

Abstract
Protein-polymer conjugates are important in diverse fields including drug delivery, biotechnology, and nanotechnology. This feature article highlights recent advances in the synthesis and application of protein-polymer conjugates by controlled radical polymerization techniques. Special emphasis on new applications of the materials, particularly in biomedicine, are highlighted.
Introduction
Conjugation of biocompatible polymers to therapeutic proteins produces drugs with improved pharmacokinetic properties. For example, attachment of linear poly(ethylene glycol) or PEGylation1–3 of proteins has been demonstrated to improve physical and thermal stability, as well as reduce immunogenicity.4–6 This results in lower and less frequent dosing of the drug, which increases patient compliance.7 Consequently, a number of FDA-approved therapeutics that are currently used to treat diabetes, cancer and hepatitis C consist of PEGylated-proteins.1, 2
The preparation of well-defined molecular architectures is of particular interest because the properties of the bioconjugates are dependent on the homogeneity of the structure.7–12 Furthermore, the location of the polymer chain on the protein can have a large affect on the activity of the conjugate. Therefore, precise control over placement of the polymer is crucial in the design of these drugs.7, 10, 12 In an effort to address these issues, researchers have worked to develop genetically engineered proteins with single conjugation sites. Typically, proteins are engineered to contain a single free cysteine or lysine residue for conjugation to a polymer. Recombinant techniques have also been employed in order to introduce unnatural amino acids into protein and peptides for site-specific conjugations.13 These recombinant techniques, as well as activation of specific amino acids for bioconjugations have been reviewed elsewhere.14–17
“Smart” polymer conjugates make up another important class of bioconjugates. The response to external stimuli such as changes in pH, temperature, and light can invoke changes in the physical properties of the polymer.18, 19 These changes are subsequently transferred to the attached biomolecule. Such conjugates have been used for enzyme switching, microfluidics and affinity separations.19–21 The ability to tune or switch the physical properties of materials has potential pharmaceutical applications in reversible protein capture, reversible drug targeting, or reversible binding of small molecule factors. Thus, there is a growing need for versatile synthetic strategies to prepare well-defined, “smart” conjugates.20, 22
Controlled radical polymerizations (CRPs) have emerged as powerful techniques to prepare well-defined protein- and peptide-polymer conjugates.23–25 These biomolecule-polymer hybrids have been achieved by three main methods (Scheme 1): conjugation of the polymer to the biomolecule through ligand-protein interaction or covalent attachment to amino acid side chains,26, 27 polymerization directly from biomolecules,28, 29 and side-chain polymers synthesized by polymerizing peptide monomers30, 31 or by postpolymerization conjugation of multiple biomolecules to a polymer chain.32, 33 Recent reviews have covered general design criteria for bioconjugates,24, 34–36 the synthesis of giant ampiphiles,37 responsive bioconjugates,38 and enzymatic activity of bioconjugates.39
Scheme 1.

Methods to prepare protein-polymer conjugates.
This highlight will focus on recent reports of CRP techniques to produce protein-polymer conjugates. We seek to highlight works that showcase new approaches and innovative applications for protein-polymer conjugates. The synthesis of bioreactive-polymers by CRP and conjugation of these polymers to proteins will be covered. Approaches to directly polymerize from protein macroinitiators will also be discussed. Techniques for conjugation of multiple proteins via homo-, hetero-, and star-telechelic polymers will be presented. Applications or CRP techniques to conjuate formation on surfaces is also of increasing interest for applications in molecular biology, materials science and nanomedicine. Finally, a discussion of the cytotoxicity of polymers prepared by atom transfer radical polymerization (ATRP) and reversible addition-fragmentation chain transfer (RAFT) polymerization, the two most widely employed methods to prepare well-defined constructs for biomaterial and therapeutic applications, will be summarized.
End-Group Reactive Polymers
A common approach to produce protein-polymer conjugates entails the synthesis of polymers with protein-reactive handles. These polymers can be designed to react with either the C- or N-terminus or amino acid side-chains of the protein. The following sections will highlight recent examples utilizing these strategies.
Amine Reactive Polymers
Amine side chains of lysine residues and the N-terminus are the most common sites for polymer conjugations. This approach is desirable for conjugate formation because these residues react with a variety of functional groups, including activated esters, thioimido esters, isocyanates, chlorotriazines, aldehydes and ketones.40–42 A disadvantage to this approach is that many proteins contain more than one solvent-exposed amine residue, which makes it difficult to control the site of conjugation. However, due to the ease of synthesis and conjugation of these materials to proteins, this approach is still pursued and results in materials with improved pharmacokinetics over unmodified proteins. Some examples by CRP are provided below.
Braslau and coworkers reported the synthesis of carboxylic acid functionalized alkoxyamine to be used as an initiator in nitroxide-mediated polymerization (NMP).43 In this report, N,N-dimethylacrylamide was polymerized using the carboxylic acid-functionalized NMP initiator. Postpolymerization, the carboxylic acid was transformed into an N-hydroxysuccimide (NHS) ester and conjugated to hen egg white lysozyme. Sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) confirmed that the NHS-modified polymer had reacted with lysozyme under mild conditions to create conjugates modified at one to seven of the available amines.
Recently, Davis and coworkers have demonstrated the utility of thiazolidine-2-thione (TT) for polymer conjugation via amine residues on lysozyme (Fig. 1).44 A TT-chain transfer agent (CTA) was synthesized and utilized in the RAFT polymerization of 2-hydroxypropyl methacryate (HPMA). The CTA allowed for the synthesis of well-defined pHPMA with targeted molecular weights. Postpolymerization, the polymer chains maintained the TT functional group. Conjugation of these polymers to lysozyme at two different pHs demonstrated the ability to control protein activity based upon the number of polymer chains conjugated to lysozyme.
Fig. 1.

A) Synthesis of thiazolidine-2-thione polymers and B) conjugation to lysozyme (protein structure from PBD # 132L).44
In another recent example, a poly(vinyl pyrrolidone) (PVP) (a blood plasma expander) was conjugated to the amine residues of lysozyme via an activated ester45 as well as by forming a Schiff base followed by reduction.46 An NHS-ester terminated CTA mediated the RAFT polymerization of vinyl pyrrolidone resulting in α-NHS, ω-thiocarbonylthio-PVP. The conversion was taken to 90% resulting in polymers with narrow molecular weight distributions. SDS-PAGE and gel permeation chromatography (GPC) indicated conjugation of up to seven polymer chains to lysozyme. Aldehyde ω–end functional polyPVP was also prepared by conversion of a xanthate end-functional precursor prepared by macromolecular design via inter-exchange of xanthate MADIX/RAFT polymerization (Fig. 2). The thiocarbonylthio end-groups were quantitatively converted to aldehydes via hydrolysis and subsequent thermolysis. SDS-PAGE analysis confirmed conjugate formation with lysozyme via reductive amination.
Fig. 2.

A) Synthesis of ω-aldehyde-PVP. B) Conjugation of aldehyde end-functional PVP to a protein (protein structure from PBD # 132L).46
Haddleton and coworkers demonstrated the ability to target the N-terminal amine residue of Salmon calcitonin using α-aldehyde functional polymers.47 Salmon calcitonin (sCT) is a 32 amino acid peptide containing two disulfide bridges and three amine residues (two lysines and the N-terminal amine). It is a calcitropic hormone used to treat Paget’s disease, hypercalcaemia, and osteoporosis. An α-aldehyde poly(methoxyPEG methacrylate) (MW = 1,100 g/mol) (pPEGMA) was synthesized via ATRP. The aldehyde-pPEGMA was then conjugated to the N-terminus of sCT and reduced with sodium cyanoborohydride (Fig. 3). Conjugation to the N-terminus was confirmed by mass spectrometry (MS) analysis of the conjugate after tryptic digest. Bioactivity of the conjugate was determined by monitoring intracellular levels of cyclic adenosine monophosphate (cAMP) in T47D human breast cancer cells via enzyme-linked immunsorbent assay (ELISA). Native sCT versus sCT-pPEGMA elevated cAMP levels by 92% versus 85% at ∼2nM, thus demonstrating that the conjugate had similar activity to native peptide. The authors also demonstrated the ability of different molecular weight polymers to decrease the rate of enzymatic digestion of the peptide. The rate of trypsin digest of two different conjugates versus the native peptide was investigated and quantified via reverse phase-high pressure liquid chromatography (RP-HPLC). The unmodified peptide was digested in less than five minutes, whereas sCT-pPEGMA (6.5 kDa) and sCT-pPEGMA (49.7 kDa) conjugates were digested in 15 min and 30 min, respectively.
Fig. 3.

