CD38, RP105, CD40 and lipopolysaccharide) on splenic B cells (Webb et al., 1991, 1998). However, the level of Bright expression remains unchanged after such induction, suggesting that post-translational modification is responsible for the enhancement of DNA-binding activity. Although Bright transactivation requires binding to the Eµ MARs, the molecular consequence is unknown. MARs and attachment to the nuclear matrix can mediate specific alterations in chromatin structure (Käs et al., 1993; Singh et al., 1994; Pemov et al., 1998; reviewed in Stein et al., 1998). Such a mechanism is implicated for Bright, based on the ARID domain sequence homology with SWI1, its association with MARs and its residence within the nuclear matrix (Herrscher et al., 1995) This notion is further supported by the finding that components of human SWI–SNF are tightly associated with the nuclear matrix (Reyes et al., 1997). This suggests that at least a fraction of the SWI–SNF complex could be involved in the chromatin organization properties associated with MARs (reviewed in Schnitzler et al., 1998).
Here, we report the identification and characterization of a Bright interaction that may provide a link between nuclear subdomains, matrix attachment and transcription. We show that interation of Bright with Sp100, a component of PML NBs tightly associated with the nuclear matrix, results in repression through loss of Eµ MAR binding. Interaction with LYSp100B, which partitions from Sp100 into a different subnuclear domain (LANDs), leads to Bright coactivation even though this interaction can also block MAR binding in vitro. Differential interaction and colocalization of Bright in these subnuclear structures suggests that nuclear topology has a direct role in MAR-mediated regulation of transcription.
Results
Isolation of Bright-interacting partners
Proteins comprising SWI and other chromatin-remodeling systems exist in large heteromeric complexes. To search for potential partners of this type for Bright, we employed a Gal4 DNA-binding domain fused to Bright (amino acids 263–701; Figure 1B) as our bait in a yeast two-hybrid screen. One cDNA was independently isolated four times and was chosen for further characterization (Figure 1A). Sequencing showed that each clone encoded overlapping regions of the same protein, Sp100 (Speckled, 100 kDa), a ubiquitious nuclear antoantigen of unknown function that colocalizes with PML in PML NBs (Szostecki et al., 1990; Xie et al., 1993). In our SDS–PAGE system, Sp100 migrates at 72 kDa, which is consistent with the results of Xie et al. (1993). To confirm specificity, we tested Sp100 against two baits unrelated to Bright in a liquid β-galactosidase assay (Figure 1B). Neither p53 nor lamin C interacted with Sp100, while Bright interacted with Sp100 with a strength two-thirds that of the p53–T-antigen interaction. Thus, the interaction between Bright and Sp100 in yeast is strong and specific.



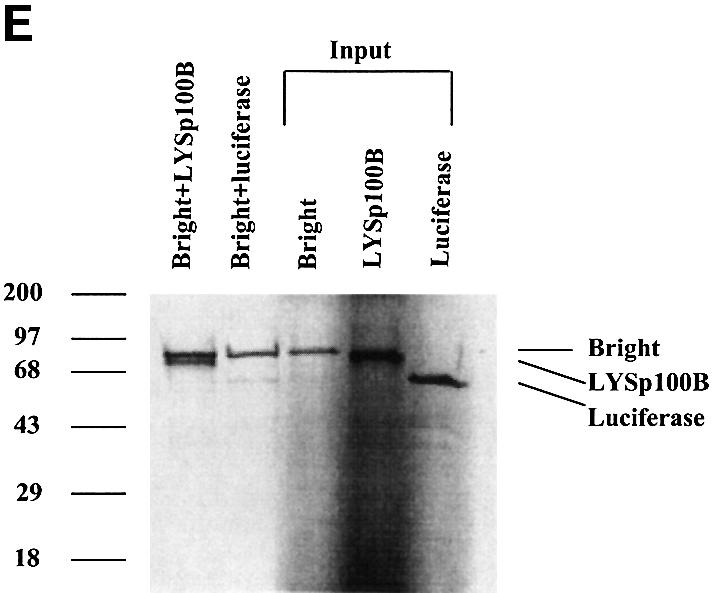
Fig. 1. The B-cell-specific transactivator Bright interacts with Sp100 and its B-cell-restricted homolog, LYSp100B. (A) Diagram of full-length Sp100, our Sp100 clones isolated from two-hybrid screening and LYSp100B. Amino acids are indicated above. Boxes indicate the position of a conserved domain (CD) with similarities to several transactivation domains, a PHD/TTC domain (PHD) and a bromodomain (Bromo). Of the 24 Bright-interacting clones obtained by yeast two-hybrid screening, Sp100 was independently isolated four times. (B) Analysis of Sp100 (clone CL16)–Bright interaction by a liquid culture β-galactosidase assay in yeast. The pACT vector and lamin C served as negative controls, while p53 and SV40 large T antigen were used as positive controls to evaluate relative binding affinity. (C) Functional domains of the transcription factor, Bright. Amino acids are indicated below. (D) Immunoprecipitation–PAGE analysis of the Bright–Sp100 interaction in vitro. Radiolabeled Bright, HA-Sp100 and luciferase were generated by in vitro translation in a rabbit reticulocyte lysate. Aliquots were incubated and HA–Sp100-bound protein complexes were immunoprecipitated with anti-HA mAb. HA–Sp100 can interact with full-length (FL) Bright (lane 1) and its REKLES domain (lane 3), but not with its ARID domain (lane 2) or luciferase (lane 4). Aliquots of input proteins (lanes 5–8) were loaded on to the gel as controls. No cross-reaction between Bright and anti-HA mAb was detected in control experiments. (E) Bright–LYSp100B interaction in vitro. Bright–LYSp100B complexes were immunoprecipitated with Bright antiserum. The negative control (luciferase) and input lanes are indicated.
Interaction of Bright with Sp100 and LYSp100
A full-length (1.6 kb) cDNA was amplified from both lymphocyte and brain libraries. Both cDNAs encoded a protein migrating at 72 kDa in SDS–PAGE. Consistent with the yeast binding analysis, hemagglutinin (HA)-tagged Sp100 protein specifically associated with Bright in vitro (Figure 1D). The in vitro interaction required the Bright REKLES domain (residues 455–561) but not the ARID (residues 226–360). The region within Sp100 that interacts with Bright is conserved in both alternatively spliced forms of its B-cell-restricted isoform, p140/LYSp100 (Bloch et al., 1996; Dent et al., 1996). While a specific interaction of Bright and LYSp100B was readily demonstrated by coimmunoprecipitation in vitro, we judged it to be significantly weaker than that of Sp100 (Figure 1E).
