

Abstract
Marine mussels anchor to a variety of surfaces by secreting liquid proteins that harden and form water-resistant bonds to a variety of surfaces. Studies have revealed that these mussel adhesive proteins contain an unusual amino acid, 3,4-dihydroxy-L-phenylalanine (DOPA), which is believed to be responsible for the cohesive and adhesive properties of these proteins. To separate the cohesive and adhesive roles of DOPA, we incorporated DOPA into the midblock of poly(methyl methacrylate)–poly(methacrylic acid)–poly(methyl methacrylate) (PMMA–PMAA–PMMA) triblock copolymers. Self-assembled hydrogels were obtained by exposing triblock copolymer solutions in dimethyl sulfoxide to water vapor. As water diffused into the solution, the hydrophobic end blocks formed aggregates that were bridged by the water-soluble midblocks. Strong hydrogels were formed with polymer weight fractions between 0.01 and 0.4 and with shear moduli between 1 and 5 kPa. The adhesive properties of the hydrogels on TiO2 surfaces were investigated by indentation with a flat-ended cylindrical punch. At pH values of 6 and 7.4, the fully protonated DOPA groups were highly adhesive to the TiO2 surfaces, giving values of ≈2 J/m2 for the interfacial fracture energy, which we believe corresponds to the cohesive fracture energy of the hydrogel. At these pH values, the DOPA groups are hydrophobic and have a tendency to aggregate, so contact times of 10 or 20 min are required for these high values of the interfacial strength to be observed. At a pH of 10, the DOPA groups were hydrophilic and highly swellable, but less adhesive gels were formed. Oxidation of DOPA groups, a process that is greatly accelerated at a pH of 10, decreased the adhesive performance of the hydrogels even further.
Introduction
There has been a growing effort to produce safe and cost-effective bioadhesives that can be applied from a low-viscosity solution and are able to self-assemble and form strong and durable bonds in wet environments. Adhesive materials inspired by marine mussels are particularly intriguing in this respect. These organisms anchor themselves to surfaces by secreting a liquid adhesive based on mussel adhesive proteins (MAPs) that harden and form water-resistant bonds within a few seconds.1,2 The most widely studied marine mussel is the blue mussel, Mytilus edulis. A range of different MAPs have been isolated from the M. edulis foot. Five of these proteins, referred to as Mefp-1 to Mefp-5, contain an unusual amino acid, 3,4-dihydroxy-L-phenylalanine (DOPA).3,4 Mefp-3 and Mefp-5 both contain large amounts of DOPA (up to 30 mol %).4 They are found to concentrate at the interface between the adhesive surface and the substrate and are generally supposed to play important roles in determining both the cohesive and the adhesive properties of the secreted material: 5,6 At high values of pH, the DOPA catechol can easily oxidize to the quinone, which can then participate in a variety of reactions, including the cross-linking reaction illustrated in Figure 1a.7–10 The adhesive role is not as well-understood, although the adsorption of catechols to metal oxide surfaces is quite strong, with Figure 1b illustrating the proposed bonding mechanisms.11
Figure 1.

Schemes showing (a) DOPA oxidation to o-quinone followed by crosslinking and (b) potential DOPA–Ti interactions.
There have been several efforts to mimic the water-resistant adhesive properties of MAPs by the incorporation of DOPA into synthetic polymers. 6,12–17 In these studies, DOPA oxidation is the trigger to form a gel network via the cross-linking reactions mentioned above. However, the unoxidized form of DOPA is believed to possess stronger adhesive properties, specifically to metallic surfaces.5 This conjecture has been confirmed recently by the single molecule AFM experiments of Lee et al.18 Furthermore, the use of DOPA oxidizing reagents (such as NaIO4 and H2O2)5 may complicate the future in vivo applications of these materials. To better understand the adhesive role of the DOPA and to develop water-resistant adhesives based on DOPA chemistry, alternative gelation mechanisms that do not rely on DOPA oxidation are highly desirable.
The most widely studied stimuli for gel formation are temperature, pH, ion concentration, and UV and/or visible light. Many of these gelation mechanisms have been applied to DOPA-containing materials. For example, Huang et al. incorporated DOPA into a thermoreversible hydrogel,19 and Lee et al. recently reported a DOPA-containing hydrogel formed by photopolymerization.20 Neither of these systems requires any oxidizing reagents for gel formation. However, the cross-linking reactions used to form strong chemical gels can be inhibited either by oxygen or by the DOPA itself. These issues are particularly important when one is interested in the adhesive properties, which can be severely degraded by the presence of a weak boundary layer. Self-assembled gels, which rely on physical association as opposed to chemical cross-linking, are a useful alternative to chemically cross-linked gels for this reason.
