

Abstract
A novel class of 2,4-disubstituted pyrimidines (7a–u, 8a–f, 9a–e) that possess substituents with varying steric and electronic properties at the C-2 and C-4 positions, were designed, synthesized and evaluated as dual cholinesterase and amyloid-β (Aβ)-aggregation inhibitors. In vitro screening identified N-(naphth-1-ylmethyl)-2-(pyrrolidin-1-yl)pyrimidin-4-amine (9a) as the most potent AChE inhibitor (IC50 = 5.5 μM). Among this class of compounds, 2-(4-methylpiperidin-1-yl)-N-(naphth-1-ylmethyl)pyrimidin-4-amine (9e) was identified as the most potent and selective BuChE inhibitor (IC50 = 2.2 μM, Selectivity Index = 11.7) and was about 5.7-fold more potent compared to the commercial, approved reference drug galanthamine (BuChE IC50 = 12.6 μM). In addition, the selective AChE inhibitor N-benzyl-2-(4-methylpiperazin-1-yl)pyrimidin-4-amine (7d), exhibited good inhibition of hAChE-induced aggregation of Aβ1–40 fibrils (59% inhibition). Furthermore, molecular modeling studies indicate that a central pyrimidine ring serves as a suitable template to develop dual inhibitors of cholinesterase and AChE-induced Aβ aggregation thereby targeting multiple pathological routes in AD.
Keywords: Cholinesterase inhibitors ChEIs; Acetylcholinesterase AChE; Butyrylcholinesterase BuChE; Human acetylcholinesterase hAChE; Structure-activity relationship SAR; Amyloid-β Aβ; 5,5'-dithiobis(2-nitrobenzoic acid), DTNB; Thioflavin T ThT; 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide MTT
1. Introduction
Neurological disorders inflict a heavy burden on healthcare costs, affected individuals and their caregivers. Alzheimer's disease (AD) is classified as a progressive, neurodegenerative disease that affects the cholinergic regions of the central nervous system (CNS) that associate with cognitive function and spatial awareness.1 This devastating neurological disease targets the elderly populations and its prevalence is on the rise.2,3 The hallmark characteristics of AD include the rapid loss of cholinergic neurotransmission, accelerated aggregation of amyloid-□(Aβ) peptides and formation of neurofibrillary tangles (NFTs) of hyperphosphorylated tau protein.2–6 These characteristics establish the basis for the cholinergic, amyloid and tau hypotheses for AD pathology respectively.
According to the cholinergic hypothesis, the pathology of AD is attributed to the rapid decline in neurotransmission in the cholinergic regions of the CNS that house acetylcholine (ACh) utilizing sympathetic and parasympathetic neurons.7 The acetyl and butyrylcholinesterase (AChE and BuChE, respectively) enzymes act on ACh to terminate its actions in the synaptic cleft by hydrolyzing the neurotransmitter to choline and acetate.8,9 Deficiencies in the ACh synthesizing enzyme (choline acetyltransferase – ChAT) also contributes to the overall decline of ACh concentration in the cortical regions of the brain.9–12 Furthermore, recent studies have shown that the ratio of AChE to BuChE is dependant on the stage of pathogenesis. In the CNS, AChE plays a vital role in the early stages of AD. However, as the disease progresses and cholinergic neurons are depleted, BuChE, which has a wider distribution within the body, acts as the major degrading enzyme.13,14
In contrast, the amyloid hypothesis suggests that the progression of AD is attributed to the accelerated accumulation of toxic forms of self-induced and/or AChE-promoted toxic aggregates of Aβ peptides.15–17 These toxic peptides arise from the cleavage of the amyloid-precursor protein (APP) by the β-secretase (BACE-1) and γ-secretase enzymes. In this regard, recent studies indicate a link between the cholinergic and amyloid hypotheses.18,19 AChE is known to induce amyloid-β (Aβ) formation leading to the highly toxic AChE-Aβ peptide complexes.20–22 These multiple factors in AD pathology mandate the need to develop small molecule therapies that exhibit dual ChE inhibition as well as reduce the formation of neurotoxic Aβ-aggregates. Research into the cholinergic hypothesis has led to the development of several fused and non-fused ring systems as ChE inhibitors (ChEIs) (Chart 1). For example, tacrine (1), an acridine derivative, was one of the earliest ChEI developed to treat AD.23 Bis-7-tacrine (2) is a potent dual AChE and BuChE inhibitor.24 Propidium (3) binds specifically to the peripheral anionic site (PAS) of AChE.25 The anti-Alzheimer drug donepezil (4), is highly selective toward AChE and its binding conformation spans both the catalytic and peripheral sites of AChE.26 Recent work by DeLisa and co-workers examined s-triazine based ring templates (5) for their ability to inhibit the aggregation of Aβ1–42 plaques.27
As part of our research program, we previously reported the development of a group of heterocyclic, non-fused ChEIs based on a 2,4-disubstituted pyrimidine ring template.28 Here we report the design, synthesis and evaluation of heterocycles 7a–u, 8a–f, 9a–e containing a 2,4-disubstituted pyrimidine ring scaffold as novel small molecules possessing dual activity as cholinesterase inhibitors and as inhibitors of hAChE-induced Aβ1–40 aggregation.
2. Results and Discussion
2.1 Chemistry
The synthesis of target 2,4-disubstituted pyrimidine derivatives (7a–r, 8a–f and 9a–e) was accomplished in two steps. In the first step, the N-benzyl, N-phenethyl and naphth-1-ylmethyl-2-chloropyrimidin-4-amine intermediates (7, 8 and 9 respectively) were synthesized from the 2,4-dichloropyrimidine starting material (6) by a nucleophilic aromatic substitution reaction at C-4 using a base such as N,N-diisopropylethylamine (DIPEA). The reaction was run in EtOH at 75–85 °C and refluxed for 3 hours. The intermediates 7, 8 and 9 were obtained in moderate to good yield ranging from 60–75% (Scheme 1).28,29
Scheme 1.

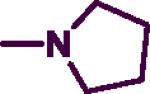
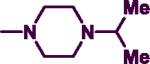
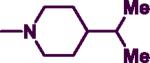
Reagents and conditions: (a) DIPEA, phenylmethanamine (7), 2-phenylethanamine (8) or (naphthalene-1-yl)methanamine (9), EtOH, 0 °C to 75–85 °C and reflux for 3 h.; (b) R2 = pyrrolidine, morpholine, thiomorpholine, 1-methylpiperazine, 4-methylpiperidine, acetylpiperazine, t-butyl piperazine-1-carboxylate, cyclohexylpiperazine, isopropyl piperazine, isopropylpiperidine, N-propylpiperazine, N-hydroxyethyl piperazine, N-methoxyethylpiperazine, 4-chloro, bromo, fluoro and trifluoromethylbenzylpiperazine, 4-amino-1-benzylpiperidine, n-BuOH, 145–150 °C, 30–40 min.
In the second step, the C-2 chlorine was displaced by various cyclic amines (R2 = pyrrolidine, morpholine, thiomorpholine, 1-methylpiperazine, 4-methylpiperidine, acetylpiperazine, t-butyl piperazine-1-carboxylate, cyclohexylpiperazine, isopropylpiperazine, isopropylpiperidine, N-propylpiperazine, N-hydroxyethylpiperazine, N-methoxyethylpiperazine, 4-chloro, bromo, fluoro and trifluoromethylbenzylpiperazine, 4-amino-1-benzylpiperidine, Scheme 1). This reaction was run under rigorous conditions (145–150 °C) for 30–40 minutes in a sealed pressure vessel (PV) using n-butanol as a solvent to afford the target 2,4-disubstituted pyrimidine derivatives (7a–r, 8a–f, 9a–e) in moderate to good yield (50–90%) (Scheme 1).28,30
N-benzyl-thiomorpholinopyrimidin-4-amine (7c) was oxidized to 4-[4-(benzylamino)pyrimidin-2-yl]thiomorpholine 1-oxide (7s) in good yield (75%) using meta-chloroperoxybenzoic acid (MCPBA) (Scheme 2) and to 4-[4-(benzylamino) pyrimidin-2-yl]thiomorpholine 1,1-dioxide (7t) in good yield (75%) using potassium peroxymonosulfate (Oxone®) as the oxidizing agent (Scheme 2). The deprotection of the tert-butoxycarbonyl (t-Boc) group of 7g [tert-butyl 4-(4-(benzylamino)pyrimidin-2-yl)piperazine-1-carboxylate] was accomplished by using 50% v/v trifluoroacetic acid (TFA) to yield N-benzyl-2(piperazin-1-yl)pyrimidin-4-amine (7u) in good yield (75%) (Scheme 3).
Scheme 2.

