

Abstract
The RET (rearranged during transfection) proto-oncogene encodes a tyrosine kinase receptor involved in both multiple endocrine neoplasia type 2 (MEN 2), an inherited cancer syndrome, and Hirschsprung disease (HSCR), a developmental defect of enteric neurons. We report here that the expression of RET receptor induces apoptosis. This pro-apoptotic effect of RET is inhibited in the presence of its ligand glial cell line-derived neurotrophic factor (GDNF). Furthermore, we present evidence that RET induces apoptosis via its own cleavage by caspases, a phenomenon allowing the liberation/exposure of a pro-apoptotic domain of RET. In addition, we report that Hirschsprung-associated RET mutations impair GDNF control of RET pro-apoptotic activity. These results indicate that HSCR may result from apoptosis of RET-expressing enteric neuroblasts.
Keywords: apoptosis/caspase/Hirschsprung/RET/tyrosine kinase receptor
Introduction
RET (rearranged during transfection) is a proto-oncogene that encodes a tyrosine kinase receptor (RTK) (Takahashi et al., 1985). As for other RTKs, RET is a cell-surface protein that possesses an extracellular ligand-binding domain, a single hydrophobic transmembrane region and an intracytoplasmic tyrosine kinase domain (Takahashi and Cooper, 1987). RET also contains a cadherin-like domain in its extracellular region, suggesting a role for RET in cell–cell interaction (Takeichi, 1991). Recently, it has been shown that RET is the signaling component of a multisubunit complex acting as a receptor for the glial cell line-derived neurotrophic factor (GDNF) (Jing et al., 1996), neurturin, artemin and persephin (Kotzbauer et al., 1996), four homologous neurotrophic factors related to the transforming growth factor-β family. These four ligands require glycosylphosphatidylinositol (GPI)-linked glycoprotein receptors (GFRα-1 to -4) to allow RET dimerization (Baloh et al., 1997). The GDNF signal mediated through RET and GFRα-1 plays a critical role in the development of the enteric nervous system and kidney, as attested by the similar and striking phenotype of mice with null mutations in GDNF, RET or GFRα-1 genes (Schuchardt et al., 1994; Sanchez et al., 1996; Cacalano et al., 1998). Following interaction with its ligands, RET undergoes autophosphorylation and then interacts with multiple effectors such as phospholipase Cγ, Shc, Enigma, Grb2, Grb7/Grb10, Src kinase and Ras-GAP (Santoro et al., 1994; Arighi et al., 1997; Lorenzo et al., 1997). As described extensively for several RTKs, the generation of a RET-mediated downstream signal depends on the transient assembly of multimolecular complexes including various adapter proteins and enzymes (Pawson and Scott, 1997).
Mutations of the RET gene have been associated with multiple endocrine neoplasia type 2 (MEN 2), an autosomal dominant inherited cancer syndrome (Mulligan et al., 1993), and with Hirschsprung disease (aganglionosis; HSCR), a frequent congenital intestinal malformation (1 in 5000 live births) characterized by the absence of neural crest-derived parasympathetic neurons of the hindgut (Edery et al., 1994; Romeo et al., 1994). In vitro, MEN 2-associated mutations lead to ligand-independent constitutive activation of the RET kinase activity through either covalent dimerization of the receptor (MEN 2A) (Santoro et al., 1995) or direct structural changes in its kinase domains (MEN 2B) (Songyang et al., 1995). In contrast, the mechanisms leading to the absence of intramural ganglion cells of the hindgut observed in HSCR remain incompletely understood. Although we and others have shown previously that some HSCR-associated mutations may impair the RET function, whether aganglionosis results from a defect of either migration or maintenance of neural crest-derived enteric neuroblasts, or both, is still unknown (Pasini et al., 1995; Pelet et al., 1998).
The observation that RET is involved in both cancer progression and nervous system development is strikingly similar to the behavior of the so-called dependence receptors p75NTR and DCC (deleted in colorectal cancer) (Rabizadeh et al., 1993; Mehlen et al., 1998). These transmembrane receptors have recently been shown to induce cell death in the absence of their ligands while they transduce a completely different signal in the presence of their ligands. In the case of DCC, this receptor was shown to induce apoptosis mediated by its intracellular domain via its cleavage by caspases or possibly other proteases, a crucial phenomenon leading to the exposure or release of a pro-apoptotic domain (Mehlen et al., 1998). Here we describe RET as a new dependence receptor. It induces apoptosis in the absence of ligand, and the addition of GDNF is then sufficient to block RET pro-apoptotic activity. Moreover RET is cleaved in vitro by caspase-3 and the caspase cleavage sites are crucial for RET-induced cell death, probably by allowing the release of a pro-apoptotic fragment lying from amino acid 708 to 1017. Finally, we suggest that HSCR may result from apoptosis of RET-expressing enteric neuroblasts, since five different HSCR-associated RET mutants are constitutive inducers of cell death, independent of the presence of RET ligand.