In this study, we have prepared a series of DOPA-modified triblock copolymer gels and have assessed their adhesive and mechanical properties. Gelation in these materials occurs by the formation of glassy end block aggregates during a simple solvent exchange process. Gels were prepared by synthesizing a DOPA-modified ABA type triblock copolymer, in which the hydrophobic end blocks consist of poly(methyl methacrylate) (PMMA) (A), and poly(methacrylic acid) (PMAA) is the water-soluble midblock (B). DOPA is incorporated into the hydrophilic PMAA midblock. These block copolymers self-assemble to form strong hydrogels when solutions in N-methylpyrrolidone (NMP) or dimethyl sulfoxide (DMSO) are exposed to water vapor. We studied the underwater adhesion of these hydrogels to TiO2 surfaces, using an axisymmetric adhesion test, and correlated the adhesive properties of these materials to the pH of the surrounding solution.
Experimental Section
Materials
Methyl methacrylate (MMA) and tert-butyl methacrylate (tBMA) were degassed by freezing in liquid nitrogen after exposure to trioctylaluminum. After it was degassed, tBMA was distilled under reduced pressure and refrigerated. MMA was distilled directly into the reaction chamber prior to polymerization. Lithium (Li) and naphthalene were reacted in distilled tetrahydrofuran (THF) at room temperature for 24 h under a nitrogen atmosphere. A persistent dark green color was observed as the reaction proceeded. LiCl was baked in the reaction chamber at 130 °C under vacuum for 16 h. Diphenylethylene (DPE) was purified by distillation under reduced pressure after addition of a small amount of s-butyllithium. DOPA methyl ester hydrochloride, o-benzotriazole-N,N,N,N′-tetramethyl-uronium-hexafluoro-phosphate (HBTU), N-hydroxybenzotriazole (HOBT), triethylamine, NMP, DMSO, dimethylformamide (DMF), and dichloromethane (DCM) were used as received from Sigma-Aldrich.
Synthesis and DOPA Modification of Methacrylic Triblock Copolymer
The overall synthesis, shown schematically in Figure 2, includes three steps: (i) sequential living anionic polymerization of MMA and tBMA at −78 °C in THF with the presence of LiCl by using a difunctional initiator, (ii) hydrolysis of poly(tBMA) (PtBMA) midblock to PMAA, and (iii) DOPA incorporation into PMAA midblock. Details of anionic synthesis of PMMA–PtBMA–PMMA triblock copolymer (i) and hydrolysis reaction (ii) were explained previously.21 In brief, Li and naphthalene were reacted in THF at room temperature for 24 h under a nitrogen atmosphere. This initiator solution was reacted with DPE to form the difunctional initiator (red color). tBMA and MMA monomers were added sequentially. Before MMA was introducing into the reaction chamber, an aliquot of the reaction mixture was withdrawn for analysis by gel permeation chromatography (GPC) (Waters 717plus autosampler with a Waters 2410 refractive index detector) to determine the molecular weight of the PtBMA midblock. The solution was then precipitated in a methanol/water mixture (90:10). The polymer was dried under vacuum overnight. The compositions of the polymers were determined by GPC and nuclear magnetic resonance spectroscopy (NMR) (1H NMR, P-Inova 500MHz) in d-chloroform. Hydrolysis of the PtBMA midblock was done by dissolving 1 g of polymer in 40 mL of dioxane, adding 4 mL of 37% HCl and heating to 80 °C for 6 h. The polymer was then precipitated in hexane. The polymer was dried under vacuum after being washed in hexane and water several times. The tert-butyl signal (1.43 ppm) in the NMR spectrum completely disappeared after the hydrolysis reaction, indicating that the midblock had been successfully converted to methacrylic acid.
Figure 2.
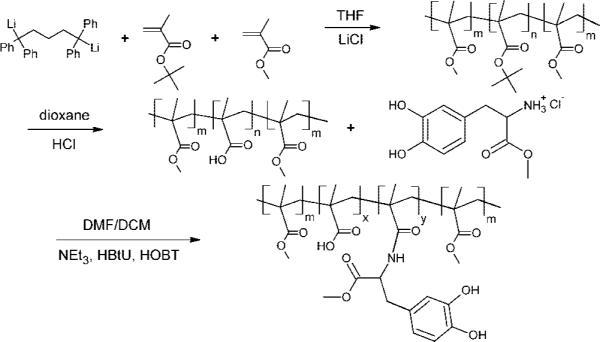
Anionic polymerization of tBMA and MMA in the presence of a difunctional initiator followed by hydrolysis of the PtBMA midblock to PMAA and incorporation of DOPA to the PMAA midblock.
