

Abstract
Background
Bidirectional ventricular tachycardia (BVT), characterized by an alternating beat-to-beat electrocardiographic QRS axis, is a rare but intriguing arrhythmia associated with digitalis toxicity, familial catecholaminergic polymorphic ventricular tachycardia (CPVT), and several other conditions which predispose cardiac myocytes to delayed afterdepolarizations (DADs) and triggered activity. Evidence from human and animal studies attributes BVT to alternating ectopic foci originating from the distal His-Purkinje system in the left and/or right ventricles, respectively.
Objective
To evaluate a simple “ping pong” model of reciprocating bigeminy to explain BVT.
Methods
We constructed a 2D anatomic model of the rabbit ventricles with a simplified His-Purkinje system, in which different sites in the His-Purkinje system had different heart rate thresholds for DAD-induced bigeminy.
Results
When the heart rate exceeded the threshold for bigeminy at the first site in the His-Purkinje system, ventricular bigeminy developed, causing the heart rate to accelerate and exceed the threshold for bigeminy at the second site. Thus, the triggered beat from the first site induced a triggered beat from the second site. The triggered beat from the second site next reciprocated by inducing a triggered beat from the first site, and so forth. Bigeminy from two sites produced BVT, and from 3 or more sites, polymorphic VT.
Conclusions
This ping pong mechanism of reciprocating bigeminy readily produces the characteristic electrocardiographic pattern of BVT, and its degeneration to polymorphic VT if additional sites develop bigeminy.
Keywords: action potentials, arrhythmia (mechanisms), catecholaminergic polymorphic ventricular tachycardia, electrophysiology, ventricular tachycardia
Introduction
Bidirectional ventricular tachycardia (BVT) is characterized by beat-to beat alternation of the QRS axis on the electrocardiogram (ECG). Although uncommon, it has fascinated clinicians since its original description in 1922 as a manifestation of digitalis toxicity 1–4. It has also been reported in the setting of hypokalemic periodic paralysis 5, Andersen-Tawil syndrome 6, and fulminant myocarditis 7. More recently, BVT has also been recognized as a hallmark of catecholaminergic polymorphic ventricular tachycardia (CPVT) syndrome, a familial disorder with a high risk of sudden cardiac death during sympathetic stimulation 8. During exercise testing, these patients typically develop premature ventricular complexes (PVCs) and sometimes ventricular bigeminy 9–11 in association with BVT, which can self-terminate or degenerate into polymorphic VT and ventricular fibrillation (VF) 8, 11, 12. Although the most characteristic electrocardiographic pattern of BVT is right bundle branch (RBB) block with an alternating QRS axis 8, other patterns, such as alternating RBB and left bundle branch (LBB) block or alternating QRS axis with a narrow QRS 13 have also been observed. Recent genetic studies have attributed familial CPVT syndromes to defects in the cardiac ryanodine receptor (RyR2) or calsequestrin, an associated regulatory protein, in the sarcoplasmic reticulum (SR), and CPVT has been recapitulated in genetically-engineered mouse models incorporating the analogous RyR2 mutations14, 15. In these mice, abnormal RyR2 regulation predisposes myocytes to delayed afterdepolarizations (DADs) and triggered activity, analogous to digitalis toxicity. A recent optical mapping study in a mouse CPVT model due to the mutation R4496C in RyR2 (RyR2- R4496C) showed that BVT was caused by two foci in the distal His-Purkinje system (HPS), one in the right ventricle (RV) and the other in the left ventricle (LV), alternately activating the ventricles 16. Moreover, ablation of the RV HPS with Lugol s solution converted BVT to monomorphic VT. These authors went on to show in a computer model of the anatomic mouse ventricles that alternate pacing from the RV and LV septum produced a characteristic electrocardiographic pattern of BVT. However, they did not address the mechanism by which triggered activity spontaneously generated this pattern.
Based on the observation that PVCs and ventricular bigeminy are common precursors to BVT 9–11, it occurred to us that a simple “ping pong” mechanism, which we call reciprocating bigeminy, could readily account for the pattern of alternating foci observed during BVT. Although a number of mechanisms for BVT have been suggested 10, 11, 17–19, the straightforward, conceptually intuitive mechanism of reciprocating bigeminy has not, to our knowledge, been explicitly proposed previously, yet is fully consistent with well-known properties of triggered activity caused by DADs. We demonstrate its plausibility by incorporating these experimentally well-documented properties into computer simulations to reproduce the characteristics of BVT.