To examine the interaction of Bright with Sp100 in vivo, HeLa cells were cotransfected with expression vectors for Bright and HA-tagged Sp100. The interaction was subsequently analyzed by coimmunoprecipitation. Using Bright antiserum (Figure 2A), Bright was found to interact with HA-tagged Sp100 (lanes 2 and 3). No HA–Sp100 was precipitated by preimmune serum (lane 1). In the reverse experiment employing anti-Sp100 antibody (Figure 2B), Sp100 was found to interact with full-length Bright (lanes 1–4) and/or the REKLES domain (lane 4), but not with the ARID domain (lane 3). These results were consistent with what we observed in vitro (Figure 1D). The interaction was confirmed in untransfected J558 B cells by detection of an endogenous Bright–Sp100 interaction (Figure 2C). We did not detect Bright–LYSp100B physical association by immunoprecipitation (data not shown). The probable reason for this is addressed below.

Fig. 2. Bright interacts with Sp100 in vivo. (A) Forty-eight hours after cotransfection (1 µg each) into HeLa cells, putative complexes were immunoprecipitated with anti-Bright, resolved by SDS–PAGE, western blotted and probed with Bright antiserum (top) or anti-HA mAb (bottom). (B) Immunoprecipitation with anti-HA mAb and western blotting with Bright antiserum. (C) BCL1 and J558 B cells lysates were immunoprecipitated with Bright antiserum, then western blotted with anti-Sp100. ‘d/BCL1’ (lane 3) indicates that the putative Bright–Sp100 complexes were disassociated by prior heating of the BCL1 lysate.
Repression of Sp100 on Bright transactivation
To determine functional consequences on IgH expression, we examined whether Bright–Sp100 interactions could influence µ enhancer-driven transcription in the context of integrated chromatin. An Eµ–chloramphenicol acetyltransferase (CAT) construct (Figure 3A, right) was stably transfected into J558 B cells, and the cells were selected with G-418 before transient transfection with Bright and Sp100 constructs. Transfection of Bright alone led to a 2- to 4-fold increase in CAT expression over basal Eµ levels (Figure 3A–C). Overexpression of Bright and Sp100 decreased Eµ–CAT activity relative to transfection with Bright alone (Figure 3A). That this repression by Sp100 is mediated through Bright interaction is consistent with the finding that Sp100 alone did not alter Eµ–CAT activity.



Fig. 3. Bright transactivation is repressed by Sp100 and stimulated by LYSp100B. J558 B cells, which contained a stably integrated, Eµ-driven CAT reporter (as indicated in the figure), were transfected transiently by electroporation with 10 µg of Bright or its empty expression vector (pBK/CMV) or with 10 or 15 µg of Sp100 or LYSp100B. (A) Sp100 repression of Bright. Empty vector (lane 1), Bright (lane 2), Bright with 10 or 15 µg of Sp100 (lanes 3 and 4, respectively) or of 10 or 15 µg of Sp100 alone (lanes 5 and 6, respectively). (B) LYSp100B activation of Bright. Empty vector (lane 1), Bright (lane 2), Bright plus 10 or 15 µg of LYSp100B (lanes 3 and 4, respectively). (C) LYSp100B activation of endogenous Eµ activity. Empty vector (lanes 1 and 2), Bright (lanes 3 and 4) and LYSp100B (lanes 5 and 6). Average fold increase was calculated from CAT conversation values averaged from four independent experiments following normalization of all TLC loads to luciferase cotransfection. (D) LYSp100B (10 or 15 μg) does not coactivate Bright in Jurkat T cells. (E) LYSp100B (10 or 15 μg) does not coactivate Bright in HeLa cells. (F) LYSp100A activation of Bright in J558B cells. This construct (5, 10 and 15 μg; lanes 6 and 7) expresses a truncated LYSp100 lacking the PHD and bromodomains as indicated in Figure 1A. Lanes 1 and 2 and lanes 3 and 4 are duplicate experiments.
Activation of LYSp100 on Bright transactivation
We next examined the effect of LYSp100B on Bright transactivition in an identical fashion. Instead of repression, LYSp100B contransfection produced significant stimulation: 6- to 10-fold increase compared with basal Eµ activities (Figure 3B). Transfection of LYSp100B alone also stimulated expression, possibly due to an interaction with endogenous Bright in J558 B cells, although the effect required high levels of transfected LYSp100B (Figure 3C). Sp100 and the N-terminus of LYSp100B share extensive (49%) sequence identity, but the extended C-terminus of LYSp100B contains two regions, a PHD and a bromodomain, which in other systems have been shown to affect transcription, DNA binding and/or protein–protein interaction (Haynes et al., 1992; Gibbons et al., 1997). We anticipated that LYSp100B might stimulate Bright transactivation because of a property endowed by these domains. However, deletion of the LYSp100B C-terminus had only modest effects (Figure 3F).
Specificity of repression and coactivation
We utilized mutations both in trans and in cis to verify the specificity of the Sp100 and LYSp100B effects. No repression of Bright was found either by employing a Sp100 mutant expression construct lacking the ‘conserved’ region (CD in Figure 1A) required for Bright interaction (Figure 4A) or with an Eµ–CAT reporter carrying mutations within the four Bright-binding sites (Figure 4B). Neither repression nor stimulation was observed when an irrelevant transcription factor, Oct1 or OcaB, was cotransfected with Bright (Figure 4C), indicating that the targeting specificity of Bright was unaltered. In our system, LYSp100B coactivation was similar in magnitude to that observed for the strong octamer coactivator OcaB (Gstaiger et al., 1995) on Oct2 (Figure 4C, lanes 9 and 10). Finally, the coactivator effect of LYSp100B was not observed in transfections of Jurkat and HeLa cells, which do not express endogenous Bright (Figure 3D and E).