Reagents and conditions: (a) mCPBA, 7c, 1,4-dioxane, 0 °C to r.t., 3 hours; (b) Oxone®, 7c, MeOH, H2O and 1,4-dioxane, 0 °C to 70–75 0 °C, 1 h. then r.t., 4 h.
Scheme 3.

Reagents and conditions: (a) TFA, 7g, DCM, 0 °C to r.t., 2 h
2.2 Cholinesterase Inhibition and SAR Studies
The ability of the synthesized derivatives (7a–u, 8a–f and 9a–e) to inhibit both hAChE and equine BuChE was evaluated in vitro (IC50 values, Table 1).28, 31 The structure-activity relationship (SAR) studies indicated that the cholinesterase inhibition and selectivity were sensitive to steric and electronic parameters at C-2 and C-4 positions of the central pyrimidine ring. They exhibited a broad range of inhibition (AChE IC50 = 5.5 to 28.8 μM range; BuChE IC50 = 2.2 to > 100 μM range).
Table 1.
AChE and BuChE Inhibitory Activities and C LogP data for 2,4-disubstituted pyrimidines (7a–u, 8a–f and 9a–e)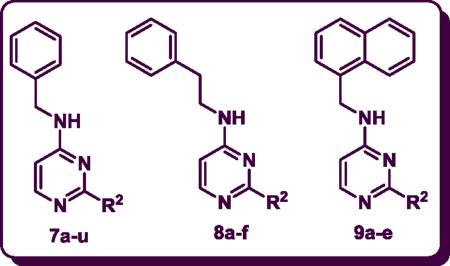
| IC50(μM)a,b |
Selectivity Index (SI)b | C logPc | |||
|---|---|---|---|---|---|
| Compd | R2 | AChE | BuChE | ||
| 7a |
|
8.70 | 26.40 | 0.33 | 2.97 |
| 7b |

|
14.0 | 68.30 | 0.21 | 2.14 |
| 7c |

|
23.20 | 6.10 | 3.80 | 2.98 |
| 7d |

|
24.90 | >100 | <0.25 | 2.71 |
| 7e |

|
18.40 | 3.40 | 5.41 | 4.05 |
| 7f |

|
16.60 | >100 | <0.17 | 1.73 |
| 7g |

|
18.80 | >100 | <0.19 | 4.12 |
| 7h |
|
22.90 | 7.60 | 3.01 | 4.65 |
| 7i |
|
25.0 | 3.40 | 7.35 | 3.54 |
| 7j |
|
14.20 | 6.50 | 2.19 | 4.97 |
| 7k |
|
15.30 | 59.90 | 0.26 | 3.76 |
| 71 |
|
26.40 | >100 | <0.26 | 2.13 |
| 7m |
|
26.70 | >100 | <0.27 | 2.89 |
| 7n |
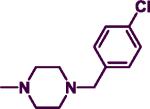
|
20.20 | 10.70 | 1.89 | 5.14 |
| 7o |
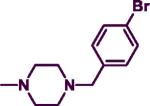
|
25.50 | 14.30 | 1.78 | 5.29 |
| 7p |
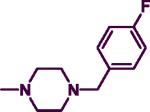
|
21.60 | 7.30 | 2.96 | 4.57 |
| 7q |
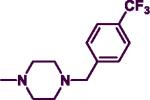
|
28.80 | >100 | <0.29 | 5.31 |
| 7r |
|
12.40 | 8.20 | 1.51 | 4.60 |
| 7s |