Results
Expression of RET induces apoptosis, which is blocked by GDNF
The full-length human RET was expressed in 293T human embryonic kidney (HEK) cells and in immortalized olfactory neuroblasts 13.S.24 (Mehlen et al., 1999). Figure 1A and B (insets) shows RET expression 48 h after transfection in 293T and 13.S.24 cells. To address the involvement of RET in cell death, populations transfected either with a mock or RET-expressing vector were analyzed for trypan blue staining as an indication of cell death. As shown in Figure 1A, a marked increase of trypan blue staining was detected in the RET-transfected population. Furthermore, co-expression of RET co-receptor GFRα-1 induced an increase in RET-induced cell death (Figure 1B). GFRα-1 not only increased the percentage of cell death but also accelerated this phenomenon, since while RET expression led to detectable cell death only after 48 h of transfection, its co-expression with GFRα-1 induced cell death after only 18 h (not shown). Virtually identical results were obtained when RET was co-expressed with GFRα-1 in the immortalized olfactory neuroblasts 13.S.24 (Figure 1B). To determine whether RET induced apoptotic cell death, caspase activation was measured by following cleavage of Ac-DEVD [although Ac-DEVD is preferentially cleaved by caspase-3, other caspases may also cleave this substrate (Stennicke and Salvesen, 1997)]. Figure 1C shows that RET expression leads to a marked increase in caspase activation. As a second assay for apoptosis, DNA fragmentation was measured by TUNEL reactivity. As shown in Figure 1D, RET expression in 13.S.24 cells is correlated with an increased number of TUNEL-positive cells. Taken together, these data demonstrate that RET has a pro-apoptotic effect on both 293T and 13.S.24 cells.

Fig. 1. RET induces apoptosis in the absence of GDNF. 293T or 13.S.24 cells were transiently transfected with either the RET expression plasmid pJ7Ω-ret9 (RET) or pCMV control plasmid (cont.). (A) 293T cell death induced by RET expression monitored by trypan blue staining. The percentage of cell death is presented as the percentage of trypan blue-positive cells in the different transfected cell populations. (B) 293T or 13.S.24 cells were co-transfected with the GFRα-1 expression plasmid (GFRα-1) and either pJ7Ω-ret9 (RET) or pCMV control plasmid (cont.). Cell death was estimated as in (A). Insets (A) and (B): 293T or 13.S.24 cells transfected with either a mock plasmid (cont.) or the RET-expressing plasmid (RET) were analyzed for RET expression by western blot assay using anti-RET antibody. While RET is not endogenously expressed in 293T or 13.S.24 cells, transfection assays allow the detection of RET in both cell lines. (C and D) RET induces caspase activation and DNA fragmentation. RET expression plasmid or a mock plasmid were co-transfected in the presence of the GFRα-1 expression plasmid into 293T cells (C) or 13.S.24 cells (D), and 48 h after transfection, caspase activity or DNA fragmentation was determined as described in Materials and methods. In (C), relative caspase activity was determined as the ratio between the caspase activity of the co-transfected RET+GFRα-1 cells and that measured in cells transfected with GFRα-1; for all samples, the background remaining after inhibition by Ac-DEVD-CHO was subtracted. In (D), the index of TUNEL-positive cells is presented as the ratio between the number of TUNEL-positive cells in the RET+GFRα-1-transfected population versus the GFRα-1-transfected population. Standard deviations are indicated (n = 3).
We then analyzed the effect of the RET ligand, GDNF, on RET-induced apoptosis. As shown in Figure 2, RET-induced cell death was blocked when 0.1 µg/ml GDNF was added to the culture medium 24 h after transfection (Figure 2). A similar inhibition of cell death was monitored in the presence of lower doses of GDNF with a correlation between the amount of GDNF and the quality of the block of RET-induced cell death (Figure 2). The addition of GDNF also led to a marked reduction in the number of TUNEL-positive 13.S.24 cells transfected with RET (not shown). Hence, RET induced apoptosis in the absence of its ligand, GDNF, while RET pro-apoptotic activity was blocked by GDNF.

Fig. 2. GDNF blocks RET-induced cell death. 293T cells were co-transfected with the GFRα-1 expression plasmid in the presence or absence of the RET expression plasmid. Twenty-four hours after transfection, 25, 50 or 100 ng/ml GDNF was added to the culture medium. Cell death was then measured by trypan blue staining as described in Materials and methods. Standard deviations are indicated (n = 3).
To monitor whether endogenously expressed RET (as opposed to vector-driven expression of RET) displayed a similar ligand-dependent pro-apoptotic activity, we analyzed the effect of GDNF withdrawal in the N2a derived neuroblastoma cell line endogenously expressing RET. These cells were maintained in culture in the presence of GDNF for 2 days, and pro-apoptotic signals were measured 12 h after withdrawal of GDNF, either by caspase activation assay or quantitation of TUNEL reactivity. Interestingly, Figure 3 reveals that the withdrawal of GDNF led, respectively, to a 2- and 3-fold increase in caspase activity and TUNEL-positive cells. Hence, GDNF withdrawal induced a pro-apoptotic signal in cells endogenously expressing RET.

Fig. 3. GDNF withdrawal induces apoptotic death in RET-expressing N2a cells. RET- and GFRα-1-expressing N2a neuroblastoma cells were cultivated in the presence of GDNF for 48 h. Cells were then allowed to grow for 12 h either in the presence or the absence of GDNF. Apoptosis was then quantified either by caspase activity measurement (A) or by TUNEL assay (B) as described in Figure 1C and D. Standard deviations are indicated (n = 3). The relative ratio of TUNEL-positive cells is also indicated. In (B), an example of a TUNEL-positive cell is shown in the absence of GDNF.
RET requires caspase activation to induce cell death
Tyrosine kinase receptors are known to work mainly by transducing a signal dependent on their kinase activities (Pawson and Scott, 1997). To analyze whether RET-induced cell death was dependent on a kinase-dependent signal transduction, we checked the ability of RET to induce cell death when mutated in its docking site Y1062, which is required to initiate kinase-dependent signal transduction (Segouffin-Cariou and Billaud, 2000). As shown in Figure 4A, expression of the RET C634R/Y1062F mutant induced 293T cell death. Similar results were obtained with a kinase-dead RET mutant E921K (Pelet et al., 1998) (not shown). These results strongly suggest that kinase activity and subsequent kinase-dependent signal transduction are not required to induce cell death.