Fig. 4. Specificity of Sp100 and LYSp100 effects on Bright activity. (A) The Bright-interacting region of Sp100 is required to repress Eµ-mediated Bright transactivation. Co-transfections were performed in J558 cells as described in the legend to Figure 3, employing 5′ Sp100 (amino acids 1–350; see Materials and methods) at 5, 10 or 15 μg. (B) Mutation of Bright-binding sites within the Eµ MARs abrogates coactivation. J558 cells were stably transfected with an Eµ CAT reporter (ΔEµ CAT) in which all four P-sites had been deleted. (C) Cotransfection of Eµ octamer transcription factors (Oct2 or OcaB) does not coactivate Bright. Lanes 1 and 2, 3 and 4, 5 and 6, and 7 and 8 depict duplicate experiments.
Both Sp100 and LYSp100B can block MAR DNA-binding activity of Bright
We reasoned that repression by Sp100 may, at least in part, result from direct inhibition of Bright MAR binding. To address this, we examined the effect of Bright interaction with Sp100 or LYSp100B on the DNA binding of Bright in vitro by gel shift assays. Under these conditions, neither Sp100 nor LYSp100B alone bound to MARs or to other DNAs examined (data not shown). As shown in Figure 5, preincubation of Sp100 with Bright disrupted Bright DNA binding at equivalent concentrations (lanes 3 and 4). At these concentrations, LYSp100B did not perturb or supershift the Bright–DNA complex (lane 5). Unexpectedly, in light of the coactivation results, higher concentrations of LYSp100 also blocked Bright binding (lane 6). These results suggest that Sp100 can repress Bright transactivation by interfering with Bright MAR binding, whereas the coactivator properties of LYSp100B derive from a different mechanism. As anticipated from Figure 3D, the C-terminal truncation of LYSp100B had no effect on gel shifts (data not shown).
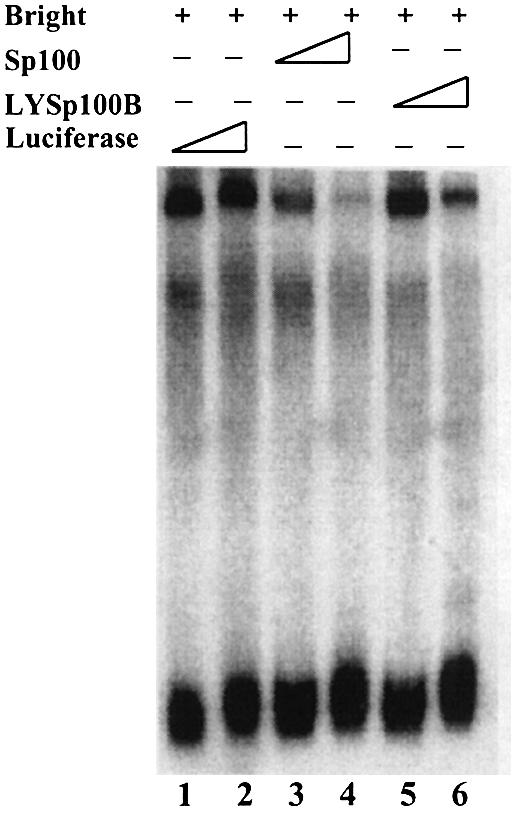
Fig. 5. Differential effects of Sp100 and LYSp100B on MAR-binding activity of Bright. A single MAR-containing 125 bp fragment from the murine S107 VH promoter (TX125) was used as a probe for a gel-shift assay, performed as previously described (Zong and Scheuermann, 1995). Bright, Sp100 and LYSp100B were translated in vitro in a rabbit reticulocyte lysate. In each reaction, ∼10 ng of Bright (+) were preincubated for 30 min at 30°C with 10 or 30 ng of luciferase, Sp100 or LYSp100B (10 or 30 ng) before 32P-labeled probe (∼10 ng) was added.
Colocalization of Bright with Sp100 in PML NBs
The PML transcription factor was identified as part of a fusion gene with the retinoic acid receptor (RAR) in patients suffering from acute PML (Doucas and Evans, 1996). PML is known to colocalize with Sp100 in PML NBs (Dyck et al., 1994; Weis et al., 1994). To seek evidence for Bright and Sp100 interaction in mammalian cells and to determine whether the association of Bright with Sp100 results in colocalization of this complex in the nucleus, we performed two-color immunofluorescence experiments. As previously observed (Dent et al., 1996), both Sp100 and LYSp100B localized strictly to dot-like structures in the nucleus of HeLa (Figure 7A and B) or J558 B cells (Figure 7K and L). As further anticipated, the majority of Sp100 and LYSp100B did not colocalize in the nucleus (Figure 7C). In Sp100 cotransfections, we observed a dot-like green fluorescence pattern for Bright (Figure 6A). Remarkably, when distribution of Sp100 (Figure 6B) was examined in the same sample by immunostaining, its red distribution appeared identical to that of green fluorescent protein (GFP)–Bright. The merged image shown in Figure 6C confirmed the colocalization of Bright and Sp100 in PML NBs in J558 B cells. Identical results were obtained in HeLa cells (data not shown).

Fig. 7. Bright induces redistribution of LYSp100B from PML NB or LANDs. (A–C) Cotransfection of Sp100 and LYSp100B in HeLa cells. GFP–Sp100, green; LYSp100B, red; overlapping Sp100 and LYSp100, yellow. (D–F) Cotransfection of Bright and LYSp100B into HeLa cells. GFP–Bright, green; LYSp100B, red; Bright with LYSp100B, yellow. (G and H) Cotransfection of Bright and LYSp100B into J558 B cells. GFP–Bright, green; LYSp100B, red; GFP–Bright with LYSp100B, yellow. (J–L) Single transfection of Bright, LYSp100 and Sp100 into J558 B cells. Bright, green; LYSp100B, red; Sp100, red.

Fig. 6. Bright colocalizes with Sp100 within PML NBs. Vector constructions and transfections are described in Materials and methods. (A–C) Colocalization of GFP–Bright (green) and Sp100 (red) by overlapping red and green fluorescence (yellow). (D–F) Colocalization of Bright with endogenous Sp100. J558 B cells were transiently transfected with GFP–Bright (D) and then stained with an anti-Sp100 mAb (E). Colocalization was demonstrated in (F). Specificity of the anti-Sp100 antibody reaction was confirmed by the lack of cross-reaction against GFP or Bright (data not shown). (G–I) Nuclear localization of Bright. GFP–Bright, green; phase-contrast view to delineate the nuclear boundaries.