N-terminal conjugation of branched PEG to sCT via Schiff base formation and subsequent reduction (protein structure from PBD # 2GLH).47
Aminooxy/Aldehyde Reactive Polymers
As demonstrated above, proteins and peptides contain amine side chains that form Schiff bases with aldehydes and ketones. However, these are typically unstable in aqueous solutions. This is remedied by reduction of the imine. As an alternative, use of hydrazone/oxime bond formation provides a convenient method to produce stable site-specific conjugation reactions in one step without reduction.9, 48, 49 Additionally, aldehyde, ketone, and aminooxy groups may be incorporated into peptides, proteins, and polymers, thus restricting conjugation to the desired site.9, 50, 51 An N-terminal transamination reaction using pyridoxal-5-phosphate (PLP) has been reported to readily transform the N-terminal amino acids of proteins; although the reaction is typically not high yielding.52 Furthermore, recombinant methods can be utilized to introduce the appropriate reactive groups into proteins and peptides site-specifically.15, 17, 53 Finally, the introduction of Nε-levulinyl lysine has also been accomplished by solid phase peptide synthesis or native chemical ligation.9, 53
We reported the use of oxime chemistry to prepare conjugates of BSA.54 In this report, two aminooxy end-functionalized ATRP initiators were synthesized. The bromoisobutyrate based initiator was used to polymerize methacrylate monomers, and the chloropropionate was used to polymerize acrylamide monomers. For example, the chloropropionate initiator was used to polymerize poly(N-isopropylacrylamide) (poly(NIPAAm)) by ATRP, and the aminooxy polymer was subsequently conjugated to BSA modified with Nε-levulinyl lysine residues. This methodology was then extended to RAFT polymerization of NIPAAm with an N-tert-butoxycarbonyl (BOC)-protected aminooxy CTA.55 Postpolymerization, the aminooxy was deprotected and conjugated to levulinic functionalized BSA. The conjugates were thermally precipitated and analyzed by SDS-PAGE demonstrating the thermoresponsiveness of the conjugates.
Thiol-Reactive Polymers
Cysteine residues are attractive targets for polymer conjugation to proteins. This chemoselective strategy is advantageous because surface-exposed, free cysteines are rare in native proteins. Moreover, if a protein does not contain a natural free cysteine residue, genetic engineering strategies have been developed to synthesize site-specific cysteine mutants without significantly altering the activity of the protein. A large number of cysteine-reactive functional groups have been introduced into polymers via CRP techniques including activated disulfides, maleimides, and vinyl sulfones being the most widely reported.
The incorporation of pyridyl disulfide functional group into ATRP initiators and RAFT CTAs has been extensively employed to prepare protein reactive polymers.24, 26, 56 For example, we were the first to demonstrate the synthesis of a pyridyl disulfide functionalized ATRP initiator for the CRP.26 Postpolymerization reaction of the activated disulfide-polymer with C34 in BSA afforded the amphiphillic BSA-poly(hydroxyethyl methacrylate) (pHEMA) conjugate. The Centre for Advanced Macromolecular Design (CAMD) group was the first to synthesize a pyridyl disulfide containing CTA for the direct synthesis of pyridyl disulfide containing polymers.56 CAMD as well as the Stayton group and our group have further elaborated on this approach to synthesize well-defined protein-, peptide- and olgionucleotide-polmyer conjugates.57–60 The CAMD group has also demonstrated the ability to transform the thiocarbonylthio group into a pyridyl disulfide postpolymerization via aminolysis and trapping of the free thiol with pyridyl disulfide.61 The activated disulfide polymers were then conjugated to thiol containing biomolecules. This approach of postpolymerization functionalization of RAFT polymers with activated disulfides has since been elaborated upon by Theato and coworkers.62 In this case the thiol was trapped with methane thiosulfonate, instead of pyridyl disulfide, to generate activated disulfide terminated polymers.
Haddleton and coworkers were the first to report the application of protected-maleimide polymers synthesized by ATRP.63 Recently, we adapted this approach to RAFT polymerization. A furan protected maleimide CTA was synthesized and mediated the RAFT polymerization of poly(ethylene glycol) methyl ether acrylate (PEGA) (MW = 454 g/mol).64 Polymers of 20,000 Da and 39,000 Da were prepared, with PDIs of 1.25 and 1.36, respectively. Retro-Diels Alder affored the ω-maleimide end-group. Subsequent conjugation to V131C T4 lysozyme produced conjugates that were characterized by SDS PAGE and size exclusion chromatography.
Vinyl sulfones have received attention for their enhanced stability in aqueous and alkaline conditions compared to maleimide.40 We have prepared vinyl sulfone end-functionalized polymers by RAFT polymerization.65 In this work, poly(ethylene glycol) methyl ether acrylate (PEGA) was polymerized in the presence of a 1-phenylethyl dithiobenzoate CTA to produce well-defined poly(PEGA)s (pPEGAs) with number average molecular weights ranging from 4.2 kDa to 26.5 kDa and PDIs ranging from 1.10–1.23. Aminolysis of the polymer and subsequent in situ trapping of the resulting thiol with divinyl sulfone gave the semi-telechelic, ω-vinyl sulfone-functionalized polyPEGA (Fig. 4) with high efficiencies. The resulting BSA-pPEGA conjugates exhibited 92% esterase activity of p-nitrophenyl acetate compared to the unmodified protein.
Fig. 4.

Synthesis and in situ aminolysis of dithiobenzate-pPEGA followed by trapping of resulting thiol with divinyl sulfone and conjugation to BSA.65 Reproduced with permission from Macromolecules 2009, 42, 7657–7663. Copyright 2009 American Chemical Society.
Overall these examples demonstrate the ability to synthesize a range of well-defined, bioreactive polymers by CRP techniques. Utilization of naturally occuring amino acids is a powerful tool to easily functionalize proteins with polymers. Conjugation of polymers to the biomolecule transfers the properties of the polymer to the protein such as thermoresponsiveness (pNIPAAm) or bio-passivation (branched PEGs). Often the protein-polymer conjugates maintains similar activity to that of the unmodified protein. These approaches will allow for the design and synthesis of new drugs with improved pharmacokinetic properties.
Novel Thiol-Reactive Functional Groups
Recently, novel thiol-reactive functional groups have been developed. Although they have not yet been applied to polymers these methods would compliment the above approaches. Baker and coworkers have recently used monobromomaleimides for reversible conjugation to cysteines.66 They demonstrated the utility of this functional group to react selectively with thiols in the presence of amines, as well as reversibility of this conjugation upon introduction of another nucleophile. Baker, Caddick and coworkers then elaborated upon this approach with dibromomaleimides to reversibly bridge a disulfide in a peptide and for reversible site-specific glycosylation of a protein.67 The disulfide bond of somatostatin (growth hormone-inhibiting hormone) was bridged with dibromomaleimide. The disulfide bond was also bridged with a N-fluoresceindibromomaleimide. This approach was then extended to the reversible site-specific glycosylation of a L111C mutant of the Src homology 2 (SH2) domain of growth factor receptor-bound protein 2 (Grb2) with thioglucose and glutathione (a ligand for glutathione S-transferase) (Fig. 5).
Fig 5.