|
12.60 | >100 | <0.13 | 1.26 |
| 7t |
|
24.20 | >100 | <0.24 | 1.18 |
| 7u |

|
15.50 | >100 | 0.16 | 2.13 |
| 8a |
|
9.80 | 13.80 | 0.71 | 3.62 |
| 8b |

|
19.70 | >100 | <0.20 | 2.79 |
| 8c |

|
26.40 | >100 | <0.26 | 3.63 |
| 8d |

|
20.40 | >100 | <0.20 | 3.35 |
| 8e |

|
8.80 | 17.70 | 0.50 | 4.69 |
| 8f |

|
19.90 | >100 | <0.20 | 2.37 |
| 9a |
|
5.50 | 8.90 | 0.62 | 4.14 |
| 9b |

|
14.70 | 28.0 | 0.53 | 3.32 |
| 9c |

|
12.80 | 34.70 | 0.37 | 4.15 |
| 9d |

|
17.50 | 2.60 | 6.73 | 3.88 |
| 9e |

|
25.80 | 2.20 | 11.73 | 5.22 |
| Tacrine. HCl | Chart 1 (1) | 0.093 | 0.019 | 9.11 | 3.27 |
| Bis(7)-tacrine | Chart 1 (2) | 0.004 | 0.010 | 0.171 | 10.10 |
| Galanthamine.HBr | − | 3.80 | 12.60 | 0.274 | 1.03 |
| Donepezil | 0.032 | 3.60 | |||
The in vitro test compound concentration required to produce 50% inhibition of hAChE and equine BuChE
The result (IC50) is the mean of two separate experiments (n = 4) and the deviation from the mean is < 10% of the mean value.
Selectivity Index = hAChE IC50/BuChE IC50.
C LogP was determined using ChemDraw Ultra 12.0. CambridgeSoft Company.
Among the N-benzyl series of derivatives (7a–u), the substituent electronic and steric factors at C-2 position modulated ChE inhibition. The presence of 5-membered heterocycloalkyl C-2 substituent such as a pyrrolidine (7a) provided ChE inhibition with superior potency and selectivity toward AChE (AChE IC50 = 8.7 μM; BuChE IC50 = 26.4 μM). Replacing the C-2 with a 6-membered morpholine provides 7b that exhibited weak AChE (IC50 = 14 μM) and BuChE inhibition (IC50 = 68.3 μM) compared to 7a. In contrast, the presence of a C-2 thiomorpholine ring in 7c provided superior BuChE inhibition and selectivity (AChE IC50 = 23.2 μM; BuChE IC50 = 6.1 μM, SI = 3.8). This observation indicates that the differences in the electronic properties of oxygen and sulfur could modulate the binding modes within the active sites of both ChEs. This was further explored by oxidizing the sulfur in 7c to either a sulfoxide (7s) or a sulfone (7t). Both 7s and 7t exhibited AChE inhibition, with 7s exhibiting about 1.8-fold more potent AChE inhibition (IC50 = 12.6 μM) relative to 7c. Interestingly, 7s and 7t (both lack the unsubstituted terminal sulfur atom present in 7c) did not exhibit BuChE inhibition up to 100 μM.
The SAR of C-2 piperazine-substituted N-benzylpyrimidine derivatives were explored by incorporating a wide range of terminal 4-alkyl, alkoxy, acyl, cycloalkyl and substituted-benzyl rings (7d,7f–i,7k–q). The incorporation of terminal 4-alkylpiperazine and 4-alkoxy substituents (7d,7f–g, 7i and 7k–m) provided AChE inhibition. The presence of a methyl group in 7d (R2 = 4-methylpiperazine) provided moderate AChE inhibition (IC50 = 24.9 μM), whereas increasing the chain length to propyl (7k, R2 = 4-propylpiperazine) led to superior AChE inhibition (IC50 = 15.3 μM). In contrast, the presence of a branched, sterically demanding alkyl chain such as an isopropyl in 7i (R2 = 4-isopropylpiperazine) exhibited similar AChE inhibition (AChE IC50 = 25 μM) relative to 7d. However, 7i was a selective BuChE inhibitor (BuChE IC50 = 3.4 μM; SI = 7.35). Similarly, the presence of alkoxy groups such as hydroxyethyl in7l (AChE IC50 = 26.4 μM) or a methoxyethyl in 7m (AChE IC50 = 26.7 μM) exhibited comparable AChE inhibition relative to 7d (IC50 = 24.9 μM). Interestingly, C-2 piperazine R2 substituents that possessed a terminal alkyl or alkoxy group (7d,7f–g and 7k–m) generally exhibited weak or a lack of BuChE inhibition (BuChE IC50 = 59.9 μMto > 100 μM range) with the exception of 7i. Furthermore, the unsubstituted C-2 piperazine containing compound 7u exhibited selective AChE inhibition (IC50 = 15.5 μM) with no BuChE inhibition up to 100 μM. It was interesting to note that the presence of a cycloalkyl ring (7h) provided superior BuChE inhibition and selectivity (AChE IC50 = 22.9 μM; BuChE IC50 = 7.6 μM, SI = 3.01) among the piperazine derivatives. In contrast to terminal alkyl or alkoxy substituted piperazines, the presence of a C-2 benzylpiperazine possessing halogen atoms (Cl, Br or F) (7n–p) or electron withdrawing groups (CF3) (7q) at the para-position exhibited dual ChE inhibition (AChE IC50 = 20.2 to 28.8 μM range; BuChE IC50 = 7.3 to 14.3 μM range) with the exception of 7q (BuChE IC50 > 100 μM). The AChE activity was of the order: Cl > F > Br > CF3, whereas the presence of a fluorine atom at para-position provides superior BuChE inhibition (BuChE IC50 = 7.3 μM). The BuChE activity was of the order: F > Cl > Br > CF3 (inactive at 100 μM) establishing the role of increasing electronegativity with superior BuChE inhibition. In addition, replacing the C-2 4-methylpiperazine (7d) with a methylpiperidine bioisostere in 7e provided dual ChE inhibition along with superior BuChE inhibition and selectivity (AChE IC50 = 18.4 μM; BuChE IC50 = 3.4 μM, SI = 5.41) relative to 7d. In addition to its superior BuChE inhibitory potency, 7e was about 3.7-fold more potent relative to the reference drug galanthamine (BuChE IC50 = 12.6 μM),although it was not as potent as bis-tacrine (BuChE IC50 = 0.010 μM). Furthermore, the bioisosteric replacement of C-2 4-isopropylpiperazine in 7i with an isopropylpiperidine bioisostere in 7j provided dual ChE inhibition similar to 7i, although with a 2.2-fold loss in BuChE inhibitory potency (BuChE IC50 = 6.5 μM) and a 1.7-fold gain in AChE inhibitory potency (AChE IC50 = 14.2 μM) compared to 7i. In derivative 7r, the benzylpiperidine pharmacophore present in donepezil was incorporated into a pyrimidine diamine template. This modification provided dual ChE inhibition and exhibited superior BuChE inhibition (BuChE IC50 = 8.2 μM) relative to the reference drug galanthamine (BuChE IC50 = 12.6 μM).
From the N-benzyl series (7a–u), 7a was identified as the most potent AChE inhibitor (IC50 = 8.7 μM); 7s as the most selective AChE inhibitor (SI < 0.12); 7e and 7i as equally potent BuChE inhibitors (IC50 = 3.40 μM), where 7i was also the most selective BuChE inhibitor (SI = 7.35).
In the N-phenethyl C-4 substituted pyrimidines (8a–f) evaluated, the presence of a 5-membered pyrrolidine ring in 8a led to dual ChE inhibition (AChE IC50 = 9.8 μM; BuChE IC50 = 13.8 μM) and similar dual ChE inhibition was seen with 8e that possessed a 6-membered methylpiperidine ring at C-2 (AChE IC50 = 8.8 μM; BuChE IC50 = 17.7 μM; Table 1). In contrast, the presence of C-2 6-membered rings such as morpholine (8b), thiomorpholine (8c), methylpiperazine (8d) and acetylpiperazine (8f) was detrimental to BuChE inhibition (IC50 values > 100 μM; Table 1). It was interesting to note that the naphth-1-ylmethyl series of C-4 substituted pyrimidines (9a–e) evaluated exhibited dual ChE inhibition (AChE IC50 = 5.5 to 25.8 μM range; BuChE IC50 = 2.2 to 34.7 μM range; Table 1). For example, the presence of a C-2 5-member pyrrolidine in 9a provided superior BuChE inhibition (IC50 = 8.9 μM) relative to donepezil (BuChE IC50 = 12.6 μM) although it was a less potent AChE inhibitor relative to tacrine (AChE IC50 = 0.093 μM) or bis-tacrine (AChE IC50 = 4 nM). Replacing the C-2 5-membered pyrrolidine with either a 6-membered morpholine (9b, AChE IC50 = 14.7 μM; BuChE IC50 = 28 μM) or thiomorpholine (9c, AChE IC50 = 12.8 μM; BuChE IC50 = 34.7 μM; Table 1) led to a decline in ChE inhibitory potency relative to 9a. In contrast, the presence of either a C-2 4-methylpiperazine (9d) or the corresponding bioisostere methylpiperidine (9e) provided superior BuChE inhibitory potency and selectivity. Compound 9e exhibited about 5.7-fold superior BuChE inhibition (IC50 = 2.2 μM) and selectivity (SI = 11.7) relative to the reference drug galanthamine (BuChE IC50 = 12.6 μM; SI = 0.27) and was much more potent compared to donepezil (BuChE IC50 = 3.6 μM) whereas the corresponding C-2 methylpiperidine 9d exhibited similar BuChE inhibition (BuChE IC50 = 2.6 μM) and superior AChE inhibition (AChE IC50 = 17.5 μM) relative to 9e (AChE IC50 = 25.8 μM). In addition, the theoretical partition coefficient values (C log P) of various synthesized pyrimidines (Table 1) compares well with the reference compound tacrine (C log P = 3.27) favoring their use as agents to treat disorders of the central nervous system (CNS).