Fig. 4. RET induces apoptosis via a caspase-dependent pathway. (A) RET-induced cell death is independent of RET-induced kinase-dependent signal. 293T cells were transfected with RET wild type or RET mutant C634R/Y1062F in the presence of GFRα-1. Caspase activity was then monitored as described in Figure 1. (B) RET-induced cell death is blocked by p35 expression. 293T cells were transiently transfected with either the RET expression plasmid pJ7Ω-ret9 (RET) or pCMV control plasmid (cont.) in the presence or absence of the p35 expression construct pBabe-p35. Cell death was then monitored as described in Figure 1. Standard deviations are indicated (n = 3). Inset: western blots using anti-RET antibody indicating the expression of the RET protein after transfection.
To understand further how RET induces apoptosis, we analyzed the RET apoptotic pathway. Co-expression of RET and a potent and general caspase inhibitor, the baculovirus protein p35 (Fisher et al., 1999), showed an absence of RET-induced 293T cell death. Hence, RET induces apoptosis via caspases (Figure 4B). This result, reminiscent of that obtained with DCC (Mehlen et al., 1998; C.Forcet, X.Ye, L.Granger, V.Corset, H.Shin, D.E.Bredesen and P.Mehlen, submitted), led us to evaluate RET as a potential caspase substrate.
RET is a caspase substrate
In order to determine whether RET is a caspase substrate, in vitro translation of the intracellular domain of RET (RET-IC) was performed, and caspases-3 and -7 were incubated with RET-IC. Figure 5A shows that RET-IC was cleaved by caspase-3 but not by caspase-7. The presence of three main cleavage products (42, 40 and 36 kDa) of RET upon caspase-3 cleavage suggests the existence of at least two different caspase cleavage sites. The caspase-3 cleavage sites were mapped by constructing mutants based on preferred P4 and P1′ positions (Thornberry et al., 1997), and the apparent relative molecular masses of the caspase cleavage fragments (Figure 5A). Mutation of Asp707 to Asn completely suppresses the 42 and 36 kDa fragments, while the substitution D1017N resulted in the loss of the 40 and 36 kDa fragments (Figure 5B); hence, the cleavage sites for caspase-3 are located in positions D707 and D1017, with consensus sequences VSVD and DYLD, respectively (Figure 5C).
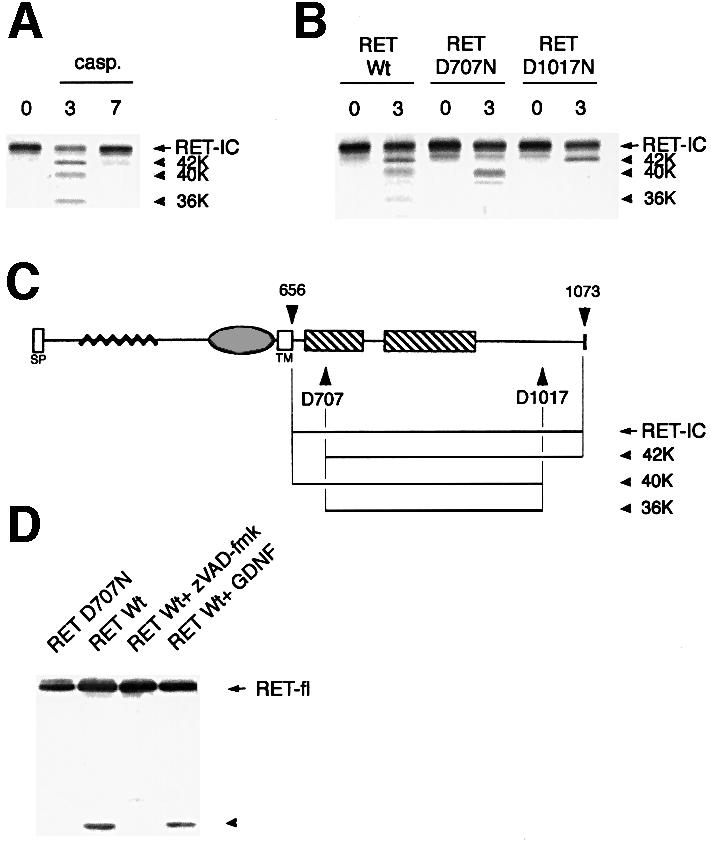
Fig. 5. RET is a caspase substrate. (A) In vitro translated RET-IC was incubated without caspase (first lane) or with purified caspase-3 (0.3 µM), or caspase-7 (1.6 µM) for 2 h. An autoradiograph is shown. (B) Similar experiment to that in (A) except that the mutants RET-IC D707N and RET-IC D1017N were translated in vitro and incubated with caspase-3. (C) Diagram of RET. (D) RET is cleaved by caspase in vivo. 293T cells were co-transfected with either the RET-expressing or the D707N-expressing construct in the presence of GFRα-1-expressing construct. Cells were then treated or not for 24 h with 20 µM zVAD-fmk or with 0.1 µg/ml GDNF. Immunoblot using anti-RET antibody is shown.