Reversible modification of the Grb2 SH2 domain (L111C) enabled by dibromomaleimide.67 Reprinted with permission from M. E. B. Smith, F. F. Schumacher, C. P. Ryan, L. M. Tedaldi, D. Papaioannou, G. Waksman, S. Caddick and J. R. Baker, J. Am. Chem. Soc., 2010, 132, 1960–1965.. Copyright 2010 American Chemical Society.
Another interesting thiol-reactive functional group was developed by Wong, Che and coworkers.68 Their approach used electron deficient alkynes for reversible conjugation to cysteine residues in peptides and proteins. Conjugation and cleavage of various thiols to alkynoic amides, alkynoic esters, and internal alkynones was demonstrated by liquid chromatography-mass spectrometry (LC-MS). The chemoselectivity of this strategy was then demonstrated with BSA (free cysteine) using lysozyme, which contains no free cysteine and seven surface amine residues, as a control. Dansyl, a fluorophore, was functionalized with an electron deficient alkyne and the authors demonstrated that BSA (C34) was tagged with an azido fluorophore and lysozyme was not tagged. This strategy was also applied for enrichment of cysteine containing peptide fragments from peptide mixtures.
Haddleton and coworkers have recently shown an example in which water-soluble organic phospines were used as a catalyst in a one pot PEGylation of the disulfide containing peptide; sCT.69 In this work, the authors demonstrate the reduction of the disulfide using tris(2-caroxyethyl)phosphine followed by subsequent in situ conjuation to poly(monomethyoxy ethylene glycol) (methy)acrylates catalyzed by free phosphine. Michael thiol-ene conjugate formation was confirmed to selectively occur at the free thiols by matrix assisted laser desorption-time of flight mass spectrometry (MALDI-TOF MS). The conjuagates were formed under mild aqueous conditions with no loss of bioactivity in less than 2 hours. These above strategies have expanded the scope of bioorthoganal reactions; however, they have not yet been applied toward the synthesis of protein-polymer conjugates by CRP.
Ligand Binding Polymers
Another route to produce well-defined, protein-reactive polymers involves polymers with ligand binding capability. The high affinity interaction between biotin and streptavidin (Ka ≈ 1015 M−1) has been widely exploited to prepare engineered surfaces as well as numerous conjugates. Biotinylated polymers have been prepared for conjugation to both avidin and streptavidin. Much of this work has been reviewed elsewhere.23, 24 Among the most interesting recent applications of ligand binding chemistries are the use of biotin/streptavidin interactions to prepare higher order conjugates. These and other examples will be discussed elsewhere in this review.
Polymers for Huisgen [3 + 2] Cycloadditions
Since the introduction of the azide-alkyne Huisgen cycloaddition reaction, researchers have worked to develop methods to create bioconjugates by introducing azides or alkynes into proteins. These groups can be incorporated site-specifically using protein engineering strategies or postranslational modifications.15, 70–72 Azide-alkyne cycloadditions to create polymer bioconjugates has been recently reviewed by Le Droumaguet and Velonia.73 Below, we highlight some interesting applications of this methodology.
Sumerlin and coworkers reported the synthesis of a BSA-polyNIPAAm conjugate synthesized by RAFT polymerization and Huisgen cycloaddition chemistry.74 This conjugate was prepared by polymerizing NIPAAm from an azide end-functionalized CTA by RAFT polymerization initiated by AIBN (Scheme 2). The polymerization proceeded with linear pseudo-first order kinetics to create a polymer with a PDI of 1.06. Introduction of an alkyne on BSA was achieved by reaction at the free cysteine with propargyl maleimide. Subsequent triazole formation occurred in phosphate buffer with a CuSO4/ascorbic acid catalyst system. Typical LCST behavior of the conjugate was observed upon heating.
Scheme 2.

A) Synthesis of polyNIPAAm-N3. B) Conjugation of polyNIPAAm-N3 with BSA-alkyne (protein structure from PBD # 1E7H).74
ATRP is also a powerful CRP technique that can easily be combined with the Huisgen cycloaddition. For example, van Hest and coworkers polymerized styrene (St) via ATRP and postpolymerization displaced the ω-bromine with an azide.75 The azide functionalized poly(styrene) (pSt) was then conjugated to a bisacetylene-PEG resulting in an α-alkyne-PEG-b-pSt. Self assembly of these block copolymers in the presence of glucose oxidase (GOx) and Candida antartica (Cal B) generated 90 nm to 180 nm polymersomes with GOx and Cal B encapsulated in the lumen and bilayer, respectively. Azido-horse radish peroxidase (HRP), prepared by diazo transfer on the lysines, was conjugated onto the actetylene coated polymersome (Fig. 6). These enzyme-functionalized polymersomes were then used to perform a cascade reaction upon incubation with 2,2’-azinobis(3-ethylbenzothiazoline-6-sulfonic acid) (ABTS) and 2,3,4,6-tetra-O-benzyl-D-glucopyranose acetylated at the anomeric position. The polymersomes demonstrated the ability to deacetylate the sugar with Cal B, followed by oxidation with GOx. The hydrogen peroxide produced then facilitated the reaction of HRP with ABTS. This work nicely demonstrates the potential of well-designed, bioreactive polymers to generate highly complex materials that fucntion to stabilize and deliver proteins.
Fig. 6.

The synthesis of three enzyme functionalized nanoreactor.75 Reprinted with permission from S.F.M. van Dongen, et. al, (2009), Chemistry-A European Journal, 15, 1107–1114. Copyright 2009 Wiley-VCH.
Modification of Proteins
Peptide and Protein Macroinitiators
While conjugation of proteins to polymers is an effective technique to form conjugates, this strategy requires the use of excess polymer, which must be removed. Characterization of polymer placement can also be difficult. We developed a technique to avoid these complications and produce well-defined conjugates via CRP directly from a protein.28, 29 The general idea was to prepare proteins in such a way that polymer growth would occur directly from the protein. To this end, maleimide and pyridyl disulfide initiators were synthesized for conjugation to V131C T4 lysozyme (Fig. 7) as well as a biotinylated ATRP initiator for conjugation to streptavidin. With both systems we were able to demonstrate high functionalization of the protein with the initiator and the conjugate maintained high activity postpolymerization compared to the unmodified protein. We and others demonstrated that narrow PDIs can be obtained and that the protein remained active postpolymerization via ATRP and RAFT polyerization.28, 29, 76–80 This work has been reviewed elsewhere, so only recent work that highlights the application of this approach will be discussed in detail.
Fig. 7.

Synthesis of V131C T4 lysozyme macroinitiator and polymerization of NIPAAm (protein structure from PBD # 132L).29
This strategy was recently used by Le Droumaguet and Velonia to prepare a giant amphiphile bionanoreactor of BSA-polystyrene (Fig. 8).81 In this work a maleimide-capped ATRP initiator was synthesized and coupled to C34 of BSA under mild conditions. MALDI-TOF MS confirmed that one molecule of maleimide initiator was present per molecule of protein. The macroinitiator was used to polymerize styrene by copper mediated emulsion ATRP. TEM confirmed formation of spherical aggregates (diameters, 20 – 100 nm) with behavior characteristic of giant amphiphiles. The authors also prepared conjugates of human serum albumin (HSA) and reduced human calcitonin. Formation of hierarchically-assembled nanocontainers was confirmed by performing the polymerizations in the presence of a fluorescently labeled non-polymerizable protein. The fluorescently labeled proteins encapsulated within the spherical superstructures. The giant amphiphile nanocontainers were determined to be permeable and may be useful as nanoreactors. This work demonstrates utility of this ATRP macroinitiator strategy for the preparation of functional polymer-conjugates and multienzyme nanoreactors. More specifically, the use of protein-macroinitiator for the ATRP of a hydrophoibic monomer facilitated the in situ synthesis of giant amphiphiles that self-assembled into particles and encapsulated another protein that maintained activity during encapsulation.
Fig. 8.

General scheme for the in situ ATRP-mediated synthesis of BSA-pS giant amphiphiles (protein structure from PBD # 1E7H).81
Depp et. al. have reported a comparison of the in vitro serum stability and enzyme activity retention for PEGylated chymotrypsin along with biocompatible chymotrypsin conjugates prepared by polymerizing from chymotrypsin covalently modified with an ATRP initiator.82 The chymotrypsin-poly(N-2-hydroxypropylmethyl methacrylamide) conjugates were determined to retain >50% of their catalytic activity in mouse serum and >25% in human serum after 5 days. Furthermore, chymotrypsin-poly(2-methacryloyloxyefhyl phosphorylcholine conjugates also retained >25% of their catalytic activity after five days in human serum. PEGylated chymotrypsin was deactivated after 24 hours in mouse serum and after four days in human serum. These results showed that the chymotrypsin initiated ATRP conjugates had higher catalytic activity compared to PEGylated chymotrypsin. These exciting results suggest that polymerizing from proteins is promising for the development of long lasting biocompatible pharmaceuticals.
Caliceti and coworkers have demonstrated improved pharmacokinetic properties upon polymerization from recombinant human growth hormone (rhGH) via ATRP.83 An NHS-tetraefhyleneglycol-bromoisobutyrate initiator was synthesized and conjugated to rhGH. A large excess of the initiator was used resulting in conjugation to 6–8 of the 10 possible free amines confirmed by MALDI-TOF MS. PEGMA (Mn 475 g mol−1) was then polymerized from the rhGH-macroinitiator. Conjugate formation was confirmed by SDS-PAGE, dynamic light scattering (radius of hydration: 10–12 nm), and size exclusion chromatography. The conjugate exhibited improved stability to proteolytic degradation by pepsin. Improved pharmacokinetics were demonstrated by administration of 120 µg doses of conjugate and unmodified hormone to female Sprague Dowley rats over nine days. The group that was administered the conjugate showed increased mass gains over an extended period versus the unmodified hormone group.
Chilkoti and coworkers have recently demonstrated similarly positive results with branched PEG via site-specific polymerization from the N-terminus of myoglobin84 and the C-terminus of green fluorescent protein (GFP).85 The N-terminus of myoglobin was oxidized to an aldehyde upon treatment with pyridoxal-5-phosphate (PLP). Oxime formation with aminooxy-functionalized bromoisobutyrate gave the macroinitiator. Functionalization of the C-terminus of GFP was facilitated by a Mycobacterium xenopi GyrA (Mxe)-intein tag that leaves a C-terminal thioester upon cleavage after which the ATRP initiator was attached via native chemical ligation (Fig. 9). Both protein-macroinitiators were then used to polymerize ologoethylene glycol methylether methacrylate (OEG-MA). Both conjugates demonstrated improved stability to enzymatic degradation in addition to improved pharmacokinetic properties compared to the unmodified proteins.
Fig. 9.