Given these results, it was critical to assess the colocalization of the endogenous proteins which, based on western blotting, are expressed at a level 3- to 5-fold lower than their transfected counterparts (data not shown). Technical difficulties precluded this for Bright. The low level of endogenous Bright in J558 B cells partitioned between dot and diffuse structures (data not shown). However, virtually all endogenous Sp100 colocalized with ectopic GFP–Bright in PML NBs (Figure 6D–F).
Redistribution of LYSp100B in the presence of over-expressed Bright
Dent et al. (1996) found that LYSp100B localized primarily to LANDs, whereas only ∼5% localized to PML NBs. Given this observation and our demonstration that Bright resides in PML NBs, we anticipated that Bright would only colocalize with LYSp100B in PML NBs. To test this, we cotransfected GFP–Bright (Figure 7D) with HA–LYSp100B (Figure 7E) into J558 cells and examined them by two-color immunofluorescence. As shown by the merged image of Figure 7F, a small percentage of Bright and LYSp100B colocalized in the nucleus. However, the majority of LYSp100B appeared to redistribute in the presence of Bright. To confirm and extend this observation, we performed the same experiments by using HeLa cells, which have smaller nuclei than J558 cells. In HeLa, Bright (Figure 7G) localized exclusively to the nucleus, as it did in J558. In contrast, LYSp100B staining (Figure 7H) was dispersed in the presence of Bright, with only a small fraction of the two proteins colocalizing within the nucleus (Figure 7I). The redistribution of LYSp100B appeared to be cytoplasmic, as demonstrated in Figure 7B and C. Further evidence that the relocalization of LYSp100B was induced by Bright was that redistribution of LYSp100B was not observed when it was transfected in the absence of Bright (Figure 7K) or when cotransfected with Sp100 (Figure 7B). The nuclear localization for these proteins was further confirmed by single transfections of Bright (Figure 7J) or Sp100 (Figure 7L).
We suggest that Bright associates with LYSp100B only in PML-containing NBs. Acquisition of (presumably) high levels of Bright in the nucleus results in delocalization of LYSp100B and, potentially, redistribution and/or disruption of LYSp100-containing LANDs. This finding is consistent with the results of our transactivation experiments, strongly suggesting that the subnuclear localization of the two proteins determines their positive or negative contribution to Bright transcriptional activity.
Association of Sp100 and LYSp100-containing nuclear dots with the nuclear matrix
Bright is associated with the nuclear matrix (Herrscher et al., 1995), so we examined whether Sp100 or LYSp100B share the same feature within the context of their subnuclear compartments. We transfected GFP, or GFP fusions of Bright, Sp100 and LYSp100B, into HeLa or J558 cells, isolated the nuclear matrix by high salt extraction (Zong and Scheuermann, 1995) and examined the preparations by fluorescence microscopy. Reminiscent of its pattern in intact nuclei, Bright localized within the HeLa matrix in a punctate distribution. Although this was more diffuse than the discrete dots observed in nuclei, the results suggest that PML NBs remain intact under the conditions employed (Figure 8A). Similarly, Sp100 and LYSp100B were detected in the nuclear matrix preparations (Figure 8B and C), indicating that both proteins are associated with the nuclear matrix. The lack of staining for GFP in the nuclear matrix preparations demonstrated the specificity of these interactions. The results support the notion that PML NBs associate tightly with the nuclear matrix. As evidenced by their similar staining patterns (Figure 8D and E), the majority of Sp100 and Bright colocalizes (Figure 8F) within the nuclear matrix of J558 B cells.

Fig. 8. Association of Bright-, Sp100- and LYSp100B-containing subnuclear domains with the nuclear matrix. Nuclear matrix preparations from transfected HeLa cells retained speckled structures (see Figure 4) containing (A) Bright, (B) Sp100 and (C) LYSp100B. Cotransfection of HA–Bright and GFP–Sp100 into J558 B cells results in a fraction of matrix-associated, PML NB colocalization. (D) Sp100 (green), (E) Bright (red) and (F) both overlapped (yellow).
Discussion
Our results provide evidence of a role for PML NBs in transcriptional regulation of immunoglobulin gene expression. We present an example in which the contribution of a transactivator of this locus is determined by its localization within this subnuclear structure. The major components of these subnuclear domains are matrix-associated and interact with a MAR-binding transcription factor. This suggests that these nuclear matrix-associated particles access the MAR regulatory elements in chromatin by interacting with a MAR-binding protein, since PML NBs do not bind directly to DNA. Therefore, identification of the interaction of a B-cell-specific transcription factor, Bright, with Sp100, the defining component of PML NBs, and with LYSp100B, the defining component of LANDs, has broad implications for understanding the function of subnuclear domains.
Sp100 is an autoantigen that was originally identified in patients suffering from the autoimmune disease, primary biliary cirrhosis (Szostecki et al., 1990). Initially it was speculated that Sp100 might play a positive role in transcriptional regulation (Xie et al., 1993). Although Sp100 does not bind DNA, weak corepressor activity was found when Gal4 DNA-binding domain fusions of Sp100 were transfected into COS cells (Xie et al., 1993). However, more recent studies found Sp100 to be a repressor when tethered to DNA in this manner (Lehming et al., 1998; Seeler et al., 1998). Its physical interaction with Bright and the functional consequences observed here provide the most direct evidence for Sp100 involvement in negative regulation of gene expression. We speculated earlier that Bright may regulate transcription through remodeling chromatin. The interaction of Bright with Sp100 further supports this notion. Sp100 also interacts with members of the heterochromatin protein 1 (HP1) family of non-histone chromosomal proteins (Seeler et al., 1998) and the human high mobility group 2 (hHMG2) family of non-histone chromosomal DNA-binding proteins (Lehming et al., 1998). Bright might contact chromatin indirectly through interaction with Sp100, which associates directly with HP1.