Methods
Ventricular and Purkinje AP cell models
For ventricular cells, we used the Mahajan et al. model 20, a detailed model of rabbit ventricular AP which includes intracellular Ca (Cai) dynamics (Fig. 1A, red trace). To model the Purkinje cell AP (Fig. 1A, black trace), we modified Ito,f, Ito,s, Ikr, Iks, and Ik1 as described by Cordeiro et al. 21. We also added a background Ca current (adopted from the LRd model 22), using the following formulation:
| (1) |
| (2) |
where Ḡca,b is the maximal current conductance, cs is submembrane Ca concentration in mM, co is extracellular Ca concentration in mM, F is Faraday s constant, R is theuniversal gas constant, and T is temperature in K. Ḡca,b was adjusted to produce physiological values of diastolic and systolic Ca in the Purkinje cells and ventricular myocytes (Fig. 1A and Table 1).
Fig. 1.

A. Comparison of simulated rabbit ventricular (dashed line) and Purkinje (solid line) APs and Cai transients, during pacing at 600 ms. B. Rate dependence of DADs and bigeminy in the Purkinje cell AP models. In the green trace a, the rate threshold for DAD-induced bigeminy was 67 bpm (PCL 900 ms), such that pacing (black arrows) at both 900 and 600 ms induced bigeminy. In purple trace b, the bigeminy rate threshold was 100 bpm (PCL 600 ms), such that pacing at 600 ms, but not 900 ms, induced bigeminy.
Table 1.
Current conductances used in Purkinje and ventricular myocyte AP models.
| Purkinje | Myocyte | |
|---|---|---|
| Ḡto,f | 0.165 | 0.11 |
| Ḡto,s | 0.06 | 0.04 |
| Ḡca,L | 364.0 | 273.0 |
| Ḡca,b | 0.0010556 | 0.001508 |
| Ḡkr | 0.0041625 | 0.0125 |
| Ḡks | 0.04615 | 0.1386 |
| Ḡk1 | 0.15 | 0.3 |
| ḠNa | 24.0 | 12.0 |
To produce DADs and triggered activity in Purkinje cells, we used the formulation of spontaneous SR Ca release described by Huffaker et al. 23, with some modifications. Spontaneous release was triggered solely by the SR Ca load (and not cytosolic Ca concentration), as follows:
| (3) |
where Jspon is the spontaneous Ca release flux, cj is the SR Ca concentration, Ḡspon is the conductance of the release and p is the gating variable obeying the following equation:
| (4) |
| (5) |
where τp 10 ms is the time constant for spontaneous SR release, and K2, K1 are the upper and lower thresholds for spontaneous SR Ca release, respectively (Table 2). With these additions, the differential equations for cj and cs become:
| (6) |
| (7) |
where Jrel is Ca-induced Ca release from the SR, Jspon is the spontaneous release from SR due to Ca overload, Jup is Ca uptake by SERCA, and Jleak is the Ca leak from the SR into the cytoplasm. Jd represents diffusion of Ca from the submembrane compartment (cs) into the cytoplasmic compartment (ci), JCa is Ca flux into the cs from the L-type Ca channel, and JNaCa is the flux through the NaCa exchanger. βs represents the time independent submembrane Ca buffering and is the time dependent binding of Ca to troponin C. Finally, vi/vs represents the cytoplasmic-to-SR volume ratio.
Table 2.
Ispon parameters for the Purkinje AP model exhibiting DADs, located in the LBB, RBB, and distal Purkinje muscle junction (PMJ), as shown in Fig. 2.
| Gspon | K1 | K2 | Ʈp | |
|---|---|---|---|---|
| RBB | 250.0 | 110.0 | 110.1 | 10.0 |
| LBB | 180.0 | 111.5 | 111.6 | 10.0 |
| PMJ | 215.0 | 111.0 | 111.1 | 10.0 |
We also modified the equation for cp, the effective Ca concentration sensed by the L-type Ca channel accounting for L-type Ca current inactivation due to spontaneous SR Ca release as follows:
| (8) |
| (9) |
where J̃SR and J̃ca are the effective fluxes from SR and Ca channel respectively and J̃spon is the effective flux from spontaneous SR calcium release and γ = 200 is the conversion factor.