Expression, purification, and synthesis of C-terminal functionalized GFP-Ppegma conjugates. A) recombinant overexpression of GFP-Mxe-intein fusion and purification by inverse transition cycling (ITC). B) cleavage of GFP from tag and subsequent conjugation to ATRP initiator. C) in situ growth PEGMA from the C-terminus of GFP.85
Peptide-polymer conjugates by CRP techniques have also been successfully investigated.30, 31, 86–93 We recently reported the introduction of an artificial amino acid functionalized with an ATRP initiator into a peptide (Scheme 3).94 In this work, tyrosine- and serine-modified ATRP initiators were prepared for polymerization of styrene and HEMA, respectively. This strategy allows for direct incorporation of amino acid functionalized initiators into peptide backbones. Kinetic studies indicated controlled polymerization with high conversions. Polymers of different molecular weight were readily targeted. In an effort to demonstrate the utility of the strategy, the serine modified initiator (S*) was incorporated into VMS*VVQTK by standard solid-phase peptide synthesis (Scheme 3). This macroinitiator was then used to polymerize a N-acetyl-D-glucosamine modified HEMA with high conversion and a narrow PDI of 1.14. This methodology is versatile because it provides a means to polymerize from a peptide at any point in the sequence. It is possible that these amino acid initiators could be recombinantly placed into proteins, thus providing a method to site-specifically modify proteins without relying on natural aminoacids.
Scheme 2.

Incorporation of an amino acid based ATRP initiator into a peptide.94 Reprinted with permission from Broyer, R. M.; Quaker, G. M.; Maynard, H. D. J. Am. Chem. Soc. 2008, 130, 1041–7. Copyright 2008 American Chemical Society.
Clearly, these examples demonstrate that bioreactive protein-polymer conjugates can be synthesized by grafting from proteins by CRP. If the synthesis of the protein-macroinitiator/CTA is efficient, purification of the conjugate is greatly simplified because all is the only purification step required is removal of small molecule reagents. This is a distinct advantage compared to the grafting to approach, in which it is often necessary to remove unconjugated polymer (and sometimes protein) from the conjugate. Another important feature of the grafting from approach is that location of the polymer on the protein is determined before polymerization. In order for this appraoch to be effective it is essential that initiator placement is site-specific; otherwise it can be difficult to remove the macroinitiators. In some cases, as demonstrated above, heterogeneity of the macro-initiator/CTA does not effect the activity of the conjugate.
Multiple Proteins
Telechelic-Bioreactive Polymers
The majority of protein-polymer conjugates contain one protein and one to several polymer chains. In the approaches discussed above, polymerization occurs from a protein/protein-reactive handle or the polymerization mediating handle is transformed into a bioreactive end-group postpolymerization. Recently, both methods have been applied to synthesize homo- and heterotelechelic bioreactive polymers. The incorporation of the same protein onto each end of a polymer generates a triblock material with a wide range of possible applications. The polymer should provide the same benefits as described above, and in addition introduce well-defined multivalency into the conjugate with a linker of tunable length. For certain systems these protein multimers should exhibit higher binding affinities than mono-functionalized conjugates with improved pharmacokinetics over dimers synthesized by small molecules.95 Additionally, the polymer can confer other properties, such as facilitating higher order self-assembly. Heterodimeric conjugates offer other interesting possibilities. The ability to tether two biomolecules would allow for surface immobilization or attachment of various tags, fluorophores, peptides, antibodies, and other proteins to be linked to the biomolecule-polymer conjugate of interest.
Homotelechelic Polymers by ATRP
Traditionally, ATRP telechelic polymers have been prepared by employing bis-functional initiators, with subsequent transformation of the halogen chain ends.96 Atom transfer radical (ATR) coupling has been demonstrated to afford protein-reactive telechelics in a highly efficient fashion. Recently, we reported the synthesis of aminooxy-end functionalized polystyrene using ATRP followed by ATR coupling.97 A 1-bromoethyl ATRP initiator with a N-hydroxyphthalimide group was used to polymerize styrene by copper-mediated ATRP with high initiator efficiency. Polymerization kinetics showed that polymers were produced with targeted molecular weights and low PDIs (as low as 1.2). Subsequent ATR coupling and deprotection with hydrazine yielded the telechelic aminooxy end-functionalized polymers. End-group reactivity was demonstrated using 4-bromobenzaldehyde as a model to confirm efficient and complete oxime bond formation. In a similar manner, we created cysteine reactive telechelic polystyrene.98 In this example, dimethylfulvene-protected maleimide-functionalized ATRP initiator was used to polymerize styrene. This approach to bis end-functionalized polymers is expected to be useful for forming protein dimers with hydrophobic polymers.
Homodimeric Protein-Polymer Conjugates by RAFT Polymerization
We also recently reported the straightforward synthesis of homodimeric protein polymer conjugates using RAFT polymerization that exploits the inherent reactivity of trithiocarbonate end-groups.99 A telechelic maleimide-end-functionalized pNIPAAm was synthesized by RAFT polymerization; a small amount of fluorescent monomer was included. Subsequent radical addition of the protected maleimide azo-initiator to the chain ends and retro-Diels Alder afforded the telechelic maleimide-end-functionalized pNIPAAm (Scheme 3). Conjugation to a V131C mutant T4 lysozyme with only one reactive cysteine gave the protein dimer.
Heterodimeric Protein-Polymer Conjugates
A variety of strategies have been extended toward the synthesis of heterodimeric protein-polymer conjugates.100–104 Davis and coworkers synthesized an α-azide, ω-pyridyl disulfide CTA.100 This bifunctional CTA was used to produce well-defined pSt, pPEGA, and pNIPAAm. The azide was utilized for a Huisgen cycloaddition with an alkyne-functionalized biotin, allowing for conjugation to SAv. The pyridyl disulfide was used for reversible conjugation of glutathione (a ligand for glutathione S-transferase) or BSA. Theato and coworkers polymerized diethyleneglycol monomethylether (DEGMA) in the presence of an activated ester CTA.104 The activated ester was used to conjugate thyroxin, a transthyretin ligand, to the polymer. The authors then transformed the thicarbonylthio via reduction with butylamine and subsequent trapping of the free thiol with N-biotinaminoethyl methanethiosulfonate. The α-thyroxin, ω-biotin-pNIPAAm was used to conjugate transthyretin-pNIPAAm conjugate to SAv coated surfaces. We demonstrated the ability to synthesize heterodimeric protein-polymer conjugates via radical coupling with a furan-protected azoinitiator to biotinylated-pNIPAAm synthesized by RAFT.101, 102 NIPAAm was polymerized in the presence of a biotinylated CTA101 and biotinylated CTA with a disulfide between biotin and the site of polymer growth.102 BSA was then conjugated to the maleimide end-group of both polymers. These conjugates were incubated with SAv coated surfaces and ELISA was performed to demonstrated the reversibility of the disulfide containing conjugate (Fig. 10).102
Fig. 10.

Synthesis of heterotelechelic polymers for reversible surface immobilization of protein-polymer conjugates.102 Heredia, et al., Polym. Chem. 2010, 1, 168–170 – Reproduced by permission of The Royal Society of Chemistry.
Protein-Polymer Conjugate Stars
As mentioned above, higher order protein-polymer conjugates should have advantageous traits over monofunctional conjugates. One of those traits could be increased binding affinity. To this end we pursued the synthesize of a star polymer with protein-reactive end-groups for the synthesis of starred protein-polymer conjugates (Fig. 11).105 A four armed CTA was synthesized and polymerization of NIPAAm to low conversion (<50 %) resulted in a mixture of three and four armed polymers characterized by gel permeation chromatography and MALDI-TOF MS. Postpolymerization the trithiocarbonate end-groups were exchanged for maleimides via radical coupling. V131C T4 lysozyme was then conjugated to the telechelic stars resulting in three lysozymes per star polymer.
Fig. 11.