To incorporate DOPA into the PMAA midblock, 1 g of the triblock copolymer (7.3 mmol of methacrylic acid), 15 mmol of DOPA methyl ester (DOPA-ME) hydrochloride, 15 mmol of HOBT, and 1 mmol of HBTU were vacuum degassed in the reaction flask for 1 h. The flask was then flushed with nitrogen gas. Forty milliliters of DMF/DCM (50:50) was injected into the flask through the rubber septum. The system was stirred for 15 min to fully dissolve all of the ingredients and to activate the methacrylic acid. To activate DOPA-ME, 7.3 mmol of triethylamine was injected into the solution. A faint yellow color was observed as the reaction proceeded. The solution was stirred for 10 h. The solution was then precipitated in hexane and washed with hexane several times before it was dried under vacuum overnight and refrigerated. After DOPA modification, significant DOPA phenyl signals at 6.6–6.8 ppm [3H–C6H3(OH2)] appeared in the NMR spectrum.
Quantification of DOPA Content
The DOPA content of the triblock copolymers was determined from a modified DOPA assay developed by Waite and Benedict.22,23 The UV absorbance of the polymer solutions in DMF at the maximum absorbance wavelength of the oxidized catechol (λmax = 500 nm) was recorded using a Hitachi U-2010 UV–vis spectrophotometer. Standard curves were constructed by using solutions containing known concentrations of oxidized DOPA methyl ester.
Self-Assembly of Hydrogels by Solvent Exchange
The DOPA-modified triblock copolymer was fully dissolved in DMSO, NMP, or DMF, which are good solvents for both the PMAA and the PMMA blocks. These solutions were then exposed to saturated water vapor for 3 h to enable solvent exchange with water. As water replaced the initial solvent, the hydrophobic end blocks (PMMA) aggregated into spherical domains. The water-soluble midblocks (PMAA) formed bridges between these domains, as was demonstrated previously for thermoreversible gels formed from similar triblock copolymers.24,25 After solvent exchange, the hydrogels were immersed in water or controlled pH buffers, to study their equilibrium swelling properties in detail. The buffers were made by mixing equal volumes of 0.1 M Tris and HEPES solutions and adjusting the pH by the addition of 1 M sodium hydroxide. Adhesion tests were performed on samples that had been immersed in these same buffer solutions. A Paar Physica MCR (TEK 150PA–C) rheometer with parallel plate geometry was used to measure the dynamic shear moduli of the hydrogels in the linear viscoelastic regime.
Mechanical Tests
The adhesive and mechanical properties of the DOPA-modified hydrogels in buffer solutions were tested using an axisymmetric indentation method.21,26 A schematic of the adhesion test apparatus is shown in Figure 3a. The geometry of a flat punch in contact with the hydrogel, shown in detail in Figure 3b, provides a well-defined contact area having a radius, a, equal to the punch radius. TiO2 was chosen as the substrate surface since Ti has been commonly used for medical alloys. The flat punch was coated with 50 nm of TiO2 by electron-beam evaporation (Edwards FL400 e-beam evaporator), using a premelted TiO2 powder evaporant (West Cerac, Inc.). Before each adhesion experiment, the punch surface was cleaned with acetone and methanol sequentially and exposed to UV-Ozone (Jelight Co. model 42 UVO-Cleaner) for 15 min. The hydrogel was placed on a TiO2-coated glass slide, which was also cleaned beforehand, and placed inside the chamber filled with one of the buffer solutions described in the previous subsection. The adhesive interactions between the hydrogel and the TiO2 surface reduced significantly or disappeared completely when the TiO2 surface was not rigorously cleaned. The flat punch was brought into contact with the fully swollen hydrogel in liquid media at a constant speed of 5 μm/s. When a maximum compressive load of 5 mN was reached, the motor was reversed and the punch was retracted at the same rate. In some cases, the contact time was increased by waiting for a specified period between the loading and the unloading portions of the test. A tensile load is developed during pull-off, which is an indication of adhesion between the gel and the TiO2-coated punch. Load and displacement data were collected with a load transducer and an optical displacement sensor.
Figure 3.

Schematic illustration of (a) the apparatus used for the indentation experiments and (b) the sample geometry, in which a and h denote the punch radius and gel thickness, respectively.