2.3 Inhibition of hAChE-induced Aβ Aggregation
The ability of some 2,4-disubstituted pyrimidines that exhibit a range of cholinesterase inhibition activities (7a, 7c, 7d, 7g, 7s, 7t, 8c and 9c) were evaluated to prevent hAChE-induced Aβ1–40 aggregation by a thioflavin T (ThT) fluorescence method.32 The anti-aggregating activity of eight 2,4-disubstituted pyrimidines along with reference compounds propidium iodide and donepezil are presented in Table 2. At a concentration of 100 μM, 2,4- disubstituted pyrimidines exhibited varying degrees of inhibition (inactive to 59% inhibition of aggregation). In compound 7a, the presence of a smaller 5-member pyrrolidine at C-2 did not provide anti-aggregation activity (Table 2). Similarly, the presence of a C-2 thiomorpholine ring in 7c, 8c and 9c provided either weak or no anti-aggregation activity. Among the 2,4-disubstituted pyrimidines evaluated, 7d with a combination of C-4 N-benzyl and a C-2 methylpiperazine substituent exhibited good activity against hAChE-induced aggregation of Aβ1–40 peptides (59% inhibition). This compound was 3.4-fold more potent compared to donepezil (17% inhibition) and was not as potent as propidium iodide (85% inhibition), which is known to bind to the PAS in AChE.20, 25. Interestingly, when the C-2 thiomorpholine substituent of 7c was oxidized to a polar sulfoxide (7s) or a sulfone (7t), it provided anti-aggregation activity of about 56% and 44% inhibition respectively (Table 1). This suggests that small molecules with polar functional groups could potentially exhibit superior binding within the AChE active which has polar pockets in its catalytic site (Ser203, His447, Glu202) as well as the active site gorge entry (Trp286, Tyr124). This justifies our results that the presence of polar functional groups at C-2 in 7d, 7s and 7t led to superior inhibition of AChE-promoted abeta aggregation compared to 7g that has a lipophilic boc group at C-2. These studies indicate that the substituent electronic and steric effects both at the C-2 and C-4 positions can be manipulated to develop 2,4- disubstituted pyrimidines that bind favorably within the PAS present in AChE. Further studies are in progress to acquire SAR data on the role of various C-2 substituents and their effect on AChE-promoted Aβ aggregation.
Table 2.
Inhibition of hAChE-Induced Aggregation of Aβ1–40 by 2,4-disubstituted pyrimidines (7a, 7c, 7d, 7g, 7s, 7t, 8c and 9c)
| Compd | Inhibition at 100 μM ± SEM (%)a |
|---|---|
| 7a | NA |
| 7c | NA |
| 7d | 59.0 ± 2.9 |
| 7g | 26.7 ± 17.0 |
| 7s | 55.9 ± 6.3 |
| 7t | 44.1 ± 11.0 |
| 8c | NA |
| 9c | 38 ± 25 |
| Donepezil | 17.4 ± 8.1 |
| Propidium iodide | 84.9 ± 4.1 |
The values are the mean of two independent measurements each performed in triplicates, SEM = standard error of mean
NA = Not active
2.4 SH-SY5Y Neuroblastoma Cell Toxicity
The cytotoxicity of 2,4- disubstituted pyrimidine derivatives 7a, 7c, 7d, 7g, 7s, 7t, 8c and 9c was evaluated using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) based colorimetric assay. The cell viability of SH-SY5Y neuroblastoma cells after exposure to 2,4-disubstituted pyrimidines derivatives was compared with reference compounds tacrine and galanthamine (Table 3). At 40 μM, all the tested pyrimidine derivatives maintained efficient cell viability as measured by MTT reduction and were relatively nontoxic (Table 3). The N-benzyl derivative 7d that exhibited selective AChE inhibition (IC50 = 24.9 μM) and excellent anti-aggregation activity (59% inhibition) was nontoxic (98% cell viability). For comparison, the viability of SH-SY5Y cells was 89% in the presence of the reference compound tacrine and 97% in the presence of the reference compound galanthamine under the same experimental conditions. In addition, the C-2 oxidized compounds 7s (56% inhibition of Aβ-aggregation) and 7t (44% inhibition of Aβ-aggregation) were only slightly toxic under these conditions, with cell viabilities of 90% and 92% respectively. These studies indicate that the 2,4-disubstituted pyrimidines were nontoxic or only slightly toxic to SH-SY5Y cells and serve as a suitable template to develop ChEIs that exhibit activity against AChE-induced aggregation of Aβ.
Table 3.
MTT reduction cytotoxicity assay in SH-SY5Y neuroblastoma cells of 2,4-disubstituted pyrimidines (7a, 7c, 7d, 7g, 7s, 7t, 8c and 9c)
| Compd | Cell Viability at 40 μM ± SEM (%)a |
|---|---|
| 7a | 100.3 ± 16.4b |
| 7c | 97.8 ± 11.5 |
| 7d | 98.3 ± 10.5 |
| 7g | 102.3 ± 0.5c |
| 7s | 89.7 ± 11.5 |
| 7t | 91.8 ± 2.2 |
| 8c | 104.5 ± 4.6d |
| 9c | 89.3 ± 2.9 |
| Tacrine | 89.0 ± 6.3 |
| Galanthamine | 97.1 ± 12.9 |
The values are the mean of measurements performed in quadruplicates, SEM = standard error of mean. 100% cell viability represents the viability of SH-SY5Y cells measured in the absence of small molecules, as determined by MTT reduction. Statistical analysis using an independent two-tailed student's t-tests were performed using Origin 7.0 (Microcal Software, Inc., Northhampton, MA) to evaluate the statistical significance of the difference between the viability of control cells (measured in the absence of test molecules) and experimental mean values. A p-value of < 0.05 was defined as statistically significant,
p-value = 0.97
p-value = 0.30
p-value = 0.55
2.5 Molecular Modeling (Docking) Studies
Among the naphth-1-yl methyl series, the enzyme-ligand binding interactions of the most potent hAChE inhibitor 9a [N-(naphth-1-ylmethyl)-2-(pyrrolidin-1-yl)pyrimidin-4-amine; hAChE IC50 = 5.5 μM; equine serum BuChE IC50 = 8.9 μM, SI = 0.62] within the active site of hAChE was investigated by molecular modeling studies (Figure 1). These investigations indicate that the central pyrimidine ring was approximately located midway through the active site gorge (~ 6 Å away from the catalytic triad His447 residue at the bottom of the active site and ~ 7 Å away from the PAS Trp286). The ring was also, a) perpendicularly oriented between Trp86 and Phe297, b) ~ 5 Å away from the hydrophobic pocket (Gly120–122) and c) equidistantly stacked between Tyr124 and Tyr337. The latter feature allowed for two hydrogen bonding interactions between the tyrosine hydroxyl groups and the C-4 NH and two between the tyrosine hydroxyl groups and the pyrimidine N-3 (distances < 3.5 Å). The C-4 naphth-1-yl ring was tightly stacked between Tyr337 and Trp86 (distance ~ 3.5 Å) and was in close proximity to His447 (distances 3.5 – 4 Å). The small C-2 pyrrolidine substituent was oriented toward an aromatic region close to the PAS (distance to Trp286 is ~ 4.5 Å) and was stacked beneath Tyr341 (distance ~ 3.5 Å). It is noteworthy that although the catalytic site is relatively exposed in this binding pattern, the presence and location of the bulky C-4 naphth-1-yl ring most likely attribute to this derivative's potency considering the role that Trp86 plays in stabilizing the substrate ACh. This binding pattern is not seen in the related N-benzyl derivative 7a (AChE IC50 = 8.7 μM), which reenforces the unique dynamics offered by the naphth-1-yl methyl series of derivatives.
Figure 1.