With these modifications, the Purkinje AP model developed DADs of sufficient amplitude to cause a single triggered AP at a critical heart rate. Parameters were adjusted such that the critical heart rate at which DADs occurred could be varied (Fig. 1B, green versus purple traces).
Tissue model
We used rabbit ventricular anatomy and fiber-orientation data obtained from the Cardiac Mechanics Research Group at the University of California (San Diego, CA), as described previously 24, 25, to extract a 2D sagittal section of the ventricles, as shown in Fig. 2A. We added a His-Purkinje network (with a thickness of 5 Purkinje cells), consisting of a His bundle dividing into a RBB and LBB, each forming further branches until inserting into the ventricular myocardium to generate a physiological activation sequence.
Fig. 2.

A. The anatomic rabbit ventricles model (green), showing a 2D section (blue) of myocardium with the HPS (red) incorporated. Regions of the HPS susceptible to DAD-induced bigeminy in the RBB (a, green) and LBB (b, purple) are indicated. In some simulations, a third region (yellow) also developed DAD-induced bigeminy. B. Voltage snapshots of normal activation when the His bundle is paced.
Wave propagation in 2D tissue was modeled using a reaction-diffusion partial differential equation:
| (10) |
where Vm is the membrane voltage, Iion is the sum of the membrane currents and Cm is the membrane capacitance. Ĩnbr is the diffusive flow from the neighbor cells as described below, which is solved by a partial differential equation. The partial differential equation was solved using operator splitting and an adaptive time step method 26, 27. We used a variable time step between 0.001 and 0.01 ms. All simulations were performed on a 128-node Beowulf cluster.
The diffusive flow between neighboring cells was defined as:
| (11) |
where D = 0.001 cm2/ms if the neighboring cell was a ventricular myocyte and D = 0.006 cm2/ms if the neighboring cell was a Purkinje cell, and dx=dy=0.015 cm. Each cell could have up to 4 neighbors. Purkinje cells were coupled to ventricular myocytes at a total of 27 sites in the terminal His-Purkinje branches (where the 5 terminal Purkinje cells were coupled to 5 adjacent ventricular cells) and at a few points within the septum (where 2 Purkinje cells were coupled to 2 adjacent ventricular cells). The diffusion at Purkinje-myocyte interfaces was defined as:
| (12) |
where Dp=0.006 and Dv =0.001 cm2/ms.
When the His bundle was paced (simulating a sinus beat), the resulting activation sequence is shown in Fig. 2B. Activation was earliest at the left septum, followed by right septum, ventricular apex and lateral ventricular walls, generally consistent with the ventricular activation sequence during sinus rhythm 28 29. Within the HPS, regions of the RBB and LBB (labeled a and b, respectively) were assigned the DAD-generating Purkinje cell AP models (corresponding to the two traces in Fig. 1B), with the remainder of the HPS comprised of the standard Purkinje cell AP model without DADs (Fig. 1A). In some simulations, a 3rd area (labeled c) was also assigned DAD-generating Purkinje cell AP models.
Pseudo-ECG calculation
A pseudo-ECG (Φe) was computed from membrane voltage using the following integral expression 30:
| (13) |
| (14) |
where ▽Vm is the spatial gradient of Vm and r is the distance from a source point (x,y,z) to the location of the “electrode” at (x′,y′,z′).
Results
The proposed “ping pong” mechanism of BVT is based on the following two commonly observed behaviors of DAD-mediated triggered activity: i) above a certain threshold heart rate, a DAD triggers a single AP after each paced AP, initiating ventricular bigeminy (Fig. 1B); ii) the threshold heart rate for bigeminy varies at different locations in the heart (Fig. 1Ba and 1Bb, respectively). From the experimental evidence indicating that BVT arises from the bundle branches of the HPS 16, we assume that the sites from which bigeminy arises are located in the HPS, where source-sink relationships are more favorable for DAD formation than in ventricular muscle 31, 32. The different heart rate thresholds for bigeminy are a natural assumption, since if the rate thresholds were the same everywhere, all rate-dependent DAD-mediated PVC s arising from the HPS would have a narrow QRS complex, which is not true. We arbitrarily chose the RBB to have the lower heart rate threshold for bigeminy, set at 67 bpm, and the LBBB to have the higher threshold, set at 100 bpm (Fig. 3, and movie in online supplement). Thus, when the heart rate exceeded 67 bpm, the paced AP with a normal activation sequence (top row) was followed by a triggered AP from the RBB, producing a QRS morphology resembling LBB block (middle row). With the onset of ventricular bigeminy, however, the average heart rate now doubled, exceeding the 100 bpm threshold for bigeminy to develop in the LBB. Thus, the triggered beat from the RBB elicited a triggered beat from the LBB, producing a QRS morphology resembling RBB block (bottom row). The arrival of this beat in the RBB then caused a triggered AP arising from the RBB, with LBB block morphology, and so forth, in a ping pong fashion. Fig. 3B shows that the computed ECG from the model closely resembles that recorded from a patient during BVT12. Thus, each triggered AP from the RBB reciprocally triggers an AP from LBB, and vice versa, producing VT with an alternating LBB/RBB block pattern, as shown by the orange symbols in Fig. 4. In the example in Fig. 3, BVT terminated spontaneously, as often occurs clinically, but sustained BVT was also simulated with different parameter settings. Had we chosen the LBB to have the lower heart rate threshold for bigeminy, the results would be the same, except that the BVT would have begun with a RBB block QRS morphology instead of LBB block.