To test whether this caspase cleavage was relevant in vivo, we analyzed the appearance of caspase-dependent fragments in 293T cells expressing RET. As shown in Figure 5D, an immunoblot assay using an antibody raised against the C-terminus of RET revealed the presence of a degradation fragment (40–50 kDa). Of interest, the presence of this fragment was strongly inhibited by the addition of the caspase inhibitor zVAD-fmk. Moreover, expression of the D707N mutant of full-length RET did not allow the detection of this fragment, strongly suggesting that this fragment is the C-terminal IC fragment of RET issued from caspase cleavage on the D707 site. However, the presence of the degradation fragment is not modulated by GDNF treatment, suggesting that the ligand has no effect on RET cleavage. Attempts to detect the smaller fragment (6 kDa) resulting from D1017 cleavage were unsuccessful (data not shown). Taken together, these results strongly suggest that both in vivo (at least for the D707 cleavage) and in vitro, RET is cleaved by a caspase-3-like caspase in positions D707 and D1017.
The caspase cleavage sites define a pro-apoptotic region of RET
To evaluate the function of the caspase cleavage of RET, the D707N, D1017N and D707N/D1017N mutants of full-length RET were expressed in 293T cells, and cell death was assessed by trypan blue exclusion. Remarkably, both the D707N and D1017N point mutants failed to induce cell death (Figure 6A). These results indicate that the caspase site mutations suppressed RET pro-apoptotic activity, arguing that caspase cleavage of RET is a crucial event in the induction of apoptosis. It was then tempting to speculate that these sites allow the exposure or the release of a pro-apoptotic region of RET. Interestingly, while expression of the complete intracellular domain of RET was unable to drive 293T cell death by itself, suggesting a requirement for RET association with the membrane for the initiation of the death process, expression of the fragment lying from amino acid 708 to 1017 was sufficient to induce cell death (Figure 6B). Therefore, the fragment lying between the two caspase sites may represent the pro-apoptotic domain of RET, which, once released by the cleavage of caspases, induces a pro-apoptotic signal leading to cell death.

Fig. 6. Cleavage of RET by caspases is crucial for RET pro-apoptotic activity. (A) Mutation of the caspase cleavage sites of RET blocks RET-induced cell death. 293T cells were transiently transfected with either the full-length RET wild type, the full-length RET-D707N, RET-D1017N or the double RET-D707N/D1017N mutant in the presence of GFRα-1 expression plasmid. Cell death was then monitored as described in Figure 1. (B) Expression of the fragment lying from amino acid 708 to 1017 drives 293T cell death. 293T cells were transiently transfected with either the complete RET intracellular domain (RET-IC) or the fragment 708–1017 (RET/708–1017) expression plasmid and cell death was monitored as in Figure 1. Standard deviations are indicated (n = 3).
RET-induced apoptosis in HSCR
Since RET induces apoptosis and is involved in HSCR, a disease related to the absence of a specific set of neural crest-derived cells in the hindgut (Edery et al., 1994; Romeo et al., 1994), it was tempting to postulate that this pro-apoptotic activity may be the direct cause of the disappearance of these cells. To test this hypothesis further, we introduced five different HSCR-associated mutations, R231H, C609W, S767R, K907E and P1039L, into the RET coding sequence by site-directed mutagenesis. As a control, the MEN 2A-associated mutation C634R was also used. Cell death analysis was performed after co-transfection of these different RET mutants with GFRα-1. RET ligand GDNF was then added (or not) 24 h after the transfection. Figure 7 shows that all five Hirschsprung-associated RET mutants displayed a similar pro-apoptotic activity in the absence of GDNF, while the C634R mutant, known to induce a constitutive dimerization of the receptor and a constitutive kinase activity, did not show any pro-apoptotic activity. All five HSCR-associated mutants also showed the fragment issued from caspase cleavage on the D707 site (Figure 5D and data not shown). Remarkably, in contrast to the wild-type RET, none of the HSCR-associated mutants demonstrated regulation of apoptosis by GDNF. Therefore, independently of whether or not the RET ligand GDNF was present, HSCR-associated mutations transformed RET into a constitutive inducer of cell death. An alternative explanation for the disappearance of enteric neuroblasts in HSCR patients could be that these cells expressing a pro-apoptotic form of RET undergo apoptosis.

Fig. 7. HSCR-associated mutations impair GDNF control of RET-induced apoptosis. 293T cells were transiently transfected with either the RET wild type or the following mutations of RET R231H, C609W, S767R, K907E, P1039L or C634R in the presence of GFRα-1 expression plasmid. As in Figure 2, 0.1 µg/ml GDNF was added 24 h after transfection. Cell death was then monitored as described in Figure 1. Standard deviations are indicated (n = 3). Insets: western blots using anti-RET antibody indicating the expression of the RET protein after transfection.