Synthesis of starred protein-polymer conjugates.105 Reproduced with permission from Macromolecules 2009, 42, 8028–8033. Copyright 2009 American Chemical Society.
The field of multimeric, protein-polymer conjugates is still in its infancy. The above examples have demonstrated that the formation of these materials is indeed possible. These materials promise to have superior properties compared to monomeric conjugates. Introduction of multivalency is also expected to generate materials with higher binding affinities. The ability to attach different biomolecules allows for efficient anchoring of proteins to surfaces, which can lead to improved biosensors. The range and scope of this methodology is actively being explored and has potential to generate polymeric materials that can target and deliver drugs or target mutliple biological processes in the same construct.
Cytotoxicity of CRP Polymers
A few groups have investigated the cytotoxicity of polymers synthesized by CRP.106, 107 This information is crucial, as many of the bioconjugates prepared using CRPs are intended for use as biomedical and/or therapeutic polymers. We investigated the cytoxicity of p(PEGA)s synthesized by ATRP and RAFT polymerization to NIH 3T3 mouse fibroblasts (NIH3T3).106 Copper mediated ATRP of PEGA was conducted to form a polymer with a PDI of 1.12 and molecular weight of 19,900 Da (Fig. 12). The polymer was purified by two different methods and the cytotoxicity was evaluated using three different polymer concentrations and the (3–4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) assay. The results were compared to PEG and media as controls. The data indicated that the method of purification of ATRP prepared polymers is critical. Polymers purified by dialysis were nontoxic compared to those purified by typical precipitation and flow through an alumina oxide column. Toxicity was attributed to residual copper from the ATRP reaction, as well as residual PEGA monomer, both of which are known to be toxic to cells. This study suggests that while precipitation is generally considered an effective way to remove excess copper and monomer, it may not be sufficient for biological applications. However, dialysis sufficiently removed toxic compounds.
Fig. 12.

Structures of pPEGAs synthesized by RAFT.106 Chang, C. W.; Bays, E.; Tao, L.; Alconcel, S. N.; Maynard, H. D. Chem. Commun. 2009, 3580–3582.-Reproduced by permission of The Royal Society of Chemistry.
Polymers prepared by RAFT polymerization were also systematically evaluated.106 Different RAFT CTAs (two dithioesters and two trithiocarbonates) were used to polymerize PEGA (Fig. 12), and the cytotoxicities of the resultant polymers were evaluated (Fig. 13). Results showed that the polymers prepared using a dithioester CTA exhibited toxicity only at a high concentration of polymer (10 mg/mL), whereas those prepared using a trithiocarbonate were nontoxic at all concentrations used in this study (Fig. 13). Further studies suggested that the hydrolytic instability of the thiocarbonylthio moiety of the dithioester was problematic. Reduction and capping of the thiol reduced the toxicity of the polymers even at high concentrations.
Fig. 13.

“Cytotoxicity of pPEGAs synthesized by RAFT using different CTAs. Cell proliferation rate of NIH 3T3 cells after A) 24 h exposure to different polymer concentrations (1, 3, and 10 mg mL−1) or B) 24 and 48 h exposure to 10 mg mL−1 of polymers. Data represent mean +/− S.E.M. (n=6). Significance indicated by: * p 0.05 vs. cells after exposure to 1, 2 or PEG 14Da.”106 Chang, C. W.; Bays, E.; Tao, L.; Alconcel, S. N.; Maynard, H. D. Chem. Commun. 2009. DOI: 10.1039/b904456f-Reproduced by permission of The Royal Society of Chemistry.
Bulmus and coworkers have also investigated the cytotoxicity of RAFT polymers.107 The toxicity of a series of similar molecular weight RAFT polymers (Table 1) to Chinese hamster ovary cells (CHO-K1), mouse macrophage cells (Raww264.7) and NIH3T3 cells was investigated at a polymer concentration of ∼10 mg/mL. The authors found that all of the dithiobenzoate- and trithiocarbonate-branched-PEG polymers did not exhibit any significant cytotoxicity over 24 hrs for all cell types. After 72 hrs the branched-PEG polymers did not exhibit cytotoxicity to CHO-K1 and NIH3T3 cells, while there was a slight dose-dependent toxicity to RAW264.7 cells. The dithiobenzoate-pHPMAs were highly toxic to all three cell lines at ∼10 mg/mL over 24 hrs. The authors found that the toxicity could be eliminated by reducing the concentration to ∼2 mg/mL or by replacing the dithiobenzoate end-group with a trithiocarbonate or reducing and capping with HPMA.
Table 1.
Polymerization conditions, characterization, and strucutures of polymers used in toxicity study. N-2-hydroxypropyl methacrylamide (HPMA), oligoethylene glycol methy ether acrylate (OEG-A), oligoethylene glycol methy ether methacrylate (OEG-MA), dithiobenzoate end-group (D), trithiocarbonate end-group (T), cumyl dithiobenzoate (CDB), and IS (aminoylsis and end capping with HPMA).107
| Polymers | Mn by GPC (g/mol) |
Mn by UV- vis and NMR (g/mol) |
PDI | Structure |
|---|---|---|---|---|
| p(OEG-A)D | 11,400 | 12,500 | 1.25 | 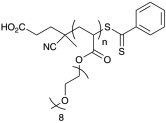 |
| p(OEG-MA)D | 10,100 13,500 |
11,200 12,000 |
1.21 1.18 |
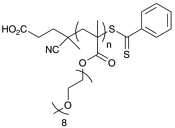 |
| p(OEG-A)T | 11,100 | 12,000 | 1.18 | 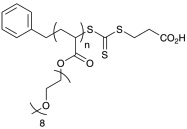 |
| p(HMPA)D | 19,300 18,800 |
10,400 9,400 |
1.03 1.04 |
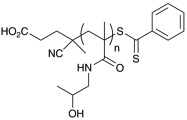 |
| p(HPMA)CDB | 18,100 | 8,900 | 1.09 | 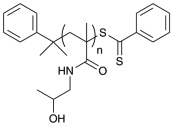 |
| p(HPMA)T | 21,200 | 12,600 | 1.25 | 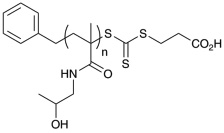 |
| p(HPMA)IS | 19,300 | 13,200 | 1.06 | 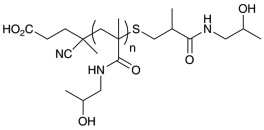 |
Together, these two studies suggest that polymer toxicity will depend on the polymer identity and/or chain end, as well as the cell type. The results also indicate that RAFT-synthesized polymers should be administered at low concentrations or that the thiolcarbonylthiol group should be removed to avoid issues with toxicity. As further biomedical applications of RAFT synthesized polymers emerge, it will be interesting to explore polymer toxicity by other measures including in vivo.
Conclusions and Outlook
The field of peptide- and protein-polymer conjugates is attracting increasing interest from researchers across many different fields. These hybrid materials are useful in applications ranging from medicine to biotechnology. While a number of postpolymerization modification strategies are in place to synthesize the conjugates, applications in nanoscience and medicine will require well-defined homogeneous conjugates. CRPs providefacile access to such conjugates. Moreover, there have been several reports of the use of protein macroinitiators to prepare conjugates in situ. These techniques provide alternative methods to PEGylation of proteins for therapeutic applications. For the later technique, incorporation of non-natural amino acids containing ATRP initiators into peptides has been demonstrated. It is envisioned that by using protein engineering it may be possible to incorporate such non-natural amino acids into proteins to provide alternate ways to access protein macroinitiators. This could provide a straightforward method to synthesize precise bioconjugates for a number of applications.
It is important to consider advantages and disadvantages when choosing to synthesize protein-polymer conjugates. The advantage of grafting to is that the polymer is synthesized seperately from the protein. This allows for additional postpolymerization modifications to be performed on the material that could have an effect on the activity of a protein. Generally, this approach is easier to synthesize multimeric conjugates from telechelic scaffolds. The disadvantage of grafting to is that difficulty can arise upon seperating free polymer from unmodified protein and conjugate. The advantage of grafting from is the ease of purification of the conjugate. Because the site of polymer growth is predefined all proteins should contain polymer thus all that needs to be removed is unused monomer and other reagents (usually by dialysis). The disadvantages of grafting from are that difficulty can arise when purifying macroinitiator from unmodified protein. Also it is essential that the polymerization conditions do not affect the activity of the protein. When chosen properly this is usually not a problem, but could be difficult for very fragile proteins.
Recent interest has also been focused on the preparation of homo- and hetero- bis-functionalized conjugates as well as starred conjugates. These strategies are expected to provide facile access to proteins linked as dimers or multimers. The ability to bring multiple biomolecules together via a polymer scaffold has powerful implications in therapeutic design and medicine. Many proteins dimerize or olgomerize to ellicit a biological response. Therefore homodimeric and starred protein-polymer conjugates could results in higher activity drugs. Additionally, for heterodimeric conjugates it is possible for these materials to target multiple steps/pathways in a biological process. Thus, multimeric conjugates are expected to have superior biological activities compared to monomeric conjugates. There is also increasing interest in the area of nanomedicine and materials science for engineered surfaces consisting of well-defined conjuages. The ability to more precisely control biomolecule absorption to surfaces using CRP teqniques may result in smart surfaces with improved properties for applications in molecular biology and materials science. ATRP and RAFT techniques are capable of achieving site-specific modifications to proteins while maintaining the bioactivity of the conjugate. Application of these techniques to surfaces may be of interest in the site-specific anchoring of proteins to surfaces for widespread applications in nanomedicine and biosensors.
Systematic investigations of the cytotoxicity of CRP polymers have also been reported. It is imperative to consider this information when designing polymer conjugates for medical applications by controlled radical polymerizations. Results showed that the purification method, end-group, and concentration may affect the toxicity of the polymers. Finally, initial studies of therapeutic protein-polymer conjugates synthesized by CRP are emerging. The data indicates that the conjugates exhibit improved stability to proteolytic degradation and improved pharmacokinetics compared to the unmodified protein. Taken together, these studies indicate that protein-polymer conjugates synthesized by CRP will have a positive impact on the fields of drug delivery and bionanotechnology.
Scheme 4.