Fig. 3. Simulated BVT.

A. Voltage snapshots showing the activation sequence at the onset of BVT. Beat #2 is the last paced beat, with normal activation. Beat #3 is the 1st beat of BVT, due to a DAD-triggered AP arising in the RBB, resulting in a QRS with LBB block pattern. Beat #4 is the 2nd beat of BVT, due to a DAD-triggered AP arising in the LBB, resulting in a QRS with RBB block pattern. Traces on right show the timing of APs recorded from the His bundle (red), RBB (green), and LBB (purple). B. The computed ECG from the simulation in A, showing BVT. C. ECG recorded in a patient during BVT 12.
Fig. 4.
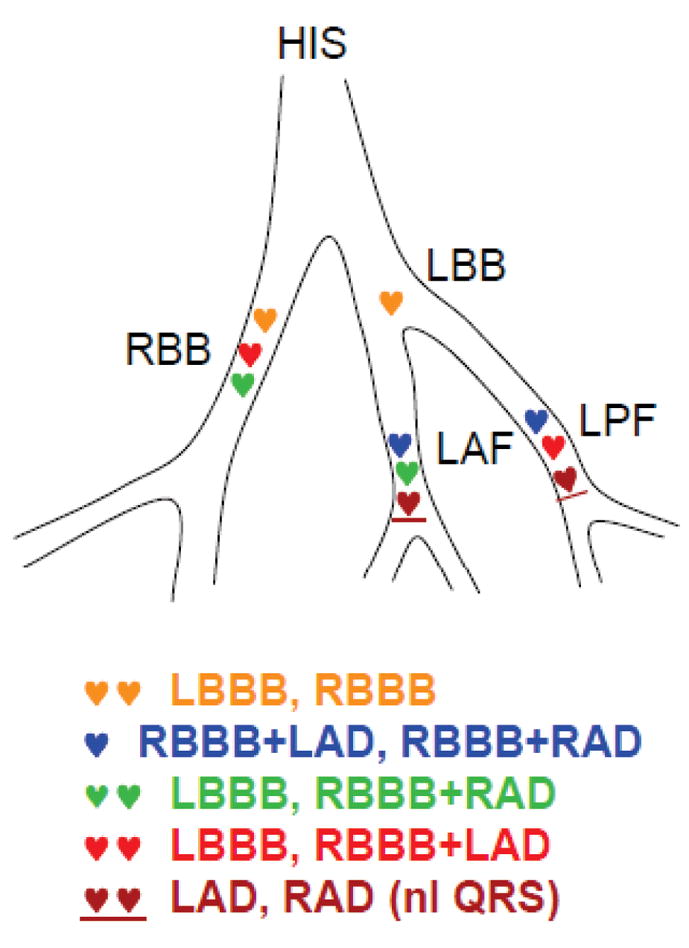
ECG patterns of BVT corresponding to locations of pairs of foci with reciprocating bigeminy. HIS, His bundle; LBB, left bundle branch; RBB, right bundle branch; LAF, left anterior fascicle; LPF, left posterior fascicle; LBBB, left bundle branch block; RBBB, right bundle branch block; LAD; left axis deviation; RAD, right axis deviation. See text for details.