Various models of repression have been proposed, including competitive binding of repressors to activator elements and the local quenching of transcriptional activators. The results presented here indicate that Sp100 can repress Bright by competing for MAR-binding sites. Presumably this would occur within PML NBs, based on our finding of an association of PML NBs with the nuclear matrix. Sp100 interacts with the domain (REKLES) within Bright required for its homotetramerization (Herrscher et al., 1995; Webb et al., 1999). Loss of this function would eliminate a property necessary for high-affinity MAR binding.
Bright and LYSp100B are expressed only in B lymphocytes, and the expression of both is maximal at the same stage in lineage differentiation (Dent et al., 1996; Webb et al., 1998). Therefore the mechanism by which the interaction stimulates Bright transactivation takes on biological significance. Our observation of Bright-induced LAND disruption suggests a possible coactivation model in which MAR-binding interference by Sp100 is relieved through subnuclear structure relocation. So, despite our observation that LYSp100B (at higher concentrations than Sp100) can complete for Bright MAR binding in vitro, LAND disruption in vivo results in physical separation of LYSp100B from Bright. This explains why we failed to detect LYSp100B–Bright interaction by coimmunoprecipitation of lysates in which Bright and Sp100 remain associated. While this is the first case for disruption and subsequent relocalization of a component of LANDs, similar phenomena for PMB NBs have been documented in DNA or RNA viral infections (see for example, Maul and Everett, 1994; Puvion-Dutilleul et al., 1995; Doucas et al., 1996; Ishov et al., 1996; Szekeley et al., 1996; Borden et al., 1998), the SCA1 neurodegenerative disorder (Matilla et al., 1997; Skinner et al., 1997) and in APL and acute malignancy of hepatocytes (Dyck et al., 1994; Koken et al., 1994; Weis et al., 1994; Terris et al., 1995). Our results differ from all these cases in that the PML transcription factor is not implicated. Our situation most resembles that of APL, where cytoplasmic redistribution of PML occurs without notable reformation of the dispersed NBs. We speculate that, following dispersion, the ‘free’ Bright remaining in the nucleoplasm is the active, MAR-binding form. An accompanying chromatin-remodeling event could be mediated by this putative free form. Alternatively, but not necessarily mutually exclusively, the Sp100-associated form of Bright still residing in PML NBs might be derepressed. As a precedent, several transcription factors, including the PML–RAR fusion protein, interact with a protein complex that includes histone deacetylase activity (Grignani et al., 1998; Lin et al., 1998). Bright-mediated disruption of LANDs may result in removal of histone deacetylase activity from PML NBs.
Materials and methods
Yeast two-hybrid screening
The Matchmaker yeast two-hybrid system was used to isolate Bright-interacting partners. The Bright coding sequence (216–701) was inserted in yeast expression vector pAS2-1 (Clontech). In-frame fusion of the protein sequence was confirmed by DNA sequence analysis. This bait was used to screen a human lymphocyte cDNA library (Clontech). Twenty-four cDNA clones were identified, one of which was isolated four times and was chosen for further characterization. Another clone, a ubiquitin-conjugated enzyme that involves the post-translation modification of Bright, will be presented elsewhere. To determine specificity, yeast mating and β-galactosidase liquid assays were performed according to the Clontech manual.
cDNA cloning and plasmid construction
To obtain full-length cDNA for Sp100, Marathon cDNA amplification was performed according to the Clontech manual. The PCR product corresponding in size to full-length Sp100 cDNA was sequenced and subcloned into the mammalian expression vector, pCR3.1 (Invitrogen). Plasmid pCR3.1/5′ Sp100 contains only the 5′ 350 amino acids of Sp100. The plasmid was constructed by PCR amplification to delete the Bright interaction region (CD in Figure 1A). CAT reporter gene constructs Eµ–CAT and ΔEµ–CAT were generated using the reporter vector pTKCAT as described by Wang et al. (1999).
Immunoprecipitations and immunoblotting
The cDNA sequences encoding Sp100 were inserted into the pCR3.1 expression vector along with an N-terminal HA epitope. The resulting constructs (pCR3.1/HA–Sp100) and pBK–CMV/Bright were used as templates for in vitro programmed transcription/translation or for transfection. For in vitro interaction experiments, [35S]methionine-labeled Bright and HA-tagged Sp100 or Bright and luciferase were incubated for 1 h, then the Bright/HA–Sp100 complex was immunoprecipitated with a monoclonal anti-HA antibody (Boehringer Mannheim). The protein complex was separated on protein A–Sepharose, washed four times and then analyzed by 10% SDS–PAGE. For Bright–LYSp100B in vitro interaction experiments, an affinity-purified LYSp100 rabbit antiserum was used to precipitate the complex as described above. In vivo interaction between Bright/HA–Sp100 was analyzed 48 h after coelectroporation (15 µg each) into HeLa cells. The transfected HeLa cells or BCL1 and J558 B cells were lysed in lysis buffer (10 mM HEPES pH 7.9, 0.5 M NaCl, 0.05% Nonidet P-40, 0.5 mM EDTA, 0.5 mM phenylmethylsulfonylfluoride), then putative complexes were immunoprecipitated on protein A–Sepharose after a 2 h incubation. Complexes were removed by boiling, resolved by 10% PAGE, then visualized by western blotting using an anti-HA monoclonal antibody or anti-Bright serum.
Cell culture and transfections
Plasmacytoma J558L was cultured in RPMI with 10% fetal calf serum (FCS). Jurkat T cells were cultured in the same medium with HEPES. HeLa cells were maintained in Dulbecco’s modified Eagle medium with 10% FCS. Stable J558L reporter lines were prepared by transfection with Eµ–CAT, a vector in which the minimal herpes simplex thymidine kinase promoter drives CAT under the influence of the MAR-dependent Eµ intronic enhancer. Twenty days after transfection and selection in G418, Eµ–CAT-bearing cells were transfected transiently by electroporation or FuGENE (Boehringer Mannheim). CAT assays were performed by thin layer chromatography of whole cell lysates 48 h after transfection and were normalized by luciferase cotransfection.