Discussion
The receptor tyrosine kinase RET as a dependence receptor
The emerging family of dependence receptors includes p75NTR, the common neurotrophin receptor (Rabizadeh et al., 1993), the androgen receptor (Ellerby et al., 1999) and DCC (Mehlen et al., 1998). To date, no RTK has been shown to belong to this family, even though several RTKs such as IGF-R, and TrkA, -B and -C have been shown to be involved in promoting or repressing cell death (Yoon et al., 1998; Peruzzi et al., 1999). We describe here the first example of a RTK inducing cell death in the absence of its ligand, a pro-apoptotic activity blocked by the presence of its ligand. Of interest, the pro-apoptotic effect of RET is drastically increased in the presence of one of its co-receptors, GFRα-1, suggesting that a RET–co-receptor complex is needed to optimize induction of cell death. In contrast to the prevailing view that the main role of RTK is to transduce a signal following ligand binding, and consequently dimerization and autophosphorylation, we present evidence here that in the absence of ligand, RET induces an active pro-apoptotic signal dependent on caspase activation. Moreover, the observation that RET mutants displaying no kinase-dependent signal transduction conserved the ability to induce cell death, strongly suggests that the RET pro-apoptotic effect is independent of the kinase activity of RET. Thus, RET, and possibly other RTKs, can display two distinct signal transduction effects: one is observed when the receptor is expressed in a context in which the ligand is unavailable and is dependent on the cleavage of the intracellular domain by caspases, allowing the exposure and/or the release of a pro-apoptotic fragment; and the other depends on ligand binding and allows (i) the block of the pro-apoptotic signal and (ii) the transduction of a kinase-dependent signal, which leads to cell proliferation and/or differentiation. This kinase-dependent signal could explain the protective effect of ligand binding per se, since the ligand-dependent activation of numerous RTKs is known to drive an anti-apoptotic signal such as phosphoinositide-3 kinase/Akt kinase activation and consequent BAD or caspase-9 inactivation (Cardone et al., 1998; Porter and Vailancourt, 1998). Along the same lines, we observed that the RET mutant C634R, which displays a constitutive kinase activity, showed no pro-apoptotic activity. However, as observed with p75NTR, the block of the cell death signal can also be linked to an alteration of receptor conformation (Bredesen et al., 1998). Such a change for RET could be due to the ligand-dependent dimerization of the receptor and/or recruitment of proteins interacting with RET and interfering for example with caspase cleavage. Consequently, it is interesting to note that phospholipase Cγ binds RET at Y1015, an amino acid included in the recognition sequence of the second caspase site. However, the caspase cleavage in position 707 does not seem to be the key switch between the induction of cell death by RET and the inhibition of RET pro-apoptotic activity by GDNF. Indeed, the in vivo RET cleavage, at least on the D707 site, was shown to be constant whether GDNF was present or not. Two models can therefore be proposed. The first is that RET constitutively initiates a pro-apoptotic pathway whether GDNF is available or not, but in the presence of GDNF it induces, at the same time, an anti-apoptotic pathway based on its kinase activity, which consequently inhibits the effect of the RET pro-apoptotic signal. The second model is that the RET pro-apoptotic signal is dependent on the availability of GDNF, but the effect of GDNF on RET is downstream of RET cleavage. Along this line, DCC, the other dependence receptor, has recently been shown to interact with caspase-9 preferentially in the absence of ligand, suggesting that the recruitment of caspase-9 to DCC is part of the switch between DCC pro-apoptotic activity and the ligand-induced block of DCC pro-apoptotic activity (C.Forcet, X.Ye, L.Granger, V.Corset, H.Shin, D.E.Bredesen and P.Mehlen, submitted). Whether RET interacts with caspase-9 or another initiator caspase, and whether GDNF inhibits this putative binding, remain to be investigated.
Caspase cleavage and RET induction of apoptosis
We have shown here that RET-induced cell death is dependent on caspase activation. Moreover, RET is a caspase substrate for caspase-3 and probably other related caspases, and the mutation of its cleavage sites suppresses RET pro-apoptotic activity. These three points resemble the behavior of DCC (Mehlen et al., 1998) and suggest that RET, like DCC, may function as a caspase amplifier. The effect of RET and DCC on caspase amplification results from the combination of the following characteristics: (i) RET and DCC are caspase substrates and (ii) RET and DCC cleavages liberate a pro-apoptotic (and hence caspase activating, either directly or indirectly) fragment, with (i) and (ii) generating a loop of caspase amplification. The fact that caspase cleavage is required for the pro-apoptotic effect of RET raises the question of whether RET plays an important or simply a passive role in apoptosis induction, since caspases are generally thought to be activated only during apoptosis (Salvesen and Dixit, 1997). However, it has become clear recently that caspase zymogens do have some activity prior to processing (Salvesen and Dixit, 1997; Yang et al., 1998); furthermore, the cellular iaps [inhibitors of apoptosis (Crook et al., 1993)] function endogenously to block active caspases (Deveraux et al., 1997). Both of these observations argue that the cellular modulation of caspase activation is likely to represent an important control for cell survival. Thus, caspase amplification by proteins such as RET and others may be very important in the cellular life/death decision, and therefore in the development of disease states.