A) Synthesis of telechelic trithiocarbonate-pNIPAAm. B) Synthesis of telechelic maleimide-pNIPAAm.99 Tao, L.; Kaddis, C. S.; Loo, R. R. O.; Grover, G. N.; Loo, J. A.; Maynard, H. D. Chem. Commun. 2009, 2148–2150-Reproduced by permission of The Royal Society of Chemistry.
Aknowledgements
The authors are grateful to the National Science Foundation (CHE-0809832) for funding. GNG thanks the Christopher S. Foote Graduate Research Fellowship in Organic Chemistry and the NIH Biotechnology Training Grant. HDM appreciates the Alfred P. Sloan Foundation for additional funding.
Biographies

Dr. Rebecca M. Broyer
Rebecca Broyer received a B.S. from UC San Diego (2000). She worked in the biotechnology industry for several years before moving to UCLA (Ph.D., 2009). Her graduate work focused on using controlled polymerizations to make synthetic modifications to peptides, as well as fabrication of multicomponent protein nanoarrays. She then moved to the LA County Museum of Art where she worked as a Mellon Postdoctoral Fellow to develop polymeric materials for the preservation of art. Dr. Broyer joined the faculty at the University of Southern California as a Lecturer (2010) where her current interests include chemical education and curriculum development.

Gregory N. Grover
Greg graduated received a honors B.S. from Gettysburg College, PA (2006) where he worked with Dr. Darren MacFarland on the synthesis and application of asymmetric phase transfer catalysts. His research earned him a Merck Summer Undergraduate Research Fellowship and the John B. Zinn Chemistry Research award. He is currently working toward his Ph.D. in organic chemistry in the Maynard group (UCLA). Gregory holds a Christopher S. Foote Graduate Fellowship in Organic Chemistry and is a trainee in the NIH-sponsored Biotechnology Training Grant. His research focuses on the application of CRP techniques for drug-delivery and the construction of multimeric, protein-polymer conjugates.