Immunofluorescence and confocal microscopy
HeLa cells were transfected by lipofection with full-length expression plasmids of Bright, Sp100, LYSp100B or GFP–Bright. For J558 B cells, only GFP–Bright was transfected to monitor green fluorescence. Controls included blue fluorescent protein (BFP) fusions of Bright or Sp100. Forty-eight hours after transfection, cells were fixed with 3.7% paraformaldehyde for 20 min and then permeabilized with 0.5% Triton X-100 for 15 min. For immunostaining, proteins were detected with affinity-purified polyclonal rabbit antibodies against Bright, Sp100 and LYSp100B and a rhodamine-conjugated secondary antibody (KPL). For endogenous Sp100 detection, we employed mouse monoclonal antibody 1550 (Maul et al., 1995). Stained cells were analyzed using confocal laser scanning microscopy and Nikon fluorescence microscopy.
Electrophoretic gel-shift assays
Nuclear extracts from various cell lines were prepared according to Dignam et al. (1983). In vitro-translated proteins were generated in the TNT coupled reticulocyte lysate system (Promega) according to the manufacturer’s instructions. The DNA fragments employed as probes were MARs from the VHS107 IgH promoter (TX125 or Bf150) or the Eµ enhancer. The gel-shift assay was performed as described by Herrscher et al. (1995).
Accession number
The DDBJ/EMBL/GenBank accession number of Bright is U60335.
Acknowledgments
Acknowledgements
We thank Drs A.Dent and L.Staudt for kindly providing LYSp100B constructs, antibodies and unpublished observations. We thank Dr G.Maul for mAb 1550. We thank Mr I.Ghosh for DNA sequencing, Ms N.Xiao for oligonucleotide synthesis, Dr J.Mendenhall for assistance with confocal microscopy and Mr L.Probst for assistance in the two-hybrid screen. We thank Drs L.Staudt, M.Kaplan, R.Gaynor and R.Scheuermann for comments on the manuscript and Mr U.Das for help in preparing it. The work is supported by NIAD and NCI grants to P.W.T.
References
- Alcalay M., Tomassoni,L., Colombo,E., Stoldt,S., Grignani,F., Fagioli,M., Szekely,L., Hellin,K. and Pelicci,P.G. (1998) The promyelocytic leukemia gene product (PML) forms stable complexes with the retinoblastoma protein. Mol. Cell. Biol., 18, 1084–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ascoli C.A. and Maul,G.G. (1991) Identification of a novel nuclear domain. J. Cell Biol., 112, 785–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell A., Boyes,J., Chung,J., Pikaart,M., Prioleau,M.-N., Recilias,F., Saitoh,N. and Felsenfeld,G. (1998) The establishment of active chromatin domains. Cold Spring Harb. Symp. Quant. Biol., 63, 509–514. [DOI] [PubMed] [Google Scholar]
- Bloch D.B., de la Monte,S.M., Guigaouri,P., Fllippov,M.M. and Bloch,K.D. (1996) Identification and characterization of a leukocyte-specific component of the nuclear body. J. Biol. Chem., 46, 29198–29204. [DOI] [PubMed] [Google Scholar]
- Bloch D.B., Chiche,J.D., Orth,D., de la Monte,S.M., Rosenzweig,A. and Bloch,K.D. (1999) Structural and functional heterogeneity of nuclear bodies. Mol. Cell. Biol., 19, 4423–4430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boddy M.N, Howe,K., Etkin,L.D., Solomon,E. and Freemon,P.S. (1996) PIC-1, a novel ubiquitin-like protein which interacts with the PML component of a mulitprotein complex that is disrupted in acute promyelocytic leukaemia. Oncogene, 13, 971–982. [PubMed] [Google Scholar]
- Borden K.L.B., Dwyer,E.J.C. and Salvato,M.S. (1998) An arenavirus RING (zinc-binding) protein binds the oncoprotein promyelocyte leukamia protein (PML) and relocates PML nuclear bodies to the cytoplasm. J. Virol., 72, 758–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang K., Fan,Y., Andreeff,M., Liu,J. and Mu,Z. (1995) The PML gene encodes a phosphoprotein associated with the nuclear matrix. Blood, 85, 3646–3653. [PubMed] [Google Scholar]
- Dent A.L., Yewdell,J., Puvion-Dutilleul,F., Koken,M.H.M., de The,H. and Staudt,L.M. (1996) LYSp100-associated nuclear domains (LANDs): description of a new class of subnuclear structures and their relationship to PML nuclear bodies. Blood, 88, 1423–1436. [PubMed] [Google Scholar]
- Desbois C., Rousset,R., Bantignies,F. and Jalinot,P. (1996) Exclusion of Int-6 from PML nuclear bodies by binding to the HTLV-I Tax oncoprotein. Science, 273, 951–953. [DOI] [PubMed] [Google Scholar]
- Deshaies R.J. (1995) Make it or break it: the role of ubiquitin-dependent protein lysis in cellular regulation. Trends Cell Biol., 5, 428–434. [DOI] [PubMed] [Google Scholar]
- Desterro J.M.P., Rodriguez,M.S. and Hay,R.T. (1998) SUMO-1 modification of IκBα inhibits NF-κB activation. Mol. Cell, 2, 233–239. [DOI] [PubMed] [Google Scholar]
- Dignam J.D., Lebovitz,R.M. and Roeder,R.G. (1983) Accurate transcription initiation by RNA polymerase II in a soluble extract isolated from mammalian nuclei. Nucleic Acids Res., 11, 1475–1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doucas V. and Evans,R.M. (1996) The PML nuclear compartment and cancer. Biochim. Biophys. Acta, 1288, M25–M29. [DOI] [PubMed] [Google Scholar]
- Doucas V., Ishov,A.M., Romo,A., Juguilon,J., Weltzman,M., Evans,R.M. and Maul,G.G. (1996) Adenovirus replication is coupled with the dynamic properties of the PML nuclear structure. Genes Dev., 10, 196–207. [DOI] [PubMed] [Google Scholar]
- Dyck J.A., Maul,G.G., Miller,W.H.J., Chen,J.D., Kakizuka,A. and Evans,R.M. (1994) A novel macromolecular structure is a target of the promyelocytic–retinoic acid receptor oncoprotein. Cell, 76, 333–343. [DOI] [PubMed] [Google Scholar]