Results and Discussion
Characterization of DOPA-Modified Acrylic Polymers
The PMMA–PMAA–PMMA triblock copolymer was synthesized by sequential anionic living polymerization of tBMA and MMA, followed by the hydrolysis of the PtBMA midblock to form a water-soluble PMAA midblock. The degrees of polymerization of the PMMA–PMAA–PMMA blocks were 370–1450–370 (Mn = 200 kg/mol, PDI = 1.09). DOPA was incorporated into the water-soluble methacrylic acid block by using carbodiimide chemistry, as explained above. The amount of DOPA attached to the midblock was measured by absorbance of the oxidized catechol at a wavelength of 500 nm. In this paper, we focus on gels with three different DOPA levels, corresponding to mole fractions of DOPA incorporation into the PMAA midblock of 0, 0.2, and 0.4. We refer to these three polymers as DOPA00, DOPA20, and DOPA40.
Hydrogel Formation and Characterization
The three different triblock copolymers were dissolved in DMSO at polymer weight fractions of 0.1. These solutions were injected into circular washers, one side of which was attached to a glass slide and exposed to saturated water vapor for 24 h (Figure 4a). Gelation occurs as the changing solvent quality causes the PMMA end blocks to aggregate. The gels were carefully taken out of the washers and immersed in buffer solutions (Figure 4b), to study their equilibrium swelling properties. In Figure 5, we plot the time dependence of the polymer weight fraction in the samples. A decrease in the polymer fraction represents an increase in the swelling. We previously showed that in the absence of attached DOPA, the hydrogels swell significantly when immersed in buffer solutions with a pH greater than the pKa of methacrylic acid (≈4.8),21 a result that we attribute to the ionization of the methacrylic acid groups. This general trend is preserved with the addition of the DOPA, where the polymer volume fraction is lower when immersed in buffers with a higher pH. However, the incorporation of DOPA into the hydrogels decreases the swelling ability of the gels at pH = 6 and pH = 7.4, a result that is illustrated schematically in Figure 6. We attribute this behavior to the hydrophobic nature of the fully protonated form of DOPA. This hydrophobic character is consistent with the behavior of the polymerized polypeptide form of DOPA, which is insoluble in water at pH values below 10.5.27 For the DOPA40 material, the hydrophobic interactions are strong enough to completely deswell the polymer, resulting in nearly dry polymer aggregate, with a polymer volume fraction very close to 1.
Figure 4.
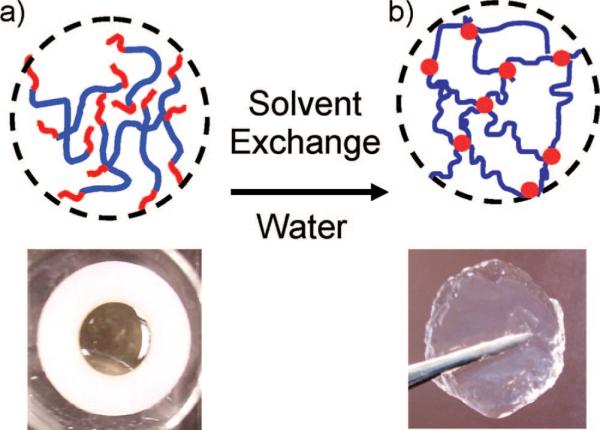
Photographs and schematic structures of the triblock copolymer: (a) triblock solution in DMSO and (b) swollen gel in water after exposure to saturated water vapor.
Figure 5.
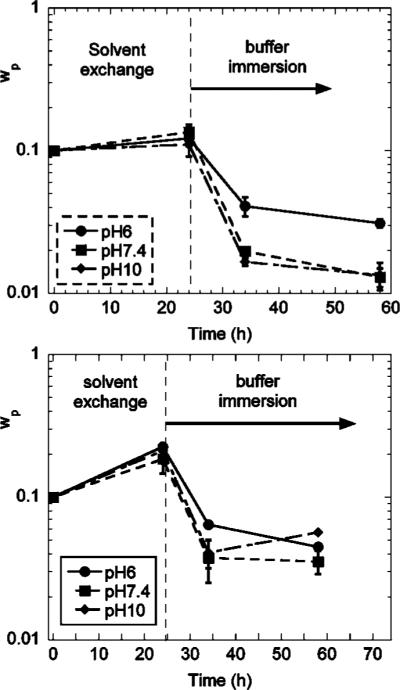
Polymer weight fractions during solvent exchange and subsequent buffer immersion for DOPA00 (top) and DOPA20 (bottom) hydrogels. The polymers were initially dissolved in DMSO at concentrations of 10 wt %, gelled by exposure to water vapor, and immersed into different buffer solutions: pH 6, pH 7.4, and pH 10. Error bars represent the standard deviation with three replications.
Figure 6.