This same scenario can also account for the classical human CPVT pattern of RBB block with alternating QRS axis, assuming that the two bigeminal foci are located in distal regions of the left anterior and left posterior fascicles of the LBB (Fig. 4, blue symbols). Thus, the reciprocating bigeminal beats arising from the distal left posterior and anterior fascicles would exhibit RBB block with left and right axis deviation, respectively. Cases of BVT have also been reported in which the axis alternates, but the QRS is narrow 13. This variant could be explained by bigeminal beats arising alternately from the anterior and posterior fascicles, but conducting retrogradely and blocking antegradely (Fig. 4, brown symbols). Thus, retrograde conduction to the contralateral fascicle and RBB would result in a narrow QRS with right or left axis deviation.
Finally, we simulated the degeneration of BVT into polymorphic VT, by assuming that when BVT further increases heart rate, additional sites in the HPS are recruited to develop bigeminy. Fig. 5 shows a scenario where an additional site exists at a Purkinje-myocyte junction in the RV free wall. When this third site developed triggered ventricular bigeminy, the earliest activation occurred irregularly among the three sites, creating a polymorphic VT pattern on the computed ECG, similar to that observed in a patient 12. Similar patterns developed when more than three sites develop spontaneous SR Ca release activity (data not shown).
Fig. 5. Simulated polymorphic VT.

A. Voltage snapshots showing the activation sequences corresponding to different QRS morphologies during polymorphic VT caused by 3 foci with reciprocating bigeminy. Red dot in each panel indicates the earliest activation site. B. The computed ECG from the simulation in A, showing polymorphic VT (with beat numbers corresponding to snapshots in A). C. ECG recorded in a patient with CPVT12.
Discussion
Both supraventricular or ventricular mechanisms of BVT have been proposed in the past, involving either focal or reentrant mechanisms 10. A supraventricular mechanism with alternating left anterior and left posterior fascicular block 17 was largely excluded with the advent of intracardiac recordings, which failed to show a His bundle potential preceding the alternating QRS complexes during BVT 18, 19. Postulated ventricular mechanisms have included a single focus in the proximal His bundle or bundle branches with alternating left fascicular block, or single or double foci in the distal HPS. In the single focus case, there is no obvious explanation for why fascicular or bundle branch block should alternate during BVT. Usually, concealed retrograde conduction perpetuates block in the fascicle/bundle branch that initially develops conduction block. Even if a conduction gap prevents concealed retrograde conduction from perpetuating block, both fascicles/bundle branches would have to exhibit the same conduction gap phenomena, which seems highly unlikely. In the double foci case, if neither focus is protected by entrance block, the faster focus should overdrive the slower focus, producing monomorphic VT rather than BVT. If both foci are protected by entrance block, then they would have to have identical cycle lengths, phase-shifted by exactly 180o to produce a constant cycle length during BVT, which also is improbable. If only one focus is protected by entrance block, then it could induce a second focus to fire at a fixed coupling interval, but the coupling interval would have to be exactly half of the first focus s cycle length to produce a constant cycle length during BVT.
In contrast to these complicated mechanisms, reciprocating bigeminy solves the puzzle of alternating QRS morphology by a simple “ping pong” mechanism in which DAD-induced triggered activity develops at different heart rate thresholds in different regions of the HPS or ventricles, consistent with known cellular properties of DAD-induced triggered activity 28, 33, 34. To produce a constant (i.e. nonalternating) cycle length during BVT requires only that the coupling intervals of the triggered beats be similar at the two sites.
Although we have modeled the bigeminy to be due to triggered activity from DADs, the same results are predicted for any mechanism inducing ventricular bigeminy (including automaticity or reentry) at more than one location in the ventricles. In addition, there is no strict requirement for the two bigeminal foci to be located in the distal HPS in opposite fascicles or ventricles. For example, two reciprocating triggered foci located in the same ventricle 8, or at sites in the endocardium and epicardium, could also produce BVT by this mechanism 35, although the QRS axis and morphology changes would be different. However, in humans, the most common BVT pattern during digitalis toxicity and CVPT is RBB block with alternating right and left axis deviation, consistent with reciprocating ectopic foci located in the distal left anterior and posterior fascicles of the left bundle. In mouse, due to its smaller heart size, the more common pattern may be foci located on opposite sides of the interventricular septum.
Finally, we show that if the increased heart rate during BVT induces a third bigeminal focus in the HPS, the interactions between the three foci can produce irregular activation patterns resulting in polymorphic VT (Fig. 5). Since the model is deterministic, the irregularity may be due to chaos, which is a common scenario observed with coupled oscillators 36. As heart rate progressively accelerates, we speculate that additional regions develop DADs, making the conversion to VF progressively more likely.