Structures of ChEIs (1, 2 and 4) and inhibitors of Aβ-aggregation (3 and 5).
A similar docking experiment was conducted to investigate the binding interactions of the most potent and selective BuChE inhibitor 9e [2-(4-methylpiperidin-1-yl)-N-(naphth-1-ylmethyl)pyrimidin-4-amine hAChE IC50 = 25.8 μM; BuChE IC50 = 2.2 μM, SI = 11.73) within the active site of hBuChE (Figure 2). These studies indicate that the central pyrimidine ring was oriented a) much closer to the catalytic active site (distance to His438 < 3.5 Å), b) was close to the hydrophobic pocket (Gly115–117; distance ~ 5 Å) and c) was also perpendicularly stacked between Trp82 and Trp231. The C-4 naphth-1-yl methylamine group was oriented toward the acyl pocket (Leu286, Ser287 and Val288; distance ~ 4 Å) and the naphth-1-yl ring was oriented toward an aromatic cage comprised of Trp231, Phe398 and Phe329. The C-4 NH was in close proximity to the catalytic residues (Ser198 and His438) and was undergoing hydrogen bonding interactions (distance > 3 Å). The C-2 4-methylpiperidine substituent was oriented in an aromatic pocket comprised of Tyr440, Tyr332, Trp430 and Trp82. The shortest distance calculated between 9e and the gorge entry residue (Ala277) was ~ 10 Å, which reiterates the proximity of 9e to the buried active site.
Figure 2.

Docking of N-(naphthalen-1-ylmethyl)-2-(pyrrolidin-1-yl)pyrimidin-4-amine (9a, ball and stick) in the active site of hAChE. Red dotted lines represent hydrogen bonding. Hydrogen atoms are not shown for clarity.
Furthermore, the similar BuChE inhibitory profiles of 9e and its N-benzyl relative (7e, BuChE IC50 = 3.4; SI = 5.4) were investigated by superimposing the binding modes of both derivatives in hBuChE (Figure 3). The superimposition revealed similar binding orientations within the active site where the larger naphth-1-ylmethyl C-4 group in 9e occupies more room in the glycine hydrophobic pocket. Also, the C-2 methylpiperidine ring is planar to the pyrimidine ring in 7e but perpendicular to the pyrimidine ring in 9e. This is consistent with the fact that 9e (volume = 231.9 Å3, supporting information) has a bulkier C-4 (naphth-1-yl) substituent whereas 7e consists of a less bulky phenyl ring at C-4 explaining the superior BuChE inhibitory potency exhibited by 9e compared to 7e.
Figure 3.