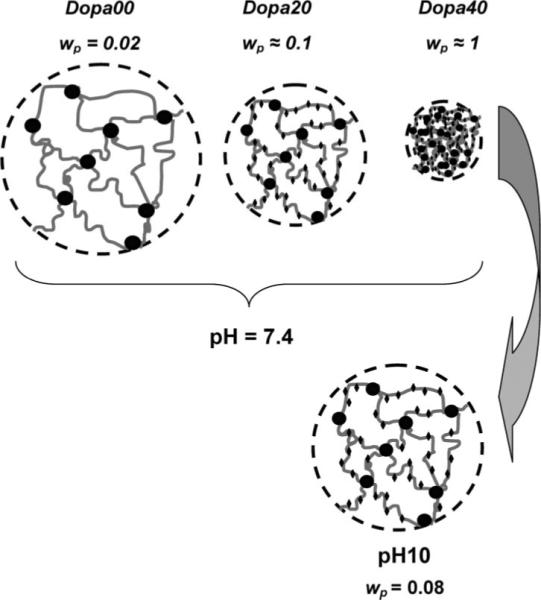
Schematics and pictures of the hydrogels equilibrated in water (top). The bottom schematic illustrates the subsequent swelling of the DOPA40 hydrogel when immersed in a solution with pH 10.
Reported values for the pKa of DOPA are pKaI = 9.4 and pKaI1 = 13.7, corresponding to the removal of the first and second protons from the catechol.28,29 If the pH of the swelling solution is increased above pKaI, the DOPA groups become ionized, and they are no longer hydrophobic. This result is most clearly observed for the DOPA40 gel. The white, deswollen solid, formed during the solvent exchange process and subsequent immersion in neutral water, forms a highly swollen gel with a polymer volume fraction of 0.08 when immersed in a buffer with pH 10. This swollen gel has a light brown color indicating the partial oxidation of DOPA, since the deprotonated form is much more prone to oxidation. These discolored gels could not be redissolved in DMSO, indicating that the oxidation was accompanied by some cross-linking, as illustrated schematically in Figure 1a.
The dynamic moduli of the swollen hydrogels, prepared from initial solution concentrations of 10 wt % in DMSO, were investigated by shear rheometry at room temperature. We previously showed that equilibrated hydrogels behave as elastic solids, having moduli near 1 kPa with a specific value that is dependent on the equilibrium polymer fraction of the hydrogel.21 There is an increase in the modulus as the concentration of DOPA in the PMAA midblock increases, which is attributed to the decrease in swelling with an increase in the DOPA content in the midblock. The largest increase was observed in the case of DOPA40, which is consistent with the high polymer fraction in this material. Shear moduli for a variety of different gels obtained by rheometry, or from the indentation experiments described in the following section, are summarized in Table 1.
Table 1.
Swelling, Mechanical, and Adhesive Properties of Hydrogels, Formed by vapor Phase Solvent Exchange from DMSO Solutions with Initial Polymer Weight Fractions, wia
| sample | wi | pH | wp | G (kPa) | σp (kPa) | Gc (mJ/m2) |
|---|---|---|---|---|---|---|
| DOPA00 | 0.1 | 6.0 | 0,031 | 0.7 ± 0,3 | 0,85 ± 0,12 | 210 ± 180 |
| DOPA00 | 0.1 | 7.4 | 0.013 | 0.4 ± 0.1 | 0,40 ± 0.27 | 65 ± 50 |
| DOPA00 | 0.1 | 10.0 | 0.013 | 0.4 ± 0.1 | 0.80 ± 0.1 | 200 ± 140 |
| DOPA20 | 0.1 | 6.0 | 0.045 | 0.33 ± 0.02 | 3.3 ± 0.3 | 2100 ± 140 |
| DOPA20 | 0.1 | 7.4 | 0.035 | 0.20 ± 0.02 | 2.5 ± 0.2 | 1780 ± 100 |
| DOPA20 | 0.1 | 10.0 | 0.04 | 0.24 ± 0.02 | 0.82 ± 0.34 | 290 ± 40 |
| DOPA20-OX | 0.1 | 7.4 | 0.05 | 0.3 ± 0.03 | 0.68 ± 0.14 | 125 ± 42 |
| DOPA20-OX | 0.1 | 10.0 | 0.09 | 1.1 ± 0.04 | 0.085 ± 0.04 | 2.8 ± 1 |
| DOPA40 | 0.2 | 10.0 | 0.08 | 0.24 | 0.62 ± 0.14 | 100 ± 40 |
Here, wp is the equilibrium polymer volume fraction after immersion in the buffer solution, G is the shear modulus of the equilibrated gel, and Gc is the critical energy release rate describing the strength of the gel/TiO2 interface after a contact time of 20 min. The mean and standard deviation are included for each measurement, with two or three replications in each case.