Dysregulation of RET pro-apoptotic activity and HSCR
The finding that RET expression can induce the death of cells that are beyond ligand availability suggests an important role for the coordinated expression of RET and GDNF. Indeed, the receptor–ligand couple, RET–GDNF, may be viewed as an important regulator of cell survival in RET-expressing cells. In support of this notion, GDNF has been shown to promote neuronal survival in vitro (Henderson et al., 1994). Furthermore, the targeted disruption of the GDNF gene results in aganglionosis in the mouse (Sanchez et al., 1996). It was then tempting to speculate that the role of RET in the development of the enteric nervous system is associated with RET pro-apoptotic activity. Indeed, the migration of RET-expressing neural crest-derived cells during development may be controlled not only at a guidance level but also at a survival level by the presence of RET ligands. In that sense, two studies recently demonstrated the role of GDNF in the control of the survival of either RET-positive primary culture cells or neural crest-derived cells isolated from rat embryo (Heuckeroth et al., 1998; Taraviras et al., 1999). It was even more tempting to postulate that in HSCR, the loss of neural crest-derived parasympathetic neurons of the hindgut might result from RET-induced cell death. We and others have shown that several HSCR-causing missense mutations disrupt RET function and that the molecular mechanisms underlying this effect are highly dependent on the location of the mutation in the protein. For instance, the R231H mutation decreases the level of RET at the cell surface, while the C609W mutation leads to a dual effect including covalent dimerization and an impairment of the translocation of RET at the plasma membrane. However, the majority of HSCR mutations (e.g. S767R and P1039L used in this study) did not have a detectable impact on RET function (Pelet et al., 1998). Here, we have shown a direct relationship between all five Hirschsprung-associated RET mutations tested and a constitutive pro-apoptotic activity of the mutant RET proteins, independent of the presence or the absence of the ligand GDNF. In other words, Hirschsprung-associated RET mutations transform RET into a constitutive inducer of cell death. Thus, even if the loss of the ligand-induced suppression of RET pro-apoptotic activity does not turn out to be the only effect leading to HSCR (i.e. the loss of RET expression via large-scale mutation), RET mutations associated with the development of HSCR may act in a dominant-negative fashion, leading to embryonic apoptosis of RET-expressing cells such as enteric neuroblasts.
Materials and methods
Cells, transfection procedures and immunoblotting
Transient transfections of HEK 293T cells were performed as described previously (Mehlen et al., 1998) according to a modified calcium phosphate procedure. Transient transfections of rat olfactory neuroblast 13.S.24 were performed using Lipofectamine according to the manufacturer’s instructions (Life Technology, Cergy Pontoise, France). The GFRα-1-expressing neuroblastoma cell line N2a was derived from an N2a cell line by the stable integration of a GFRα-1 expression construct (C.Bidaud and M.Billaud, unpublished). RET expression was analyzed by western blotting as described previously (Mehlen et al., 1998) using anti-RET antibody (Pelet et al., 1998).
Cell death analysis
Cell death was analyzed using a trypan blue (Mehlen et al., 1998) staining procedure. Caspase-3 activation was monitored using the ApoAlert caspase assay, which utilizes the Ac-DEVD-AFC substrate (Clontech, Palo Alto, CA). This activity was determined according to the manufacturer’s instructions using a Ac-DEVD-CHO-pretreated lysate as a negative control. Caspase activation is presented as the ratio between the caspase activity of the sample and that measured in 293T cells transfected with pCMV mock plasmid; for all samples, the background remaining after inhibition by Ac-DEVD-CHO was subtracted. TUNEL staining was performed on adherent 13.S.24 or cytospun N2a cells using the ApoAlert DNA fragmentation assay (Clontech). TUNEL reactivity was examined and photographed with a Zeiss Axioskop photomicroscope. The number of TUNEL-positive cells was estimated by counting at least 100 TUNEL-positive cells from six separate fields.
Site-directed mutagenesis and plasmid construction
pJ7Ω-ret9 encodes the short version of full-length RET (amino acids 1–1072) under the control of the CMV promoter (Rossel et al., 1997). pBabe-p35 has already been described elsewhere (C.Forcet, X.Ye, L.Granger, V.Corset, H.Shin, D.E.Bredesen and P.Mehlen, submitted). The RET-IC expression plasmid, pCR-ret-IC, was constructed by inserting the intracellular domain of RET into pCR3.1 (Invitrogen, Palo Alto, CA) using the PCR primers 5′-TGACCATGCACTGCTACCACAAGTTTG-3′ and 5′-TTCTAGAATCTAGTAAATGCATGGGA-3′ and pJ7Ω-ret9 as template. RET-IC D707N and RET-IC D1017N were created using the QuikChange site-directed mutagenesis system from Stratagene. Hence, pCR-ret-IC-D707N and pCR-ret-IC-D1017N were constructed using pCR-ret-IC as template and the following primers: 5′-CAGGTCTCCGTGAATGCCTTCAAG-3′, 5′-CTTGAAGGCATTCACGGAGACCTG-3′ and 5′-GAGAGACTACTTGAACCTTGCGGCGTC-3′, 5′-GACGCCGCAAGGTTCAAGTAGTCTCTC-3′. The full-length RET-D707N, RET-D1017N mutant expression plasmids, pJ7Ω-ret9-D707N and pJ7Ω-ret9-D1017N, were obtained by the same strategy using the same primers but with pJ7Ω-ret9 as template. To generate the RET-D707N/D1017N mutant, the strategy performed with the RET-D707N mutant was used, but in this case pJ7Ω-ret9-D1017N was the template. pCR-ret-IC/708–1017 was constructed by inserting the amino acid 708–1017 fragment of RET into pCR3.1 using the PCR primers 5′-TGACCATGGCCTTCAAGATCCTGGAG-3′ and 5′-GCCGCTTAGTCCAAGTAGTCTCTCCTC-3′ and pJ7Ω-ret9 as template. The different Hirschsprung-associated mutations were also obtained by the QuikChange strategy using pJ7Ω-ret9 as template and the following primers: (R231H) 5′-CGCTGGGCCCTGGACCACGAGCAGCGGGAGAAG-3′, 5′-CTTCTCCCGCTGCTCGTGGTCCAGGGCCCAGCG-3′;
(C609W) 5′-GCTATGGCACCTGGAACTGCTTCCCTG-3′, 5′-CAGGGAAGCAGTTCCAGGTGCCATAGC-3′; (S767R) 5′-GAACGCCTCCCCGAGAGAGCTTCGAGACCTG-3′, 5′-CAGGTCTCGAAGCTCTCTCGGGGAGGCGTTC-3′; (K907E) 5′-GATTCCTACGTGGAGAGGAGCCAGG-3′, 5′-CCTGGCTCCTCTCCACGTAGGAATC-3′; (P1039L) 5′-GAGGAGGAGACACTGCTGGTGGACTG-3′, 5′-CAGTCCACCAGCAGTGTCTCCTC-3′. C634R and C634R/Y1062F mutants were described previously (Pelet et al., 1998).