Docking of 2-(4-methylpiperidin-1-yl)-N-(naphthalen-1-yl methyl)pyrimidin-4-amine (9e, ball and stick) in the active site of hBuChE. Red dotted lines represent hydrogen bonding. Hydrogen atoms are not shown for clarity.
3. Conclusions
The SAR data obtained on these novel class of 2,4-disubstituted pyrimidines indicates that (i) simple and efficient synthetic methods can be used to synthesis 2,4-disubstituted pyrimidines; (ii) the cholinesterase inhibition activity was sensitive to substituent steric and electronic properties at both C-2 and C-4 positions of the central pyrimidine ring; (iii) compound 9a [N-(naphth-1-ylmethyl)-2-(pyrrolidin-1-yl)pyrimidin-4-amine] was identified as the most potent AChEI (IC50 = 5.5 μM), whereas 9e [2-(4-methylpiperidin-1-yl)-N-(naphth-1-ylmethyl)pyrimidin-4-amine] was the most potent and selective BuChEI (IC50 = 2.2 μM, S.I = 11.73); (iv) 7d containing a C-4 phenyl and a C-2 methylpiperidine acts as an AChEI and inhibits AChE-induced Aβ-aggregation; (v) 2,4-disubstituted pyrimidines could potentially serve as a suitable ring template to develop ChEIs that possess anti-Aβ aggregation activity and thus could target multiple pathological routes in AD .
4. Experimental Section
4.1. General
Melting points were determined on a Fisher-Johns apparatus and are uncorrected. Infrared (IR) spectra were recorded as films on NaCl plates using a Perkin Elmer FT-IR spectrometer. 1H-NMR spectra were recorded on a Bruker Avance 300 MHz series spectrometer (CDCl3 as solvent). Coupling constants (J values) are in hertz (Hz). The following abbreviations are used for multiplicity of NMR signals: s = singlet, d = doublet, t = triplet, q = quartet, dd = double doublet, m = multiplet, br = broad. High-resolution electron ionization mass spectral (HREIMS) analysis was recorded using a JEOL HX110 double focusing mass spectrometer. Compound 7c was synthesized from a previous literature report.28 Combustion analysis was carried out by Midwest Microlab, LLC (Indianapolis, IN) and C, H, N of tested compounds 7a–t, 8a–f and 9a–e were within ± 0.4% of theoretical values for all elements listed indicating a purity of >95%. Silica gel column purification was performed using Merck 230–400 mesh silica gel 60. Compounds 7a–u, 8a–f and 9a–e showed single spot on thin-layer chromatography (TLC) performed on Merck 60F254 silica gel plates (0.2 mm) using three different solvent systems (9:1 EtOAc: MeOH; 3:1 EtOAc: hexanes and 1:3 DCM: EtOAc) and spots were visualized with UV254nm or iodine. All other solvents and reagents were obtained from various vendors (Acros Organics, Sigma-Aldrich and Alfa Aesar) with a minimum purity of 95% and were used without further purification.
4.2. General procedure for the synthesis of 4-substituted-2-chloropyrimidin-4-amines (7–9) 28
To a mixture of 2,4-dichloropyrimidine (6) (5.00 g, 33.60 mmol) and primary amines (33.60 mmol) in 65 mL of EtOH, kept at 0 °C (ice-bath), DIPEA (6.08 mL, 36.80 mmol) was added. The reaction was allowed to stir on the ice-bath for 5 minutes and was refluxed at 75–80 °C for 3 hours. After cooling to 25 °C, the EtOH was evaporated in vacuo and the residue was re-dissolved in a solvent mixture of EtOAc and dichloromethane (DCM) in ~ 3:1 ratio and successfully washed with a concentrated NaHCO3 and NaCl solution (1 × 15 mL). Aqueous layer was washed with EtOAc (3 × 15 mL) and the combined organic layer was dried over anhydrous MgSO4 and filtered. The organic layer is evaporated in vacuo and the resulting residue was further purified using either one of the following two methods: 1) Method A: Silica gel column chromatography using EtOAc: hexanes twice (3:1 and 1:3, respectively) to afford solid products (60–65%); 2) Method B: Differential melting point separation – The collected organic layers are evaporated in vacuo and the oily residue is washed with a solution of hexanes and ether (~ 3:1) to afford a precipitate that was dried on filter paper at 80–85 °C for ~ 2 hours to afford solid products. Some physical and spectroscopy data are provided below for 7, 8 and 9.
4.2.1. N-Benzyl-2-chloropyrimidin-4-amine(7)
The product was obtained as a white/light yellow solid after coupling with phenylmethaneamine (3.67 mL, 33.60 mmol). Method A - 4.78 g, 65%; Method B - 5.31 g, 72%. mp: 130–132 °C; IR (film, CH2Cl2): 3434 cm−1 (NH); 1H-NMR (300 MHz, CDCl3): δ 8.00 (d, J = 6.0 Hz, 1H), δ 7.28–7.34 (m, 5H), δ 6.20 (d, J = 6.0 Hz, 1H), δ 4.53 (br s, 2H). HREIMS Calcd for C11H10ClN3 (M+) m/z 219.6702, observed 219.0498. Anal. Calcd for : C11H10ClN3•0.32 H2O; C, 58.55; H, 4.43, N, 18.63. Found: C, 58.61; H, 4.76; N, 18.64
4.2.2. 2-Chloro-N-phenethylpyrimidin-4-amine(8)
The product was synthesized after coupling with 2-phenylethanamine (4.35 mL, 33.60 mmol). The residue is re-dissolved in a solvent mixture of EtOAc, DCM and MeOH in ~ 4:2:1 ratio. The resulting oily residue was further purified by silica gel column chromatography (Method A) to afford a white/off-white solid (4.70 g, 60%): mp: 75–77 °C. IR (film, CH2Cl2): 3433 cm−1 (NH); 1H-NMR (300 MHz, CDCl3): δ 7.98 (d, J = 6.0 Hz, 1H), δ 7.17–7.33 (m, 5H), δ 6.16 (d, J = 6.0 Hz, 1H), δ 3.60 (br s, 2H), δ 2.88–2.92 (m, 2H). HREIMS Calcd for C12H12ClN3 (M+) m/z 233.6968, observed 233.0606.
4.2.3. 2-Chloro-N-(naphth-1-ylmethyl)pyrimidin-4-amine(9)
The product was obtained after coupling with naphthalen-1-ylmethanamine (4.95 mL, 33.60 mmol) and was a light orange/brown solid (Method A - 5.43 g, 60%; Method B - 6.83 g, 75%): mp: 158–160 °C. IR (film, CH2CL2):3432 cm−1 (NH); 1H-NMR (300 MHz, CDCl3): δ 8.01 (d, J = 6.0 Hz, 1H), δ 7.82–7.96 (m, 3H), δ 7.40–7.56 (m, 4H), δ 6.23 (d, J = 6.0 Hz, 1H) δ 4.70 (br s, 2H). HREIMS Calcd for C15H12ClN3 (M+) m/z 269.7289, observed 269.0737. C17H18ClN3•0.17 H20; C, 66.79; H, 4.48, N, 15.58. Found: C, 66.04; H, 4.56; N, 15.40.
4.3. General procedure for the synthesis of 2,4-disubstituted-pyrimidin-4-amines (7a–r, 8a–f, 9a–e)28
To a solution of 7, 8 or 9 (0.74–0.91 mmol) in 3 mL of n-BuOH kept in a PV with stirring, cyclic amines (1.12 mmol) was added. The sealed PV was placed in an oil bath at 145–150 °C and stirred for 30–40 min. n-BuOH was evaporated in vacuo and the residue was re-dissolved in 3:1 EtOAc: DCM and washed successively with saturated NaHCO3 and NaCl solution (1 × 15 mL) respectively. The aqueous layer was washed with EtOAc (3 × 5 mL) and the organic layer was dried over anhydrous MgSO4 then filtered. The solution was evaporated in vacuo to afford either solid or semisolid product. Some physical and spectroscopy data are provided below for 7a–r, 8a–f, 9a–e
4.3.1. N-Benzyl-2-(pyrrolidin-1-yl)pyrimidin-4-amine (7a)