- Forrester W.C., van Genderen,C., Jenuwein,T. and Grosschedl,R. (1994) Dependence of enhancer-mediated transcription of the immuno globulin gene on nuclear matrix attachment regions. Science, 265, 1221–1225. [DOI] [PubMed] [Google Scholar]
- Gibbons R.J. et al. (1997) Mutations in transcriptional regulator ATRX establish the functional significance of a PHD-like domain. Nature Genet., 17, 146–148. [DOI] [PubMed] [Google Scholar]
- Grignani F. et al. (1996) Effects on differentiation by the promyelocytic leukemia PML/RARα protein depend on the fusion of the PML protein dimerization and RARα DNA binding domains. EMBO J., 15, 4949–4958. [PMC free article] [PubMed] [Google Scholar]
- Grignani F. et al. (1998) Fusion proteins of the retinoic acid receptor-α recruit histone deacetylase in promyelocytic leukaemia. Nature, 391, 815–818. [DOI] [PubMed] [Google Scholar]
- Gstaiger M., Knoepfl,L., Georgiev,O., Schaffner,W. and Hovens,C.M. (1995) A B-cell coactivator of octamer-binding transcription factors. Nature, 373, 360–362. [DOI] [PubMed] [Google Scholar]
- Haynes S.R., Dollard,C., Winston,F., Beck,S., Trowsdale,J. and Dawid,I.B. (1992) The bromodomain: a conserved sequence found in human, Drosophila and yeast proteins. Nucleic Acids Res., 20, 2603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrscher R.F., Kaplan,M.H., Lelsz,D.L., Scheuermann,R.H. and Tucker,P.W. (1995) The immunoglobulin heavy-chain matrix associating regions are bound by Bright: a B cell-specific trans-activator that describes a new DNA-binding protein family. Genes Dev., 9, 3067–3082. [DOI] [PubMed] [Google Scholar]
- Ishov A.M. and Maul,G.G. (1996) The periphery of nuclear domain 10 (ND10) as a site of DNA virus deposition. J. Cell Biol., 134, 815–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Käs E., Poljak,L., Adachi,Y. and Laemmli,U.K. (1993) A model for chromatin opening: stimulation of topoisomerase II and restriction enzyme cleavage of chromatin by distamycin. EMBO J., 12, 115–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koken M.H.M. et al. (1994) The t(15;17) translocation alters a nuclear body in a retinoic acid-reversible fashion. EMBO J., 13, 1073–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korioth F., Gleffers,C., Maul,G.G. and Frey,J. (1995) Molecular characterization of NDP52, a novel protein of the nuclear domain 10, which is redistributed upon virus infection and interferon treatment. J. Cell Biol., 130, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laemmli U.K., Kas,E., Poljak,L. and Adachi,Y. (1992) Scaffold-associated regions: cis-acting determinants of chromatin structural loops and functional domains. Curr. Opin. Genet. Dev., 2, 275–285. [DOI] [PubMed] [Google Scholar]
- Lamond A.I. and Earnshaw,W.C. (1998) Structure and function in the nucleus. Science, 280, 547–553. [DOI] [PubMed] [Google Scholar]
- Lehming N., Saux,A.L., Schuller,J. and Patshne,M. (1998) Chromatin components as part of a putative transcriptional repressing complex. Proc. Natl Acad. Sci. USA, 95, 7322–7326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin R.J., Nagy,L., Inoue,S., Shao,W., Miller,W.H.,Jr and Evans,R.M. (1998) Role of the histone deacetylase complex in acute promyelocytic leukaemia. Nature, 391, 811–814. [DOI] [PubMed] [Google Scholar]
- Matilla A.B., Koshy,T., Cummings,C.J., Isobe,T., Orr,H.T. and Zoghbi,H.Y. (1998) The cerebellar leucine-rich acidic nuclear protein interacts with ataxin-1. Nature, 389, 974–978. [DOI] [PubMed] [Google Scholar]
- Maul G.G. and Everett,R.D. (1994) The nuclear location of PML, a cellular member of the C3HC4 zinc-binding domain protein family, is rearranged during herpes simplex virus infection by the C3HC4 viral protein ICP0. J. Gen. Virol., 75, 1223–1233. [DOI] [PubMed] [Google Scholar]
- Maul G.G., Guldner,H.H. and Spvack,J.G. (1993) Modification of discrete nuclear domains induced by herpes simplex virus type-1 immediate early gene 1 product (ICP0). J. Gen. Virol., 74, 2679–2690. [DOI] [PubMed] [Google Scholar]
- Maul G.G., Yu,E., Ishov,A.M. and Epstein,A.L. (1995) Nuclear domain 10 (ND10) associated proteins are also present in nuclear bodies and redistribute to hundreds of nuclear sites after stress. J. Cell. Biochem., 59, 489–513. [DOI] [PubMed] [Google Scholar]
- McKnight R.A., Shamay,A., Sankaran,L., Wall,R.J. and Henninghausen,L. (1992) Matrix-attachment regions can impart position-independent regulation of a tissue-specific gene in transgenic mice. Proc. Natl Acad. Sci. USA, 89, 6943–6947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mittnachit S. and Weinberg,R.A. (1991) G1/S phosphorylation of the retinoblastoma protein is associated with an altered affinity for the nuclear matrix. Cell, 65, 381–393. [DOI] [PubMed] [Google Scholar]
- Mu Z., Chin,K.V., Liu,J.H., Lozano,G. and Chang,K.S. (1994) PML, a growth-suppressor disrupted in acute promyelocytic leukemia. Mol. Cell. Biol., 14, 6858–6867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller S., Matunis,M.J. and Dejean,A. (1998) Conjugation with the ubiquitin-related modifier SUMO-1 regulates the partitioning of PML within the nucleus. EMBO J., 17, 61–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oancea A.E., Berru,M. and Shulman,M.J. (1997) Expression of the (recombinant) endogenous immunoglobulin heavy-chain locus requires the intronic matrix attachment regions. Mol. Cell. Biol., 17, 2658–2668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pemov A., Bavykin,S. and Hamlin,J.L. (1998) Attachment to the nuclear matrix mediates specific alterations in chromatin structure. Proc. Natl Acad. Sci. USA, 95, 14757–14762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson C.L. and Tamkun,J.W. (1995) The SWI–SNF complex: a chromatin remodeling machine? Trends Biochem. Sci., 20, 143–146. [DOI] [PubMed] [Google Scholar]