In vitro translation and caspase cleavage reactions
Purified caspases were a generous gift from Guy Salvesen (The Burnham Institute, La Jolla, CA). In vitro translation and caspase incubation were performed as described previously (Mehlen et al., 1998).
Acknowledgments
Acknowledgements
We thank G.S.Salvesen for the gift of purified caspases. We wish to thank X.Ye for critical reading of the manuscript and M.Poulat for excellent technical assistance. This work was supported by the Ligue Contre le Cancer, the FRM, the ARC (9036,9340) and ‘emmergence’ program to P.M., and by NIH grants NS33376 and CA69381 to D.E.B.
References
- Arighi E. et al. (1997) Identification of Shc docking site on Ret tyrosine kinase. Oncogene, 14, 773–782. [DOI] [PubMed] [Google Scholar]
- Baloh R.H. et al. (1997) TrnR2, a novel receptor that mediates neurturin and GDNF signaling through Ret. Neuron, 18, 793–802. [DOI] [PubMed] [Google Scholar]
- Bredesen D.E., Ye,X., Tasinato,A., Sperandio,S., Assa-Munt,N. and Rabizadeh,S. (1998) p75NTR and the concept of cellular dependence: seeing how the other half die. Cell Death Differ., 5, 365–371. [DOI] [PubMed] [Google Scholar]
- Cacalano G. et al. (1998) GFRα1 is an essential receptor component for GDNF in the developing nervous system and kidney. Neuron, 21, 53–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardone M.H., Roy,N., Stennicke,H.R., Salvesen,G.S., Franke,T.F., Stanbridge,E., Frisch,S. and Reed,J.C. (1998) Regulation of cell death protease caspase-9 by phosphorylation. Science, 282, 1318–1321. [DOI] [PubMed] [Google Scholar]
- Crook N.E., Clem,R.J. and Miller,L.K. (1993) An apoptosis-inhibiting baculovirus gene with a zinc finger-like motif. J. Virol., 67, 2168–2174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deveraux Q.L., Takahashi,R., Salvesen,G.S. and Reed,J.C. (1997) X-linked IAP is a direct inhibitor of cell-death proteases. Nature, 388, 300–304. [DOI] [PubMed] [Google Scholar]
- Edery P. et al. (1994) Mutations of the RET proto-oncogene in Hirschsprung’s disease. Nature, 367, 378–380. [DOI] [PubMed] [Google Scholar]
- Ellerby L.M. et al. (1999) Kennedy’s disease: caspase cleavage of the androgen receptor is a crucial event in cytotoxicity. J. Neurochem., 72, 185–195. [DOI] [PubMed] [Google Scholar]
- Fisher A.J., Cruz,W.D., Zoog,S.J., Schneider,C.L. and Friesen,P.D. (1999) Crystal structure of baculovirus P35: role of a novel reactive site loop in apoptotic caspase inhibition. EMBO J., 18, 2031–2039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson C.E. et al. (1994) GDNF: a potent survival factor for motoneurons present in peripheral nerve and muscle. Science, 266, 1062–1064. [DOI] [PubMed] [Google Scholar]
- Heuckeroth R.O., Lampe,P.A., Johnson,E.M. and Milbrandt,J. (1998) Neurturin and GDNF promote proliferation and survival of enteric neuron and glial progenitors in vitro. Dev. Biol., 200, 116–129. [DOI] [PubMed] [Google Scholar]
- Jing S. et al. (1996) GDNF-induced activation of the Ret protein tyrosine kinase is mediated by GDNFR-a, a novel receptor for GDNF. Cell, 85, 1113–1124. [DOI] [PubMed] [Google Scholar]
- Kotzbauer P.T., Lampe,P.A., Heuckeroth,R.O., Golden,J.P., Creedon, D.J., Johnson,E.M.,Jr and Milbrandt,J. (1996) Neurturin, a relative of glial-cell-line-derived neurotrophic factor. Nature, 384, 467–470. [DOI] [PubMed] [Google Scholar]
- Lorenzo M.J., Gish,G.D., Houghton,C., Stonehouse,T.J., Pawson,T., Ponder,B.A. and Smith,D.P. (1997) RET alternate splicing influences the interaction of activated RET with the SH2 and PTB domains of Shc and the SH2 domain of Grb2. Oncogene, 14, 763–771. [DOI] [PubMed] [Google Scholar]
- Mehlen P., Rabizadeh,S., Snipas,S.J., Assa-Munt,N., Salvesen,G.S. and Bredesen,D.E. (1998) The DCC gene product induces apoptosis by a mechanism requiring receptor proteolysis. Nature, 395, 801–804. [DOI] [PubMed] [Google Scholar]
- Mehlen P., Coronas,V., Ljubic,V., Ducasse,C., Granger,L., Jourdan,F. and Arrigo,A.-P. (1999) Small stress protein Hsp27 accumulation during dopamine-mediated differentiation of rat olfactory neurons counteracts apoptosis. Cell Death Differ., 6, 227–233. [DOI] [PubMed] [Google Scholar]
- Mulligan L.M. et al. (1993) Germ-line mutations of the RET proto-oncogene in multiple endocrine neoplasia type 2A. Nature, 363, 458–460. [DOI] [PubMed] [Google Scholar]
- Pasini B. et al. (1995) Loss of function effect of RET mutations causing Hirschsprung disease. Nature Genet., 10, 35–40. [DOI] [PubMed] [Google Scholar]