The product was obtained as a light brown solid after coupling 7 with pyrrolidine (0.15 g, 65%). mp: 103–105 °C. IR (film, CH2 Cl2 -2 2): 3435 cm−1 (NH); 1H-NMR (300 MHz, CDCl3): δ 7.87 (d, J = 6.0 Hz, 1H), δ 7.26–7.32 (m, 5H), δ 5.62 (d, J = 6.0 Hz, 1H), δ 4.86 (br s, 1H), δ 4.51 (d, J = 6.0 Hz, 2H), δ 3.49–3.53 (m, 4H), δ 1.90–1.94 (m, 4H). HREIMS Calcd for C15H18N4 (M+) m/z 254.3302, found 254.1964. Anal. Calcd for : C15H18N4•1H2O; C, 66.15; H, 7.40, N, 20.57. Found: C, 66.15; H, 7.40, N, 20.57.
4.3.2. N-Benzyl-2-morpholinopyrimidin-4-amine (7b)
The product was obtained after 7 coupling with morpholine (0.10 mL, 1.12 mmol) to afford an orange/light brown solid (0.20 g, 80%): mp: 93–95 °C. IR (film, CH2Cl2 ): 3434 cm−1(NH); 1H-NMR (300 MHz, CDCl3): δ 7.87 (d, J = 6.0 Hz, 1H), δ 7.29–7.31 (m, 5H), δ 5.69 (d, J = 6.0 Hz, 1H), δ 4.91 (br s, 1H), δ 4.50 (d, J = 6.0 Hz, 2H), δ 3.70–3.75 (m, 8H). HREIMS Calcd for C15H18N4O (M+) m/z 270.3296, found 270.1605. C15H18N4O; C, 66.64; H, 6.71, N, 20.73. Found: C, 66.45; H, 6.72; N, 20.46.
4.3.3. N-Benzyl-2-thiomorpholinopyrimidin-4-amine (7c)28
The product was obtained after 7 coupling with thiomorpholine (0.11 mL, 1.12 mmol) to afford an orange/light brown solid (0.20 g, 77%): mp: 85–87 °C. IR (film, CH2Cl2): 3258 cm1 (NH); 1H-NMR (300 MHz, CDCl3): δ 7.84 (d, J = 5.7 Hz, 1H), δ 7.24–7.33 (m, 5H), δ 5.66 (d, J = 5.7 Hz, 1H), δ 5.03 (br s, 1H), δ 4.47 (d, J = 5.6 Hz, 2H), δ 4.03–4.07 (m, 4H), δ 2.54–2.58 (m, 4H). HREIMS Calcd for C15H18N4S (M+) m/z 286.3952, found 286.1872. C15H18N4S; C, 62.91; H, 6.33, N, 19.56. Found: C, 62.62; H, 6.33; N, 19.31.
4.3.4. N-Benzyl-2-(4-methylpiperazin-1-yl)pyrimidin-4-amine (7d)
The product was obtained after coupling 7 with methylpiperazine (0.13 mL, 1.12 mmol)to afford a light yellow solid (0.22 g, 85%). mp: 150–153 °C. IR (film, CH2Cl2): 3454 cm−1 (NH); 1H-NMR (300 MHz, CDCl3): δ 7.86 (d, J = 6.0 Hz, 1H), δ 7.27–7.32 (m, 5H), δ 5.66 (d, J = 6.0 Hz, 1H), δ 4.87 (br s, 1H), δ 4.50 (d, J = 6.0 Hz, 2H), δ 3.77 (t, J = 6.0 Hz, 4H), δ 2.42 (t, J = 6.0 Hz, 4H), δ 2.31 (s, 3H). HREIMS Calcd for C16H21N5 (M+) m/z 283.3714, found 283.1804. C16H21N5•0.6 EtOAc; C, 65.74; H, 7.74, N, 20.83. Found: C, 65.64; H, 7.75; N, 20.67.
4.3.5. N-Benzyl-2-(4-methylpiperidin-1-yl)pyrimidin-4-amine (7e)
The product was obtained after coupling 7 with methylpiperidine (0.14 mL, 1.12 mmol)to afford a whitish pink solid (0.22 g, 85%). mp: 83–85 °C. IR (film, CH2Cl2): 3433 cm−1 (NH); 1H-NMR (300 MHz, CDCl3): δ 7.85 (d, J = 6.0 Hz, 1H), δ 7.28–7.32 (m, 5H), δ 5.61 (d, J = 6.0 Hz, 1H), δ 4.84 (br s, 1H), δ 4.63–4.67 (m, 2H), δ 4.50 (d, J = 6.0 Hz, 2H), δ 2.73–2.81 (m, 2H), δ 1.62–1.66 (m, 2H), δ 1.22–1.26 (m, 1H), δ 1.10–1.18 (m, 2H), δ 0.91 (d, J = 6.0 Hz, 3H). HREIMS Calcd for C17H22N4 (M+) m/z 282.3834, found 282.2376. C17H22N4; C, 72.31; H, 7.85, N, 19.84. Found: C, 72.18; H, 7.85; N, 19.60.
4.3.6. 1-[4-(4-(Benzylamino)pyrimidin-2-yl)piperazin-1-yl]ethanone (7f)
The product was obtained after coupling 7 with acetylpiperazine (0.15 g, 1.12 mmol) to afford a yellowish white solid (0.25 g, 86%). mp: 150–153 °C. IR (film, CH2Cl2): 3437 cm−1 (NH); 1H-NMR (300 MHz, CDCl3): δ 7.87 (d, J = 6.0 Hz, 1H), δ 7.28–7.31 (m, 5H), δ 5.70 (d, J = 6.0 Hz, 1H), δ 4.93 (br s, 1H), δ 4.49 (d, J = 6.0 Hz, 2H), δ 3.72–3.79 (m, 4H), δ 3.61–3.64 (m, 4H), δ 3.44–3.48 (m, 2H), δ 2.11 (s, 1H). HREIMS Calcd for C17H21N5O (M+) m/z 311.3815, found 311.1746. C17H21N5O•0.4 EtOAc; C, 64.46; H, 7.04, N, 20.21. Found: C, 64.43; H, 7.04; N, 20.15.
4.3.7. Tert-butyl 4-[4-(Benzylamino)pyrimidin-2-yl]piperazine-1-carboxylate (7g)
The product was obtained after coupling 7 with tert-buty1 piperazine-1-carboxylate (0.22 g, 1.12 mmol).Product was purified using a 3:1 EtoAc: hexanes silica gel chromatography to afford a light yellowish solid (0.31 g, 90%). mp: 115–117 °C. IR (film, CH2Cl2): 3438 cm−1 (NH); 1H-NMR (300 MHz, CDCl3): δ 7.80 (d, J = 6.0 Hz, 1H), δ 7.21–7.30 (m, 5H), δ 5.65 (d, J = 6.0 Hz, 1H), δ 4.91 (br s, 1H), δ 4.49 (d, J = 6.0 Hz, 2H), δ 3.66–3.69 (m, 4H), δ 3.37–3.40 (m, 4H), δ 1.43 (s, 9H). HREIMS Calcd for C20H27N5o (M+) m/z 369.4607, found 369.2163. C20H27N5O2; C, 65.02; H, 7.37, N, 18.96. Found: C, 65.54; H, 7.33; N, 18.76.
4.3.8. N-Benzyl-2-(4-cyclohexylpiperazin-1-yl)pyrimidin-4-amine(7h)
The product was obtained after coupling 7 with cyclohexylpiperazine (0.20 g, 1.12 mmol) and was purified using a 3:1 EtOAc: hexanes silica gel chromatography to afford an orange solid (0.21 g, 66%). mp: 60–62 °C. IR (film, CH2Cl2): 3435 cm−1 (NH); 1H-NMR (300 MHz, CDCl3): δ 7.82 (d, J = 6.0 Hz, 1H), δ 7.21–7.32 (m, 5H), δ 5.61 (d, J = 6.0 Hz, 1H), δ 5.07 (br s, 1H), δ 4.46 (d, J = 6.0 Hz, 2H), δ 3.72–3.75 (m, 4H), δ 2.54–2.57 (m, 4H), δ 2.25 (s, 1H), δ 1.86 (br s, 2H), δ 1.75 (br s, 2H), δ 1.58–1.61 (m, 1H), δ 1.13–1.19 (m, 5H). HREIMS Calcd for C21H29N5 (M+) m/z 351.4885, found = 351.2259. C21H29N5•0.5 DCM; C, 65.55; H, 7.68, N, 17.78. Found: C, 65.77; H, 7.70; N, 17.85.
4.3.9. N-Benzyl-2-(4-isopropylpiperazin-1-yl)pyrimidin-4-amine (7i)
The product was obtained after coupling 7 with isopropylpiperazine (0.17 mL, 1.12 mmol) and was purified using a 3:1 EtOAc: hexanes silica gel chromatography to afford a white solid (0.14 g, 50%). mp: 103–105 °C. IR (film, CH2Cl2): 3438 cm−1 (NH); 1H-NMR (300 MHz, CDCl3): δ 7.85 (d, J = 6.0 Hz, 1H), δ 7.24–7.34 (m, 5H), δ 5.63 (d, J = 6.0 Hz, 1H), δ 4.93 (br s, 1H), δ 4.48 (d, J = 6.0 Hz, 2H), δ 3.73–3.76 (m, 4H), δ 2.62–2.71 (m, 1H), δ 2.50–2.53 (m, 4H), δ 1.03 (d, J = 6.0 Hz, 6H). HREIMS Calcd for C18H25N5 (M+) m/z 311.4246, found 311.2552. C18H25N5; C, 69.42; H, 8.09, N, 22.49. Found: C, 69.69; H, 8.31; N, 22.15.
4.3.10. N-Benzyl-2-(4-isopropylpiperidin-1-yl)pyrimidin-4-amine (7j)
The product was obtained after coupling 7 with isopropylpiperidine (0.17 mL, 1.12 mmol) and was purified using 3:1 ether: hexanes silica gel chromatography to afford a yellow solid (0.16 g, 55%). mp: 58–60 °C. IR (film, CH2Cl2): 3438 cm−1 (NH); 1H-NMR (300 MHz, CDCl3): δ 7.86 (d, J = 6.0 Hz, 1H), δ 7.22–7.32 (m, 5H), δ 5.61 (d, J = 6.0 Hz, 1H), δ 4.84 (br s, 1H), δ 4.71–4.75 (m, 2H), δ 4.49 (d, J = 6.0 Hz, 2H), δ 2.66–2.74 (m, 2H), δ 1.65–1.69 (m, 2H), δ 1.38–1.48 (m, 1H), δ 1.06–1.21 (m, 5H), δ 0.85–0.88 (m, 6H). HREIMS Calcd for C19H26N4 (M+) m/z 310.4365, found 310.3086. C19H26N4; C, 73.51; H, 8.44, N, 18.05. Found: C, 73.51; H, 8.57; N, 17.91.
4.3.11. N-Benzyl-2-(4-propylpiperazin-1-yl)pyrimidin-4-amine (7k)
The product was obtained after coupling 7 with n-propylpiperazine (0.15 g, 1.12 mmol) and was purified using a 3:1 EtOAc: hexanes silica gel chromatography to afford a light orange solid (0.16 g, 55%). mp: 93–95 °C. IR (film, CH2Cl2 ): 3436 cm−1 (NH); 1H-NMR (300 MHz, CDCl3): δ 7.85 (d, J = 6.0 Hz, 1H), δ 7.21–7.30 (m, 5H), δ 5.64 (d, J = 6.0 Hz, 1H), δ 4.93 (br s, 1H), δ 4.48 (d, J = 6.0 Hz, 2H), δ 3.71–3.75 (m, 4H), δ 2.39–2.42 (m, 4H), δ 2.27–2.30 (m, 2H), δ 1.49–1.56 (m, 2H), δ 0.87–0.91 (m, 3H). HREIMS Calcd for C18H25N5 (M+) m/z 311.4246, observed 311.2008. C18H25N5•0.6 H2O; C, 67.09; H, 8.20, N, 21.73. Found: C, 67.11; H, 7.85; N, 21.55.