In summary, we conjecture that the full spectrum of arrhythmias described electrocardiographically in acquired and familial conditions associated with BVT can be accounted for based on the known properties of DAD-triggered arrhythmias, as follows:
Ventricular bigeminy, when a single site in the HPS or ventricular myocardium develops a single DAD-triggered beat following each sinus beat;
BVT, when a second site develops ventricular bigeminy, and reciprocally activates the first site by the ping-pong mechanism described above;
Polymorphic VT, when 3 or more sites concurrently develop bigeminy. Note that these first 3 mechanisms specifically require bigeminy, i.e. that DADs are capable of triggering only a single PVC following each sinus or paced beat, so that the subsequent beat has to arise from a different location (thereby altering the QRS morophology/axis).
Monomorphic VT, when bigeminy progresses to repetitive DADs which generate a run of triggered activity, such that the site with most rapid rate of the triggered activity overdrives the other slower sites, producing a monomorphic QRS complex.
Degeneration to VF, when any of these VT forms results in wavebreak and initiation of reentry, which is then likely to be maintained by a mixture of reentry and DAD-triggered focal activations.
Limitations
Several limitations of our study should be recognized. Our Purkinje AP model is an approximation that does not replicate all experimental details such as a lowered AP plateau 21. We did not attempt to build a complete “bottom up” model linking Ca waves and DADs at the subcellular-cellular levels to DADs and triggered activity at the tissue-whole heart levels. In particular, Ca waves and DADs at the cellular level are strongly influenced by stochastic factors. How they overcome source-sink mismatches in well-coupled tissue to produce regular behaviors such as ventricular bigeminy is not well-understood. Nevertheless, periodic arrhythmias attributed to DADs, such as ventricular bigeminy are commonly observed in cardiac tissue 28, 33, 34 in the setting of glycoside toxicity and in association with BVT 9–11. The experimental observation that PVC s with wide QRS complexes arise from the HPS during or after rapid pacing also implies that different regions of the heart must have different rate thresholds for DAD-mediated triggered activity, since otherwise, PVC s would all have a narrow QRS complex. This is not surprising, since many factors which influence the precise rate threshold at which DADs cause triggered activity, including cell properties, intercellular coupling, geometrical factors influencing the source-sink relationship in the bundle branches and HPS, etc. To illustrate the general feasibility of the reciprocating bigeminy mechanism, we therefore phenomenologically introduced these well-documented experimental features into our computer model, rather than a building a highly detailed model from the bottom up incorporating stochastic elements, etc. To simplify the computer modeling, we also simulated a ventricular slice, rather than the full 3D anatomic heart. This is not likely to change the findings, however, since the ventricular myocardium acts simply as a passive electrical conduit for the ping pong dynamics occurring in the HPS.
Supplementary Material
Acknowledgments
Funding Sources. This study was supported by NIH/NHLBI grants P01 HL078931 and R01 HL103662, and the Laubisch and Kawata Endowments.
We thank Tannaz Tebbi for technical assistance.