- Pawson T. and Scott,J.D. (1997) Signaling through scaffold, anchoring and adaptor proteins. Science, 278, 2075–2080. [DOI] [PubMed] [Google Scholar]
- Pelet A. et al. (1998) Various mechanisms cause RET-mediated signaling defects in Hirschsprung’s disease. J. Clin. Invest., 101, 1415–1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peruzzi F., Prisco,M., Dews,M., Salomoni,P., Grassilli,E., Romano,G., Calabretta,B. and Baserga,R. (1999) Multiple signaling pathways of the insulin-like growth factor 1 receptor in protection from apoptosis. Mol. Cell. Biol., 19, 7203–7215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter A.C. and Vaillancourt,R.R. (1998) Tyrosine kinase receptor-activated signal transduction pathways which lead to oncogenesis. Oncogene, 17, 1343–1352. [DOI] [PubMed] [Google Scholar]
- Rabizadeh S., Oh,J., Zhong,L., Yang,J., Bitler,C., Butcher,L. and Bredesen,D. (1993) Induction of apoptosis by the low-affinity NGF receptor. Science, 261, 345–348. [DOI] [PubMed] [Google Scholar]
- Romeo G. et al. (1994) Point mutations affecting the tyrosine kinase domain of the RET proto-oncogene in Hirschsprung’s disease. Nature, 367, 377–378. [DOI] [PubMed] [Google Scholar]
- Rossel M. et al. (1997) Distinct biological properties of two RET isoforms activated by MEN 2A and MEN 2B mutations. Oncogene, 14, 265–275. [DOI] [PubMed] [Google Scholar]
- Salvesen G.S. and Dixit,V.M. (1997) Caspases: intracellular signaling by proteolysis. Cell, 91, 443–446. [DOI] [PubMed] [Google Scholar]
- Sanchez M.P., Silos-Santiago,I., Frisen,J., He,B., Lira,S. and Barbacid,M. (1996) Renal agenesis and the absence of enteric neurons in mice lacking GDNF. Nature, 382, 70–73. [DOI] [PubMed] [Google Scholar]
- Santoro M., Wong,W.T., Aroca,P., Santos,E., Matoskova,B., Grieco,M., Fusco,A. and Di Fiore,P.P. (1994) An epidermal growth factor receptor/ret chimera generates mitogenic and transforming signals: evidence for a ret-specific signalling pathway. Mol. Cell. Biol., 14, 663–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santoro M. et al. (1995) Activation of RET as a dominant transforming gene by germline mutations of MEN 2A and MEN 2B. Science, 267, 381–383. [DOI] [PubMed] [Google Scholar]
- Schuchardt A., D’Agati,V., Larsson-Blomberg,L., Costantini,F. and Pachnis,V. (1994) Defects in the kidney and enteric nervous system of mice lacking the tyrosine kinase receptor Ret. Nature, 367, 380–383. [DOI] [PubMed] [Google Scholar]
- Segouffin-Cariou C. and Billaud,M. (2000) Transforming ability of MEN2A-RET requires activation of the phosphatidylinositol 3-kinase Akt signaling pathway. J. Biol. Chem., 275, 3568–3576. [DOI] [PubMed] [Google Scholar]
- Songyang Z. et al. (1995) Catalytic specificity of protein tyrosine kinases is critical for selective signalling. Nature, 373, 536–539. [DOI] [PubMed] [Google Scholar]
- Stennicke H.R. and Salvesen,G.S. (1997) Biochemical characteristics of caspases-3, -6, -7 and -8. J. Biol. Chem., 272, 25719–25723. [DOI] [PubMed] [Google Scholar]
- Takahashi M. and Cooper,G. (1987) Ret transforming gene encodes a fusion protein homologous to tyrosine kinases. Mol. Cell. Biol., 7, 1378–1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi M., Ritz,J. and Cooper,G.M. (1985) Activation of a novel human transforming gene, ret, by DNA rearrangement. Cell, 42, 581–588. [DOI] [PubMed] [Google Scholar]
- Takeichi M. (1991) Cadherin cell adhesion receptors as a morphogenetic regulator. Science, 251, 1451–1455. [DOI] [PubMed] [Google Scholar]
- Taraviras S. et al. (1999) Signalling by the RET receptor tyrosine kinase and its role in the development of the mammalian enteric nervous system. Development, 126, 2785–2797. [DOI] [PubMed] [Google Scholar]
- Thornberry N.A. et al. (1997) Combinatorial approach defines specificities of members of the caspase family and granzyme B. Functional relationships established for key mediators of apoptosis. J. Biol. Chem., 272, 17907–17911. [DOI] [PubMed] [Google Scholar]
- Yang X., Chang,H.Y. and Baltimore,D. (1998) Autoproteolytic activation of pro-caspases by oligomerization. Mol. Cell, 1, 319–325. [DOI] [PubMed] [Google Scholar]
- Yoon S.O., Casaccia-Bonnefil,P., Carter,B. and Chao,M.V. (1998) Competitive signaling between TrkA and p75 nerve growth factor receptors determines cell survival. J. Neurosci., 18, 3273–3281. [DOI] [PMC free article] [PubMed] [Google Scholar]