Professor Heather D. Maynard
Heather D. Maynard received a B.S. from the University of North Carolina at Chapel Hill in 1992 and a M.S. in Materials Science in 1995 from the University of California, Santa Barbara. Her Ph.D. from the California Institute of Technology was awarded in 2000. She then moved to the Swiss Federal Institute of Technology in Zurich (ETH), where from 2000–2002 she was an American Cancer Society Postdoctoral Fellow. Dr. Maynard joined the UCLA faculty as an Assistant Professor in 2002 as the first Howard Reiss Career Development Chair in the Department of Chemistry and Biochemistry and as a member of the California NanoSystems Institute. She is now an Associate Professor. Since arriving at UCLA, Maynard has given over 120 invited lectures including the WCC ACS Lecture at Southern Methodist University and the Walter F. Enz Lectures at the University of Kansas. Maynard has been selected as an Outstanding Emerging Investigator by the Journal of Materials Chemistry and has received the Amgen New Faculty Award, NSF Career Award, Seaborg Award, and an Alfred P. Sloan Fellowship for her research.
References
- 1.Duncan R. Nat. Rev. Drug Discovery. 2003;2:347–360. doi: 10.1038/nrd1088. [DOI] [PubMed] [Google Scholar]
- 2.Jain A, Jain SK. Crit. Rev. Ther. Drug Carrier Syst. 2008;25:403–447. doi: 10.1615/critrevtherdrugcarriersyst.v25.i5.10. [DOI] [PubMed] [Google Scholar]
- 3.Nucci ML, Shorr R, Abuchowski A. Adv. Drug Delivery Rev. 1991;6:133–151. [Google Scholar]
- 4.Abuchowski A, Davis FF. Biochim. Biophys. Acta. 1979;578:41–46. doi: 10.1016/0005-2795(79)90110-7. [DOI] [PubMed] [Google Scholar]
- 5.Hadley KB, Sato PH. Enzyme. 1989;42:225–234. doi: 10.1159/000469036. [DOI] [PubMed] [Google Scholar]
- 6.Savoca KV, Abuchowski A, Vanes T, Davis FF, Palczuk NC. Biochim. Biophys. Acta. 1979;578:47–53. doi: 10.1016/0005-2795(79)90111-9. [DOI] [PubMed] [Google Scholar]
- 7.Glue P, Fang JWS, Rouzier-Panis R, Raffanel C, Sabo R, Gupta SK, Salfi M, Jacobs S, Hepatitis CITG. Clin. Pharmacol. Ther. 2000;68:556–567. doi: 10.1067/mcp.2000.110973. [DOI] [PubMed] [Google Scholar]
- 8.Hannink JM, Cornelissen JJLM, Farrera JA, Foubert P, De Schryver FC, Sommerdijk NAJM, Nolte RJM. Angew. Chem. Int. Ed. 2001;40:4732–4734. [PubMed] [Google Scholar]
- 9.Kochendoerfer GG, Chen SY, Mao F, Cressman S, Traviglia S, Shao HY, Hunter CL, Low DW, Cagle EN, Carnevali M, Gueriguian V, Keogh PJ, Porter H, Stratton SM, Wiedeke MC, Wilken J, Tang J, Levy JJ, Miranda LP, Crnogorac MM, Kalbag S, Botti P, Schindler-Horvat J, Savatski L, Adamson JW, Kung A, Kent SBH, Bradburne JA. Science. 2003;299:884–887. doi: 10.1126/science.1079085. [DOI] [PubMed] [Google Scholar]
- 10.Pepinsky RB, Lepage DJ, Gill A, Chakraborty A, Vaidyanathan S, Green M, Baker DP, Whalley E, Hochman PS, Martin P. J. Pharmacol. Exp. Ther. 2001;297:1059–1066. [PubMed] [Google Scholar]
- 11.Velonia K, Rowan AE, Nolte RJM. J. Am. Chem. Soc. 2002;124:4224–4225. doi: 10.1021/ja017809b. [DOI] [PubMed] [Google Scholar]
- 12.Wang YS, Youngster S, Bausch J, Zhang RM, McNemar C, Wyss DF. Biochemistry. 2000;39:10634–10640. doi: 10.1021/bi000617t. [DOI] [PubMed] [Google Scholar]
- 13.Noren CJ, Anthonycahill SJ, Griffith MC, Schultz PG. Science. 1989;244:182–188. doi: 10.1126/science.2649980. [DOI] [PubMed] [Google Scholar]
- 14.Basle E, Joubert N, Pucheault M. Chemistry & Biology. 2010;17:213–227. doi: 10.1016/j.chembiol.2010.02.008. [DOI] [PubMed] [Google Scholar]
- 15.Connor RE, Tirrell DA. Polym. Rev. 2007;47:9–28. [Google Scholar]
- 16.Tiefenbrunn TK, Dawson PE. Biopolymers. 2010;94:95–106. doi: 10.1002/bip.21337. [DOI] [PubMed] [Google Scholar]
- 17.Wang L, Schultz PG. Angew. Chem. Int. Ed. 2005;44:34–66. doi: 10.1002/anie.200460627. [DOI] [PubMed] [Google Scholar]
- 18.Hoffman AS, Stayton PS. Macromol. Symp. 2004;207:139–151. [Google Scholar]
- 19.Hoffman AS, Stayton PS, Bulmus V, Chen GH, Chen JP, Cheung C, Chilkoti A, Ding ZL, Dong LC, Fong R, Lackey CA, Long CJ, Miura M, Morris JE, Murthy N, Nabeshima Y, Park TG, Press OW, Shimoboji T, Shoemaker S, Yang HJ, Monji N, Nowinski RC, Cole CA, Priest JH, Harris JM, Nakamae K, Nishino T, Miyata T. J. Biomed. Mater. Res. 2000;52:577–586. doi: 10.1002/1097-4636(20001215)52:4<577::aid-jbm1>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- 20.Hoffman AS, Stayton PS. Prog. Polym. Sci. 2007;32:922–932. [Google Scholar]
- 21.Stayton PS, Hoffman AS, El-Sayed M, Kulkarni S, Shimoboji T, Murthy N, Bulmus V, Lackey C. Proceedings of the IEEE. 2005;93:726–736. [Google Scholar]
- 22.Alarcon CDH, Pennadam S, Alexander C. Chem. Soc. Rev. 2005;34:276–285. doi: 10.1039/b406727d. [DOI] [PubMed] [Google Scholar]
- 23.Heredia KL, Maynard HD. Org. Biomol. Chem. 2007;5:45–53. doi: 10.1039/b612355d. [DOI] [PubMed] [Google Scholar]
- 24.Le Droumaguet B, Nicolas J. Polym. Chem. 2010;1:563–598. [Google Scholar]
- 25.Nicolas J, Mantovani G, Haddleton DM. Macromol. Rapid Commun. 2007;28:1083–1111. [Google Scholar]
- 26.Bontempo D, Heredia KL, Fish BA, Maynard HD. J. Am. Chem. Soc. 2004;126:15372–15373. doi: 10.1021/ja045063m. [DOI] [PubMed] [Google Scholar]
- 27.Bontempo D, Li RC, Ly T, Brubaker CE, Maynard HD. Chem. Commun. 2005:4702–4704. doi: 10.1039/b507912h. [DOI] [PubMed] [Google Scholar]
- 28.Bontempo D, Maynard HD. J. Am. Chem. Soc. 2005;127:6508–6509. doi: 10.1021/ja042230+. [DOI] [PubMed] [Google Scholar]
- 29.Heredia KL, Bontempo D, Ly T, Byers JT, Halstenberg S, Maynard HD. J. Am. Chem. Soc. 2005;127:16955–16960. doi: 10.1021/ja054482w. [DOI] [PubMed] [Google Scholar]
- 30.Ayres L, Adams P, Lowik D, van Hest JCM. Biomacromolecules. 2005;6:825–831. doi: 10.1021/bm049421p. [DOI] [PubMed] [Google Scholar]
- 31.Ayres L, Vos MRJ, Adams P, Shklyarevskiy IO, van Hest JCM. Macromolecules. 2003;36:5967–5973. [Google Scholar]
- 32.Hwang JY, Li RC, Maynard HD. J. Controlled Release. 2007;122:279–286. doi: 10.1016/j.jconrel.2007.04.010. [DOI] [PubMed] [Google Scholar]
- 33.Stukel JM, Li RC, Maynard HD, Caplan MR. Biomacromolecules. 2010;11:160–167. doi: 10.1021/bm9010276. [DOI] [PubMed] [Google Scholar]
- 34.Canalle LA, Lowik D, van Hest JCM. Chem. Soc. Rev. 2010;39:329–353. doi: 10.1039/b807871h. [DOI] [PubMed] [Google Scholar]
- 35.Klok HA. Macromolecules. 2009;42:7990–8000. [Google Scholar]
- 36.Gauthier MA, Klok HA. Chem. Commun. 2008:2591–2611. doi: 10.1039/b719689j. [DOI] [PubMed] [Google Scholar]
- 37.Velonia K. Polym. Chem. 2010;1:944–952. [Google Scholar]
- 38.Shakya AK, Sami H, Srivastava A, Kumar A. Prog. Polym. Sci. 2010;35:459–486. [Google Scholar]
- 39.Gauthier MA, Klok HA. Polym. Chem. 2010 [Google Scholar]
- 40.Hermanson GT. Bioconjugate Techniques. Elsevier Inc.; 2008. [Google Scholar]
- 41.Pasut G, Veronese FM. Polymer Therapeutics I: Polymers as Drugs, Conjugates and Gene Delivery Systems. 2006;192:95–134. [Google Scholar]
- 42.Roberts MJ, Bentley MD, Harris JM. Adv. Drug Delivery Rev. 2002;54:459–476. doi: 10.1016/s0169-409x(02)00022-4. [DOI] [PubMed] [Google Scholar]
- 43.Ruehl J, Morimoto C, Stevens DJ, Braslau R. React. Funct. Polym. 2008;68:1563–1577. [Google Scholar]
- 44.Tao L, Liu JQ, Xu JT, Davis TP. Org. Biomol. Chem. 2009;7:3481–3485. doi: 10.1039/b907061c. [DOI] [PubMed] [Google Scholar]
- 45.McDowall L, Chen GJ, Stenzel MH. Macromol. Rapid Commun. 2008;29:1666–1671. [Google Scholar]
- 46.Pound G, McKenzie JM, Lange RFM, Klumperman B. Chem. Commun. 2008:3193–3195. doi: 10.1039/b803952f. [DOI] [PubMed] [Google Scholar]
- 47.Sayers CT, Mantovani G, Ryan SM, Randev RK, Keiper O, Leszczyszyn OI, Blindauer C, Brayden DJ, Haddleton DM. Soft Matter. 2009;5:3038–3046. [Google Scholar]
- 48.Kochendoerfer GG. Curr. Opin. Chem. Biol. 2005;9:555–560. doi: 10.1016/j.cbpa.2005.10.007. [DOI] [PubMed] [Google Scholar]
- 49.Lemieux GA, Bertozzi CR. Trends Biotechnol. 1998;16:506–513. doi: 10.1016/s0167-7799(98)01230-x. [DOI] [PubMed] [Google Scholar]
- 50.Schlick TL, Ding ZB, Kovacs EW, Francis MB. J. Am. Chem. Soc. 2005;127:3718–3723. doi: 10.1021/ja046239n. [DOI] [PubMed] [Google Scholar]