Non-standard Abbreviations and Acronyms
- BVT
bidirectional ventricular tachycardia
- CPVT
catecholaminergic polymorphic ventricular tachycardia
- DAD
delayed afterdepolarization
- EAD
early afterdepolarization
- ECG
electrocardiogram
- HPS
His-Purkinje system
- LBB
left bundle branch
- LV
left ventricle
- PVC
premature ventricular complex
- RyR2
Ryanodine receptor
- RBB
right bundle branch
- RV
right ventricle
Footnotes
Disclosures. No conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Schwensen C. Ventricular Tachycardia as the result of the administration of digitalis. Heart. 1922;9:199–204. [Google Scholar]
- 2.Valent S, Kelly P. Digoxin-Induced Bidirectional Ventricular Tachycardia. N Engl J Med. 1997;336(8):550. doi: 10.1056/NEJM199702203360805. [DOI] [PubMed] [Google Scholar]
- 3.Kummer JL, Nair R, Krishnan SC. Bidirectional Ventricular Tachycardia Caused by Digitalis Toxicity. Circulation. 2006;113(7):e156–157. doi: 10.1161/CIRCULATIONAHA.105.557561. [DOI] [PubMed] [Google Scholar]
- 4.Menduiña MJ, Candel JM, Alaminos P, et al. Bidirectional Ventricular Tachycardia Due to Digitalis Poisoning. Revista Espanola de Cardiologia. 2005;58(8):991–993. [PubMed] [Google Scholar]
- 5.Stubbs WA. Bidirectional ventricular tachycardia in familial hypokalaemic periodic paralysis. Proc R Soc Med. 1976;69(3):223–224. [PMC free article] [PubMed] [Google Scholar]
- 6.Morita H, Zipes DP, Morita ST, et al. Mechanism of U wave and polymorphic ventricular tachycardia in a canine tissue model of Andersen-Tawil syndrome. Cardiovasc Res. 2007;75(3):510–518. doi: 10.1016/j.cardiores.2007.04.028. [DOI] [PubMed] [Google Scholar]
- 7.Berte B, Eyskens B, Meyfroidt G, et al. Bidirectional ventricular tachycardia in fulminant myocarditis. Europace. 2008;10(6):767–768. doi: 10.1093/europace/eun105. [DOI] [PubMed] [Google Scholar]
- 8.Leenhardt A, Lucet V, Denjoy I, et al. Catecholaminergic Polymorphic Ventricular Tachycardia in Children : A 7-Year Follow-up of 21 Patients. Circulation. 1995;91(5):1512–1519. doi: 10.1161/01.cir.91.5.1512. [DOI] [PubMed] [Google Scholar]
- 9.Levy S, Hilaire J, Clementy J, et al. Bidirectional tachycardia. Mechanism derived from intracardiac recordings and programmed electrical stimulation. Pacing Clin Electrophysiol. 1982;5(5):633–638. doi: 10.1111/j.1540-8159.1982.tb02298.x. [DOI] [PubMed] [Google Scholar]
- 10.Levy S, Aliot E. Bidirectional tachycardia: a new look on the mechanism. Pacing Clin Electrophysiol. 1989;12(5):827–834. doi: 10.1111/j.1540-8159.1989.tb01905.x. [DOI] [PubMed] [Google Scholar]
- 11.Sumitomo N, Harada K, Nagashima M, et al. Catecholaminergic polymorphic ventricular tachycardia: electrocardiographic characteristics and optimal therapeutic strategies to prevent sudden death. Heart. 2003;89(1):66–70. doi: 10.1136/heart.89.1.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lin YH, Lai LP, Lin TK, et al. Exercise-provoked bidirectional ventricular tachycardia in a young woman. J Formos Med Assoc. 2004;103(10):780–783. [PubMed] [Google Scholar]
- 13.Rothfeld EL. Bidirectional tachycardia with normal QRS duration. Am Heart J. 1976;92(2):231–233. doi: 10.1016/s0002-8703(76)80259-1. [DOI] [PubMed] [Google Scholar]
- 14.Cerrone M, Colombi B, Santoro M, et al. Bidirectional Ventricular Tachycardia and Fibrillation Elicited in a Knock-In Mouse Model Carrier of a Mutation in the Cardiac Ryanodine Receptor. Circ Res. 2005;96(10):e77–82. doi: 10.1161/01.RES.0000169067.51055.72. [DOI] [PubMed] [Google Scholar]
- 15.Knollmann B, Chopra N, Hlaing T, et al. Casq2 deletion causes sarcoplasmic reticulum volume increase, premature Ca release, and catecholaminergic polymorphic ventricular tachycardia. The Journal of Clinical Investigation. 2006;116(9):2510–2520. doi: 10.1172/JCI29128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cerrone M, Noujaim SF, Tolkacheva EG, et al. Arrhythmogenic mechanisms in a mouse model of catecholaminergic polymorphic ventricular tachycardia. Circ Res. 2007;101(10):1039–1048. doi: 10.1161/CIRCRESAHA.107.148064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rosenbaum MB, Elizari MV, Lazzari JO. The mechanism of bidirectional tachycardia. Am Heart J. 1969;78(1):4–12. doi: 10.1016/0002-8703(69)90250-6. [DOI] [PubMed] [Google Scholar]