- Puvion-Dutilleul F., Chelbi-Alix,M.K., Koken,M., Quignon,F., Puvion,E. and de The,H. (1995). Adenovirus infection induces rearrangements in the intranuclear distribution of the nuclear body associated PML protein. Exp. Cell Res., 218, 9–16. [DOI] [PubMed] [Google Scholar]
- Reyes J.C., Muchardt,C. and Yaniv,M. (1997) Components of the human SWI/SNF complex are enriched in active chromatin and are associated with the nuclear matrix. J. Cell Biol., 137, 263–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnitzler G.R., Sif,S. and Kingston,R.E. (1998) A model for chromatin remodeling by the SWI/SNF family. Cold Spring Harb. Symp. Quant. Biol., 63, 535–543. [DOI] [PubMed] [Google Scholar]
- Schwarz S.E., Matuschewsk,K., Liakopoulos,D., Scheffner,M. and Jentsch,S. (1998) The ubiquitin-like proteins SMT3 and SUMO-1 are conjugated by the UBC9 E2 enzyme. Proc. Natl Acad. Sci. USA, 95, 560–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeler J.-S., Marchio,A., Sitterlin,D., Transy,C. and Dejean,A. (1998) Interaction of Sp100 with HP1 proteins: a link between the promyelocytic leukemia-associated nuclear bodies and the chromatin compartment. Proc. Natl Acad. Sci. USA, 95, 7316–7321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh L., Panicker,S.G., Nagaraj,R. and Majumdar,K.C. (1994) Banded krait micro-satellite (Bkm)-associated Y chromosome-specific repetitive DNA in mouse. Nucleic Acids Res., 22, 2289–2295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skinner P.J., Koshy,B.T., Cummings,C.J., Klement,I.A., Helin,K., Servadio,A., Zoghbi,H.Y. and Orr,H.T. (1997) Ataxin-1 with an expanded glutamine tract alters nuclear matrix-associated structures. Nature, 389, 971–974. [DOI] [PubMed] [Google Scholar]
- Spector D.L. (1993) Macromolecular domains within the cell nucleus. Annu. Rev. Cell. Biol., 9, 265–315. [DOI] [PubMed] [Google Scholar]
- Stein G.S., van Wijnen,A.J., Stein,J.L., Lian,J.B., Pockwinse,S. and McNeil,S. (1998) Interrelationships of nuclear structure and transcriptional control: functional consequences of being in the right place at the right time. J. Cell. Biochem., 70, 200–212. [PubMed] [Google Scholar]
- Sternsdorf T., Grotzinger,T., Jensen,K. and Will,H. (1997) Nuclear dots: actors on many stages. Immunoblotting, 198, 307–331. [DOI] [PubMed] [Google Scholar]
- Stief A., Winter,D.M., Stratling,W.H. and Sippel,A.E. (1989) A nuclear DNA attachment element mediates elevated and position-independent gene activity. Nature, 341, 343–345. [DOI] [PubMed] [Google Scholar]
- Szekely I., Pokrovskaja,K., Jiang,W.A., de The,H., Ringertz,N., Klein,G. (1996) The Epstein–Barr virus-encoded nuclear antigen EBNA-5 accumulates in PML-containing bodies. J. Virol., 70, 2562–2568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szostecki C., Guldner,H.H., Netter,H.J. and Will,H. (1990) Isolation and characterization of cDNA encoding a human nuclear antigen predominantly recognized by autoantibodies from patients with primary biliary cirrhosis. J. Immunol., 145, 4338–4347. [PubMed] [Google Scholar]
- Terris B., Baldin,V., Dubois,S., Degott,C., Flejou,J.F., Henin,D. and Dejean,A. (1995) PML nuclear bodies are general targets for inflammation and cell proliferation. Cancer Res., 55, 1590–1597. [PubMed] [Google Scholar]
- Wang Z., Goldstein,A., Zong,R., Lin,D., Neufeld,E.J., Scheuermann,R.H. and Tucker,P.W. (1999) Cux/CDP homeoprotein is a component of NF-µNR and represses the immunoglobulin heavy chain intronic enhancer by antagonizing the Bright transcription activator. Mol. Cell. Biol., 19, 284–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webb C.F., Das,C., Eaton,S., Calame,K. and Tucker,P.W. (1991) Novel protein–DNA interactions associated with increased immunoglobulin transcription in response to antigen plus interleukin-5. Mol. Cell. Biol., 11, 5197–5205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webb C.F., Smith,E.A., Buchanan,K.L., Smithson,G. and Dou,S. (1998) Expression of Bright at two distinct stages of B lymphocyte development. J. Immunol., 160, 4747–4754. [PubMed] [Google Scholar]
- Webb C. et al. (1999) Differential regulation of immunoglobulin gene transcription via nuclear matrix associated regions. Cold Spring Harb. Symp. Quant. Biol., 64, 109–118. [DOI] [PubMed] [Google Scholar]
- Weis K., Rambaud,S., Lavau,C., Jansen,J., Carvalho,T., Carmofonseca,M., Lamond,A. and Dejean,A. (1994) Retinoic acid regulates aberrant nuclear localization of PML-RARα in acute promyelocytic leukemia cells. Cell, 76, 345–356. [DOI] [PubMed] [Google Scholar]
- Xie K., Lambie,E. and Snyder,M. (1993) Nuclear dot antigens may specify transcriptional domains in the nucleus. Mol. Cell. Biol., 13, 6170–6179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zong R. and Scheuermann,R.H. (1995) Mutually exclusive interaction of a novel MAR-binding protein and the NF–µNR enhancer repressor: implications for regulation of immunoglobulin heavy-chain expression. J. Biol. Chem., 270, 24010–24018. [DOI] [PubMed] [Google Scholar]
- Zhu Z., Cai,W. and Schaffer,P.A. (1994) Cooperativity among herpes simplex virus type 1 immediate-early regulatory proteins: ICP4 and ICP27 affect the intracellular localization of ICP0. J. Virol., 68, 3027–3040. [DOI] [PMC free article] [PubMed] [Google Scholar]