Adhesion Tests
Adhesion of DOPA-modified hydrogels with TiO2 was quantified with an axisymmetric indentation method. The TiO2 surface was cleaned prior to the experiment as described above. The TiO2-coated flat punch was brought into contact with a hydrogel that was immersed in aqueous solution. Experiments were conducted where the surfaces were separated immediately after a compressive load of 5 mN had been attained and after the surfaces were held in contact at this maximum load for contact times of 10, 20, and 30 min before they were separated. Typical plots, generally referred to as “tack” curves in the adhesion science community, are shown in Figure 7. Here, we plot the average stress (σavg ≡ P/πa2) as a function of the normalized displacement (δ/a) for experiments conducted in buffer solutions with pH 6 (Figure 7a) and pH 7.4 (Figure 7b). For these experiments, the surfaces were kept in contact for 20 min.
Figure 7.

Average stress vs strain data for DOPA00 and DOPA20 hydrogel in contact with TiO2-coated flat punch. Gels were produced from 10 wt % solutions in DMSO by exposing them to saturated water vapor, and gels were equilibrated in (a) pH 6 and (b) pH 7.4 buffer solutions. The DOPA00 data are shifted along the horizontal axis for clarity.
The modulus of the layers is determined from the slope of the nominal stress/strain relationship. When the thickness of the elastic layer, h, is not substantially larger than the punch radius, a, the sample compliance depends on both a and h.26 For an incompressible elastic layer with a Poisson's ratio equal to 0.5, the relationship between σavg and δ/a for relatively small displacements is given by the following expression:30
| (1) |
where G is the shear modulus of the elastic layer.
The critical energy release rate, Gc, characterizing the adhesion of the gel to the TiO2 surface, is obtained from the maximum tensile stress, σp, illustrated schematically in Figure 7a. For our geometry, the critical energy release rate is given by the following expression:30
| (2) |
Values of h for the hydrogels ranged from 1 to 4 mm. Two punch sizes were employed in the experiments: a = 1.17 and 0.78 mm. The shear moduli and critical energy release rates for contact times of 20 min are listed in Table 1. Eq 2 is based on the assumption that the contact radius remains fixed at the punch radius up to the point of failure, which corresponds to the point where the applied energy release rate exceeds the critical energy release rate Gc. In this case, a linear relationship between nominal stress and strain would be obtained up to the maximum tensile stress, at which point the interface fails and the stress returns to zero, provided that a/h is less than 0.45.26 This behavior is generally observed in our experiments, and the critical energy release rate is an appropriate measure of the adhesive interaction between the gel and the punch.
The dependence of the adhesion on the contact time between the loading and the unloading portions of the experiment is summarized in Figure 8. For the lowest contact times, the gels are actually more adhesive at a pH value of 10 than they are at the lower pH values. Our interpretation of this result is that the aggregation of the hydrophobic DOPA groups at low values of the pH inhibits their ability to quickly interact with the surface. As a result, longer contact times are required to establish contact between the DOPA groups and the titania surface at the lower values of the pH. For longer contact times, we obtain values of the adhesion that we feel are more representative of the equilibrated gel/titania interface. In Figure 9, we show values of the critical energy release rate for a contact time of 20 min, at pH values of 6, 7.4, and 10. The DOPA00 hydrogels show relatively weak adhesion to the TiO2 surfaces, with Gc less than 250 mJ/m2 or less in all cases. Because of the collapse of the DOPA40 gel at pH values of 6 and 7.4, we were only able to quantify the adhesion of this material at a pH of 10, where a value of Gc close to 100 mJ/m2 is obtained. The most interesting results were obtained for the DOPA20 gels, which for long contact times adhere much more strongly at pH values of 6 and 7.4 than they do at a pH value of 10. In fact, the DOPA/titania interface is strong enough so that failure occurs adhesively within the gel but in very lose proximity to the gel/punch interface. We believe that the plateau value of ≈2 J/m2, obtained for Gc at long contact times under conditions where the DOPA catechol is in its fully protonated form, corresponds to the fracture strength of the gel. The magnitude of this toughness is consistent with measured values for other physically cross-linked gels, such as gelatin.31 The values of the average stress at which failure occurs are listed in Table 1 and are several times the shear modulus of the gel for the samples that are expected to fail cohesively.
Figure 8.

Critical energy release rate as a function of contact time for DOPA20 gels in different buffer solutions. Error bars represent the standard deviation, with three or four measurements at each time.