- 18.Cohen SI, Deisseroth A, Hecht HS. Infra-His bundle origin of bidirectional tachycardia. Circulation. 1973;47(6):1260–1266. doi: 10.1161/01.cir.47.6.1260. [DOI] [PubMed] [Google Scholar]
- 19.Morris SN, Zipes DP. His bundle electrocardiography during bidirectional tachycardia. Circulation. 1973;48(1):32–36. doi: 10.1161/01.cir.48.1.32. [DOI] [PubMed] [Google Scholar]
- 20.Mahajan A, Shiferaw Y, Sato D, et al. A rabbit ventricular action potential model replicating cardiac dynamics at rapid heart rates. Biophys J. 2008;94(2):392–410. doi: 10.1529/biophysj.106.98160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cordeiro JM, Spitzer KW, Giles WR. Repolarizing K currents in rabbit heart Purkinje cells. The Journal of Physiology. 1998;508(3):811–823. doi: 10.1111/j.1469-7793.1998.811bp.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Luo CH, Rudy Y. A dynamic model of the cardiac ventricular action potential .1. Simulations of ionic currents and concentration changes. Circ Res. 1994;74:1071–1096. doi: 10.1161/01.res.74.6.1071. [DOI] [PubMed] [Google Scholar]
- 23.Huffaker RB, Samade R, Weiss JN, et al. Tachycardia-induced early afterdepolarizations: insights into potential ionic mechanisms from computer simulations. Comput Biol Med. 2008;38(11–12):1140–1151. doi: 10.1016/j.compbiomed.2008.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xie F, Qu Z, Yang J, Baher A, et al. A simulation study of the effects of cardiac anatomy in ventricular fibrillation. J Clin Invest. 2004;113:686–693. doi: 10.1172/JCI17341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Baher A, Qu Z, Hayatdavoudi A, et al. Short-term cardiac memory and mother rotor fibrillation. Am J Physiol Heart Circ Physiol. 2007;292(1):H180–189. doi: 10.1152/ajpheart.00944.2005. [DOI] [PubMed] [Google Scholar]
- 26.Qu Z, Garfinkel A. An advanced algorithm for solving partial differential equation in cardiac conduction. IEEE Trans Biomed Eng. 1999;46(9):1166–1168. doi: 10.1109/10.784149. [DOI] [PubMed] [Google Scholar]
- 27.Qu Z, Weiss JN, Garfinkel A. Cardiac electrical restitution properties and stability of reentrant spiral waves: a simulation study. Am J Physiol. 1999;45:269–H283. doi: 10.1152/ajpheart.1999.276.1.H269. [DOI] [PubMed] [Google Scholar]
- 28.Braunwald E. Heart Disease: A Textbook of Cardiovascular Medicine. 1. Philadelphia, PA: W.B. Saunders Company; 1980. [Google Scholar]
- 29.Azarov JE, Shmakov DN, Vityazev VA, et al. Activation and repolarization patterns in the ventricular epicardium under sinus rhythm in frog and rabbit hearts. Comp Biochem Physiol A Mol Integr Physiol. 2007;146(3):310–316. doi: 10.1016/j.cbpa.2006.10.036. [DOI] [PubMed] [Google Scholar]
- 30.Gima K, Rudy Y. Ionic current basis of electrocardiographic waveforms: a model study. Circ Res. 2002;90(8):889–896. doi: 10.1161/01.res.0000016960.61087.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Maruyama M, Joung B, Tang L, et al. Diastolic intracellular calcium-membrane voltage coupling gain and postshock arrhythmias: role of purkinje fibers and triggered activity. Circ Res. 2010;106(2):399–408. doi: 10.1161/CIRCRESAHA.109.211292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xie Y, Sato D, Garfinkel A, Qu Z, et al. So little source, so much sink: requirements for afterdepolarizations to propagate in tissue. Biophys J. 2010;99(5):1408–1415. doi: 10.1016/j.bpj.2010.06.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ferrier GR, Saunders JH, Mendez C. A cellular mechanism for the generation of ventricular arrhythmias by acetylstrophanthidin. Circ Res. 1973;32(5):600–609. doi: 10.1161/01.res.32.5.600. [DOI] [PubMed] [Google Scholar]
- 34.Garfinkel A, Spano ML, Ditto WL, et al. Controlling cardiac chaos. Science. 1992;257:1230–1235. doi: 10.1126/science.1519060. [DOI] [PubMed] [Google Scholar]
- 35.Nam GB, Burashnikov A, Antzelevitch C. Cellular mechanisms underlying the development of catecholaminergic ventricular tachycardia. Circulation. 2005;111(21):2727–2733. doi: 10.1161/CIRCULATIONAHA.104.479295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hilborn RC. Chaos and Nonlinear Dynamics. Vol. 6. New York: Oxford University Press; 1994. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.