- 51.Shao H, Crnogorac MM, Kong T, Chen SY, Williams JM, Tack JM, Gueriguian V, Cagle EN, Carnevali M, Tumelty D, Paliard X, Miranda LP, Bradburne JA, Kochendoerfer GG. J. Am. Chem. Soc. 2005;127:1350–1351. doi: 10.1021/ja043096w. [DOI] [PubMed] [Google Scholar]
- 52.Gilmore JM, Scheck RA, Esser-Kahn AP, Joshi NS, Francis MB. Angew. Chem. Int. Ed. 2006;45:5307–5311. doi: 10.1002/anie.200600368. [DOI] [PubMed] [Google Scholar]
- 53.Muir TW. Annu. Rev. Biochem. 2003;72:249–289. doi: 10.1146/annurev.biochem.72.121801.161900. [DOI] [PubMed] [Google Scholar]
- 54.Heredia KL, Tolstyka ZP, Maynard HD. Macromolecules. 2007;40:4772–4779. [Google Scholar]
- 55.Vazquez-Dorbatt V, Tolstyka ZP, Maynard HD. Macromolecules. 2009;42:7650–7656. doi: 10.1021/ma9013803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Liu J, Bulmus V, Barner-Kowollik C, Stenzel MH, Davis TP. Macromol. Rapid Commun. 2007;28:305–314. [Google Scholar]
- 57.Boyer C, Liu J, Wong L, Tippett M, Bulmus V, Davis TP. J. Polym. Sci., Part A: Polym. Chem. 2008;46:7207–7224. [Google Scholar]
- 58.Duvall CL, Convertine AJ, Benoit DSW, Hoffman AS, Stayton PS. Mol. Pharmaceutics. 2010;7:468–476. doi: 10.1021/mp9002267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Heredia KL, Nguyen TH, Chang CW, Bulmus V, Davis TP, Maynard HD. Chem. Commun. 2008:3245–3247. doi: 10.1039/b804812f. [DOI] [PubMed] [Google Scholar]
- 60.Vazquez-Dorbatt V, Tolstyka ZP, Chang CW, Maynard HD. Biomacromolecules. 2009;10:2207–2212. doi: 10.1021/bm900395h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Boyer C, Bulmus V, Davis TP. Macromol. Rapid Commun. 2009;30:493–497. doi: 10.1002/marc.200800708. [DOI] [PubMed] [Google Scholar]
- 62.Roth PJ, Kessler D, Zentel R, Theato P. J. Polym. Sci., Part A: Polym. Chem. 2009;47:3118–3130. [Google Scholar]
- 63.Mantovani G, Lecolley F, Tao L, Haddleton DM, Clerx J, Cornelissen JJLM, Velonia K. J. Am. Chem. Soc. 2005;127:2966–2973. doi: 10.1021/ja0430999. [DOI] [PubMed] [Google Scholar]
- 64.Bays E, Tao L, Chang CW, Maynard HD. Biomacromolecules. 2009;10:1777–1781. doi: 10.1021/bm9001987. [DOI] [PubMed] [Google Scholar]
- 65.Grover GN, Alconcel SN, Matsumoto NM, Maynard HD. Macromolecules. 2009;42:7657–7663. doi: 10.1021/ma901036x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tedaldi LM, Smith MEB, Nathani RI, Baker JR. Chem. Commun. 2009:6583–6585. doi: 10.1039/b915136b. [DOI] [PubMed] [Google Scholar]
- 67.Smith MEB, Schumacher FF, Ryan CP, Tedaldi LM, Papaioannou D, Waksman G, Caddick S, Baker JR. J. Am. Chem. Soc. 2010;132:1960–1965. doi: 10.1021/ja908610s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Shiu HY, Chan TC, Ho CM, Liu Y, Wong MK, Che CM. Chem. Eur. J. 2009;15:3839–3850. doi: 10.1002/chem.200800669. [DOI] [PubMed] [Google Scholar]
- 69.Jones MW, Mantovani G, Ryan SM, Wang XX, Brayden DJ, Haddleton DM. Chem. Commun. 2009:5272–5274. doi: 10.1039/b906865a. [DOI] [PubMed] [Google Scholar]
- 70.Kiick KL, Saxon E, Tirrell DA, Bertozzi CR. Proc. Nat. Acad. Sci. U.S.A. 2002;99:19–24. doi: 10.1073/pnas.012583299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Neumann H, Wang KH, Davis L, Garcia-Alai M, Chin JW. Nature. 2010;464:441–444. doi: 10.1038/nature08817. [DOI] [PubMed] [Google Scholar]
- 72.van Dongen SFM, Teeuwen RLM, Nallani M, van Berkel SS, Cornelissen JJLM, Nolte RJM, van Hest JCM. Bioconjugate Chem. 2009;20:20–23. doi: 10.1021/bc8004304. [DOI] [PubMed] [Google Scholar]
- 73.Le Droumaguet B, Velonia K. Macromol. Rapid Commun. 2008;29:1073–1089. [Google Scholar]
- 74.Li M, De P, Gondi SR, Sumerlin BS. Macromol. Rapid Commun. 2008;29:1172–1176. [Google Scholar]
- 75.van Dongen SFM, Nallani M, Cornelissen J, Nolte RJM, van Hest JCM. Chem. Eur. J. 2009;15:1107–1114. doi: 10.1002/chem.200802114. [DOI] [PubMed] [Google Scholar]
- 76.Lele BS, Murata H, Matyjaszewski K, Russell AJ. Biomacromolecules. 2005;6:3380–3387. doi: 10.1021/bm050428w. [DOI] [PubMed] [Google Scholar]
- 77.Nicolas J, San Miguel V, Mantovani G, Haddleton DM. Chem. Commun. 2006:4697–4699. doi: 10.1039/b609935a. [DOI] [PubMed] [Google Scholar]
- 78.Boyer C, Bulmus V, Liu JQ, Davis TP, Stenzel MH, Barner-Kowollik C. J. Am. Chem. Soc. 2007;129:7145–7154. doi: 10.1021/ja070956a. [DOI] [PubMed] [Google Scholar]
- 79.De P, Li M, Gondi SR, Sumerlin BS. J. Am. Chem. Soc. 2008;130:11288–11289. doi: 10.1021/ja804495v. [DOI] [PubMed] [Google Scholar]
- 80.Liu JQ, Bulmus V, Herlambang DL, Barner-Kowollik C, Stenzel MH, Davis TR. Angew. Chem. Int. Ed. 2007;46:3099–3103. doi: 10.1002/anie.200604922. [DOI] [PubMed] [Google Scholar]
- 81.Le Droumaguet B, Velonia K. Angew. Chem. Int. Ed. 2008;47:6263–6266. doi: 10.1002/anie.200801007. [DOI] [PubMed] [Google Scholar]
- 82.Depp V, Alikhani A, Grammer V, Lele BS. Acta Biomater. 2009;5:560–569. doi: 10.1016/j.actbio.2008.08.010. [DOI] [PubMed] [Google Scholar]
- 83.Magnusson JP, Bersani S, Salmaso S, Alexander C, Caliceti P. Bioconjugate Chem. 2010;21:671–678. doi: 10.1021/bc900468v. [DOI] [PubMed] [Google Scholar]
- 84.Gao WP, Liu WG, Mackay JA, Zalutsky MR, Toone EJ, Chilkoti A. Proc. Nat. Acad. Sci. U.S.A. 2009;106:15231–15236. doi: 10.1073/pnas.0904378106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Gao W, Liu W, Christensen T, Zalutsky MR, Chilkoti A. Proc. Nat. Acad. Sci. U.S.A. 2010 doi: 10.1073/pnas.1006044107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Becker ML, Liu JQ, Wooley KL. Chem. Commun. 2003 doi: 10.1039/b209557b. 802-802. [DOI] [PubMed] [Google Scholar]
- 87.Becker ML, Liu JQ, Wooley KL. Biomacromolecules. 2005;6:220–228. doi: 10.1021/bm049551y. [DOI] [PubMed] [Google Scholar]
- 88.Couet J, Biesalski M. Macromolecules. 2006;39:7258–7268. [Google Scholar]
- 89.Couet J, Samuel JD, Kopyshev A, Santer S, Biesalski M. Angew. Chem. Int. Ed. 2005;44:3297–3301. doi: 10.1002/anie.200462993. [DOI] [PubMed] [Google Scholar]
- 90.Mei Y, Beers KL, Byrd HCM, Vanderhart DL, Washburn NR. J. Am. Chem. Soc. 2004;126:3472–3476. doi: 10.1021/ja039583d. [DOI] [PubMed] [Google Scholar]
- 91.Rettig H, Krause E, Borner HG. Macromol. Rapid Commun. 2004;25:1251–1256. [Google Scholar]
- 92.ten Cate MGJ, Rettig H, Bernhardt K, Borner HG. Macromolecules. 2005;38:10643–10649. [Google Scholar]
- 93.Venkataraman S, Wooley KL. Macromolecules. 2006;39:9661–9664. doi: 10.1021/ma0619583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Broyer RM, Quaker GM, Maynard HD. J. Am. Chem. Soc. 2008;130:1041–1047. doi: 10.1021/ja0772546. [DOI] [PubMed] [Google Scholar]
- 95.Choi SK, Mammen M, Whitesides GM. J. Am. Chem. Soc. 1997;119:4103–4111. [Google Scholar]
- 96.Coessens V, Pintauer T, Matyjaszewski K. Prog. Polym. Sci. 2001;26:337–377. [Google Scholar]
- 97.Kopping JT, Tolstyka ZP, Maynard HD. Macromolecules. 2007;40:8593–8599. [Google Scholar]
- 98.Tolstyka ZP, Kopping JT, Maynard HA. Macromolecules. 2008;41:599–606. [Google Scholar]
- 99.Tao L, Kaddis CS, Loo RRO, Grover GN, Loo JA, Maynard HD. Chem. Commun. 2009:2148–2150. doi: 10.1039/b822799c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Boyer C, Liu J, Bulmus V, Davis TP, Barner-Kowollik C, Stenzel MH. Macromolecules. 2008;41:5641–5650. [Google Scholar]
- 101.Heredia KL, Grover GN, Tao L, Maynard HD. Macromolecules. 2009;42:2360–2367. doi: 10.1021/ma8022712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Heredia KL, Tao L, Grover GN, Maynard HD. Polym. Chem. 2010;1:168–170. doi: 10.1039/B9PY00369J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Liu JQ, Liu HY, Bulmus V, Tao L, Boyer C, Davis TP. J. Polym. Sci., Part A: Polym. Chem. 2010;48:1399–1405. [Google Scholar]
- 104.Roth PJ, Jochum FD, Zentel R, Theato P. Biomacromolecules. 2010;11:238–244. doi: 10.1021/bm901095j. [DOI] [PubMed] [Google Scholar]
- 105.Tao L, Kaddis CS, Loo RRO, Grover GN, Loo JA, Maynard HD. Macromolecules. 2009;42:8028–8033. doi: 10.1021/ma901540p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Chang CW, Bays E, Tao L, Alconcel SNS, Maynard HD. Chem. Commun. 2009:3580–3582. doi: 10.1039/b904456f. [DOI] [PubMed] [Google Scholar]
- 107.Pissuwan D, Boyer C, Gunasekaran K, Davis TP, Bulmus V. Biomacromolecules. 2010;11:412–420. doi: 10.1021/bm901129x. [DOI] [PubMed] [Google Scholar]