Figure 9.
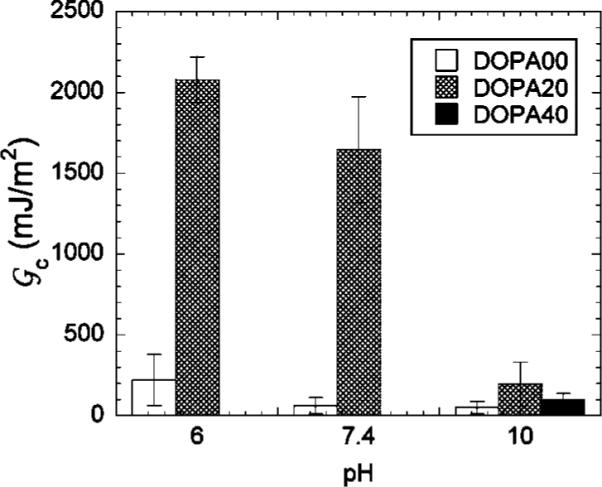
Critical energy release rate at a contact time of 20 min for DOPA20 gels in different buffer solutions. Error bars represent the standard deviation, with three or four measurements for each condition.
Our results are consistent with measurements of catechol adsorption to TiO2 surfaces, where adsorption is shown to decrease for pH values greater than 9.2, corresponding to pKaI for the catechol.11 In addition, single molecule measurements of the DOPA/TiO2 interaction indicate that a stronger interaction is observed at low pH than at high pH.18 A further complicating factor for high pH values is that DOPA oxidation to the quinone form occurs more readily under these conditions, producing forms that are no longer adhesive. These oxidation reactions are presumably responsible for the diminished strength of the DOPA/TiO2 interaction observed at high pH values in the single molecule experiments18 and are observed in our experiments as well. A DOPA20 gel that had been immersed in the pH 10 buffer solution for 14 days had a brownish-red color that is indicative of oxidation of the catechol to the quinone form, which can lead to intra- or intermolecular cross-linking reactions (Figure 1a) or to other side reactions. The adhesiveness of this gel was reduced dramatically, in comparison to the gel that had remained in the pH 10 buffer for only 3 h. The results for this oxidized gel are shown as the DOPA20-OX sample in Figure 8 and Table 1.
Conclusions
We have synthesized a DOPA-functionalized PMMA–PMAA–PMMA triblock copolymer that can self-assemble and form a hydrogel in situ by vapor phase solvent exchange from water-miscible solvents such as DMSO and NMP. As water replaces the original solvent, the hydrophobic PMMA end blocks form aggregates, with the water-soluble PMAA midblocks bridging these aggregates to form an elastic network. To study the effect of DOPA on the mechanical and adhesive properties of the hydrogel, we have compared the properties of three polymers with 0, 20, and 40 mol % DOPA incorporation into the midblock. The swelling and mechanical properties of these hydrogels were investigated, as were the adhesive properties of these hydrogels in contact with TiO2 surfaces that were immersed in aqueous buffers. Our results can be summarized as follows:
Highly swollen transparent gels are easily formed by solvent exchange from triblock solutions with attached DOPA groups, giving gels with shear moduli in the kilopascal range.
Incorporation of DOPA decreases the hydrophilicity of the midblock for pH values where the DOPA catechol is completely protonated (pH < ≈9.4).
At sufficiently long contact times (typically 20 min), the DOPA-containing gels adhere most strongly to titania surfaces when they are in the fully protonated form. Under these conditions, the strength of the gel/titania interface is limited by the cohesive strength of the gel (≈2 J/m2).
Oxidation of DOPA by prolonged exposure to high pH solutions reduces the adhesion significantly.
Acknowledgment
This work was supported by the Northwestern University Materials Research Center through the NSF MRSEC program (DMR-0520513) and by the NIH (R01 DE14193). We also acknowledge Dr. Bruce Lee for a variety of helpful discussions.
References and Notes
- (1).Waite JH. Int. J. Adhes. Adhes. 1987;7:9. [Google Scholar]
- (2).Waite JH. Ann. N. Y. Acad. Sci. 1999;875:301. doi: 10.1111/j.1749-6632.1999.tb08513.x. [DOI] [PubMed] [Google Scholar]
- (3).Waite JH. J. Biol. Chem. 1983;258:2911. [PubMed] [Google Scholar]
- (4).Papov VV, Diamond TV, Biemann K, Waite JH. J. Biol. Chem. 1995;270:20183. doi: 10.1074/jbc.270.34.20183. [DOI] [PubMed] [Google Scholar]
- (5).Yu ME, Hwang JY, Deming TJ. J. Am. Chem. Soc. 1999;121:5825. [Google Scholar]
- (6).Yu ME, Deming TJ. Macromolecules. 1998;31:4739. doi: 10.1021/ma980268z. [DOI] [PubMed] [Google Scholar]
- (7).Haemers S, Koper GJM, Frens G. Biomacromolecules. 2003;46:32. doi: 10.1021/bm025707n. [DOI] [PubMed] [Google Scholar]
- (8).Burzio LA, Waite JH. Biochemistry. 2000;39:11147. doi: 10.1021/bi0002434. [DOI] [PubMed] [Google Scholar]
- (9).Waite JH, Qin X. Biochemistry. 2001;40:2887. doi: 10.1021/bi002718x. [DOI] [PubMed] [Google Scholar]
- (10).Monahan J, Wilker JJ. Langmuir. 2004;20:3724. doi: 10.1021/la0362728. [DOI] [PubMed] [Google Scholar]
- (11).Rodriguez R, Blesa MA, Regazzoni AE. J. Colloid Interface Sci. 1996;177:122. doi: 10.1006/jcis.1996.0012. [DOI] [PubMed] [Google Scholar]
- (12).Tatehata H, Mochizuki A, Ohkawa K, Yamada M, Yamamoto H. J. Adhes. Sci. Technol. 2001;15:1003. [Google Scholar]
- (13).Yamamoto H, Kuno S, Nagai A, Nishida A, Yamauchi S, Ikeda K. Int. J. Biol. Macromol. 1990;12:305. doi: 10.1016/0141-8130(90)90019-7. [DOI] [PubMed] [Google Scholar]
- (14).Yu M, Deming TJ. Polym. Mater. Sci. Eng. 1998;79:248. [Google Scholar]
- (15).Deming TJ. Curr. Opin. Chem. Biol. 1999;3:100. doi: 10.1016/s1367-5931(99)80018-0. [DOI] [PubMed] [Google Scholar]
- (16).Deming TJ, Yu M, Hwang J. Polym. Mater. Sci. Eng. 1999;80:471. [Google Scholar]
- (17).Dalsin JL, Hu B-H, Lee BP, Messersmith PB. J. Am. Chem. Soc. 2003;125:4253. doi: 10.1021/ja0284963. [DOI] [PubMed] [Google Scholar]
- (18).Lee H, Scherer NF, Messersmith PB. Proc. Natl. Acad. Sci. U.S.A. 2006;103:12999. doi: 10.1073/pnas.0605552103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Huang K, Lee BP, Ingram DR, Messersmith PB. Biomacro-molecules. 2002;3:397. doi: 10.1021/bm015650p. [DOI] [PubMed] [Google Scholar]
- (20).Lee BP, Dalsin JL, Messersmith PB. Biomacromolecules. 2002;3:1038. doi: 10.1021/bm025546n. [DOI] [PubMed] [Google Scholar]
- (21).Guvendiren M, Shull KR. Soft Matter. 2007;3:619. doi: 10.1039/b615412c. [DOI] [PubMed] [Google Scholar]
- (22).Waite JH, Benedict CV. Methods Enzymol. 1984;107:397. doi: 10.1016/0076-6879(84)07028-2. [DOI] [PubMed] [Google Scholar]
- (23).Lee BP, Chao C-Y, Nunalee FN, Motan E, Shull KR, Messersmith PB. Macromolecules. 2006;39:1740. [Google Scholar]
- (24).Flanigan CM, Crosby AJ, Shull KR. Macromolecules. 1999;32:7251. [Google Scholar]
- (25).Seitz ME, Burghardt WR, Faber KT, Shull KR. Macromolecules. 2007;40:1218. [Google Scholar]
- (26).Webber RE, Shull KR, Roos A, Creton C. Phys. Rev. E. 2003;68:021805. doi: 10.1103/PhysRevE.68.021805. [DOI] [PubMed] [Google Scholar]
- (27).Yamamoto H, Hayakawa T. Polymer. 1977;18:979. [Google Scholar]
- (28).Saleem MMM, Wilson MT. Biochem. J. 1982;201:433. doi: 10.1042/bj2010433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Liu B, Burdine L, Kodadek T. J. Am. Chem. Soc. 2006;128:15228. doi: 10.1021/ja065794h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Shull KR. Mater. Sci. Eng., R. 2002;36:1. [Google Scholar]
- (31).Baumberger T, Caroli C, Martina D. Nat. Mater. 2006;5:552. doi: 10.1038/nmat1666. [DOI] [PubMed] [Google Scholar]