4.3.12. 2-[4-(4-(Benzylamino)pyrimidin-2-yl)piperazin-1-yl]ethanol (7l)
The product was obtained after coupling 7 with hydroxyethylpiperazine (0.14 mL, 1.12 mmol). Residue was re-dissolved in 1:1 EtOAc: DCM and was purified using a 3:1 EtOAc: hexanes silica gel chromatography to afford a brownish yellow solid (0.14 g, 50%). mp: 103–105 °C. IR (film, CH2CL2): 3439 cm−1 (NH); 1H-NMR (300 MHz, CDCl3): δ 7.84 (d, J = 6.0 Hz, 1H), δ 7.24–7.34 (m, 5H), δ 5.65 (d, J = 6.0 Hz, 1H), δ 5.01 (br s, 1H), δ 4.48 (d, J = 6.0 Hz, 2H), δ 3.73–3.76 (m, 4H), δ 3.60–3.64 (m, 2H), δ 2.94 (s, 1H), δ 2.51–2.55 (m, 2H), δ 2.47–2.50 (m, 4H). HREIMS Calcd for C17H23N5O (M+) m/z 313.3974, found 313.1637. C17H23N5O•0.3 DCM; C, 60.20; H, 6.78, N, 20.65. Found: C, 60.03; H, 6.89; N, 20.10.
4.3.13. N-Benzyl-2-[4-(2-methoxyethyl)piperazin-1-yl]pyrimidin-4-amine (7m)
The product was obtained after coupling 7 with methoxyethylpiperazine (0.17 mL, 1.12 mmol) to afford an orange semi-solid (0.17 g, 57%). mp: 63–65 °C. IR (film, CH2CL2): 3433 cm−1 (NH); 1H-NMR (300 MHz, CDCl3): δ 7.82 (d, J = 6.0 Hz, 1H), δ 7.21–7.31 (m, 5H), δ 5.62 (d, J = 6.0 Hz, 1H), δ 5.06 (br s, 1H), δ 4.46 (d, J = 6.0 Hz, 2H), δ 3.74–3.77 (m, 4H), δ 3.48–3.52 (m, 2H), δ 3.32 (s, 3H), δ 2.54–2.58 (m, 2H), δ 2.45–2.48 (m, 4H). HREIMS Calcd for C18H25N5O (M+) m/z 327.4240, found 327.2040.
4.3.14. N-Benzyl-2-[4-(4-chlorobenzyl)piperazin-1-yl]pyrimidin-4-amine (7n)
The product was obtained after coupling 7 with 4-chlorobenzylpiperazine (0.22 mL, 1.12 mmol). The sample was purified using a 9:1 EtOAc:DCM silica gel column to afford a yellowish solid (0.20 g, 55%). mp: 88–90 °C. IR (film, CH2Cl2): 3437 cm−1 (NH); 1H-NMR (300 MHz, CDCl3) δ 7.85 (d, J = 6.0 Hz, 1H), δ 7.24–7.30 (m, 9H), δ 5.65 (d, J = 6.0 Hz, 1H), δ 4.87 (br s, 1H), δ 4.48 (d, J = 6.0 Hz, 2H), δ 3.73–3.76 (m, 4H), δ 3.47 (s, 2H), δ 2.41–2.44 (m, 4H). HREIMS Calcd for C22H24ClN5 (M+) m/z 393.9125, found 393.2443. C22H24ClN5; C, 67.08; H, 6.14, N, 17.78. Found: C, 67.44; H, 6.13; N, 17.68.
4.3.15. N-Benzyl-2-[4-(4-bromobenzyl)piperazin-1-yl]pyrimidin-4-amine (7o)
The product was obtained after coupling 7 with 4-bromobenzylpiperazine (0.29 g, 1.12 mmol) to afford a yellowish solid (0.20 g, 50%). mp: 90–93 °C. IR (film, CH2Cl2): 3434 cm−1 (NH); 1H-NMR (300 MHz, CDCl3) δ 7.85 (d, J = 6.0 Hz, 1H), δ 7.41–7.44 (m, 2H), δ 7.24–7.34 (m, 5H), δ 7.19–7.22 (m, 2H), δ 5.65 (d, J = 6.0 Hz, 1H), δ 4.87 (br s, 1H), δ 4.48 (d, J = 6.0 Hz, 2H), δ 3.73–3.76 (m, 4H), δ 3.45 (s, 2H), δ 2.41–2.44 (m, 4H). HREIMS Calcd C22H24BrN5 (M+) m/z 438.3635, found 438.2430. C22H24BrN5•0.2 EtOAc; C, 60.06; H, 5.66, N, 15.36. Found: C, 60.07; H, 5.65; N, 15.39.
4.3.16. N-Benzyl-2-[4-(4-fluorobenzyl)piperazin-1-yl]pyrimidin-4-amine (7p)
The product was obtained after coupling 7 with 4-fluorobenzylpiperazine (0.22 g, 1.12 mmol) to afford an orange semi-solid (0.22 g, 65%). IR (film, CH2Cl2): 3439 cm−1 (NH); 1HNMR (300 MHz, CDCl3): δ 7.84 (d, J = 6.0 Hz, 1H), δ 7.26–7.29 (m, 7H), δ 6.96–7.02 (m, 3H), δ 5.63 (d, J = 6.0 Hz, 1H), δ 5.17 (br s, 1H), δ 4.47 (d, J = 6.0 Hz, 2H), δ 3.74–3.76 (m, 4H), δ 3.46 (s, 2H), δ 2.40 (t, J = 6.0 Hz, 4H). HREIMS Calcd for C22H24FN5 (M+) m/z 377.4579, found 377.1980. C22H24FN5•0.5 DCM; C, 64.36; H, 6.00, N, 16.68. Found: C, 63.95; H, 5.97; N, 16.54.
4.3.17. N-Benzyl-2-[4-(4-trifluoromethylbenzyl)piperazin-1-yl]pyrimidin-4-amine (7q)
The product was obtained after coupling 7 with 4-trifluoromethybenzylpiperazine (0.24 mL, 1.12 mmol) to afford a yellowish orange solid (0.25 g, 64%). mp: 88–90 °C. IR (film, CH2Cl2): 3437 cm−1 (NH). 1H-NMR (300 MHz, CDCl3): δ 7.85 (d, J = 6.0 Hz, 1H), δ 7.55–7.58 (m, 2H), δ 7.44–7.47 (m, 2H), δ 7.24–7.34 (m, 5H), δ 5.65 (d, J = 6.0 Hz, 1H), δ 5.00 (br s, 1H), δ 4.48 (d, J = 6.0 Hz, 2H), δ 3.75–3.78 (m, 4H), δ 3.55 (s, 2H), δ 2.43–2.46 (m, 4H). HREIMS Calcd C23H24F3N5 (M+) m/z 427.4654, found 427.2203. C23H24F3N5; C, 64.62; H, 5.66, N, 16.38. Found: C, 64.39; H, 5.64; N, 16.12.
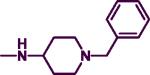
4.3.18. N4-Benzyl-N2-(1-benzylpiperidin-4-yl)pyrimidine-2,4-diamine (7r)
The product was obtained after coupling 7 with 4-amino-1-benzylpiperidine (0.23 mL, 1.12 mmol) to afford a dark orange solid (0.20 g, 59%). mp: 88–90 °C. IR (film, CH2Cl2): 3438 cm−1 (NH);. 1H-NMR (300 MHz, CDCl3): δ 7.78 (d, J = 6.0 Hz, 1H), δ 7.21–7.34 (m, 10H), δ 5.66 (d, J = 6.0 Hz, 1H), δ 4.92 (br s, 1H), δ 4.46 (d, J = 6.0 Hz, 2H), δ 3.76–3.82 (m, 1H), δ 3.48 (s, 2H), δ 2.76–2.80 (m, 2H), 2.09–2.16 (m, 2H), 1.95–1.99 (m, 3H), 1.42–1.55 (m, 2H). HREIMS Calcd C23H27N5 (M+) m/z 373.4940, found 373.2008. C23H27N5•DCM; C, 62.88; H, 6.38, N, 15.28. Found: C, 62.88; H, 6.38; N, 15.28.
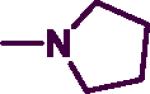
4.3.19. N-Phenethyl-2-(pyrrolidin-1-yl)pyrimidin-4-amines (8a)
The product was obtained by coupling 8 with pyrrolidine to afford a light yellowish brown solid (0.15 g, 65%). mp: 85–87 °C. IR (film, CH2Cl2): 3436 cm−1 (NH); 1H-NMR (300 MHz, CDCl3): δ 7.86 (d, J = 6.0 Hz, 1H), δ 7.18–7.32 (m, 5H), δ 5.59 (d, J = 6.0 Hz, 1H), δ 4.56 (br s, 1H), δ 3.55 (t, J = 6.0 Hz, 2H), δ 3.50 (t, J = 6.0 Hz, 4H), δ 2.86–2.91 (m, 2H), δ 1.90–1.93 (m, 4H). HREIMS C16H20N4 (M+) m/z 268.3568, found 268.2125. C16H20N4•0.5 H2O; C, 69.29; H, 7.63, N, 20.2. Found: C, 69.51; H, 7.32; N, 20.11.
4.3.20. 2-Morpholino-N-phenethylpyrimidin-4-amine (8b)
The product was obtained after coupling 8 with morpholine (0.10 mL, 1.11 mmol) to afford a light brown solid (0.21 g, 86%). mp: 93–95 °C. IR (film, CH2Cl): 3433 cm−1 (NH); 1HNMR (300 MHz, CDCl3): δ 7.85 (d, J = 6.0 Hz, 1H), δ 7.18–7.32 (m, 5H), δ 5.65 (d, J = 6.0 Hz, 1H), δ 4.59 (br s, 1H), δ 3.68–3.75 (m, 8H), δ 3.55–3.59 (m, 2H), δ 2.86–2.90 (m, 2H). HREIMS Calcd C16H20N4O (M+) m/z 284.3562, found 284.1725. C16H20N4O; C, 67.58; H, 7.09, N, 19.70. Found: C, 67.71; H, 7.14; N, 19.42.
4.3.21. N-Phenethyl-2-thiomorpholinopyrimidin-4-amine (8c)
The product was obtained after coupling 8 with thiomorpholine (0.11 mL, 1.11 mmol) to afford a brown solid (0.21 g, 81%). mp: 60–62 °C. IR (film, CH2Cl2 ): 3437 cm−1 (NH); 1H-NMR (300 MHz, CDCl3) δ 7.84 (d, J = 6.0 Hz, 1H), δ 7.18–7.30 (m, 5H), δ 5.61 (d, J = 6.0 Hz, 1H), δ 4.58 (br s, 1H), δ 3.98 (t, J = 6.0 Hz, 4H), δ 3.54 (t, J = 6.0 Hz, 2H), δ 2.86–2.90 (m, 2H), δ 2.61 (t, J = 6.0 Hz, 4H). HREIMS Calcd C16H20N4 (M+) m/z 284.3562, found 284.1725. C16H20N4S; C, 63.97; H, 6.71, N, 18.65. Found: C, 64.12; H, 6.85; N, 18.46.
4.3.22. 2-(4-Methylpiperazin-1-yl)-N-phenethylpyrimidin-4-amine (8d)
The product was obtained after coupling 8 with methylpiperazine (0.12 mL, 1.11 mmol) to afford a light yellow semi-solid (0.18 g, 69%). mp: 58–60 °C IR (film, CH2CL2): 3436 cm−1(NH); 1H-NMR (300 MHz, CDCl3): δ 7.85 (d, J = 6.0 Hz, 1H), δ 7.18–7.30 (m, 5H), δ 5.62 (d, J = 6.0 Hz, 1H), δ 4.57 (br s, 1H), δ 3.76–3.79 (m, 4H), δ 3.52–3.56 (m, 2H), δ 2.86–2.90 (m, 2H), δ 2.41–2.44 (m, 4H), δ 2.31 (s, 3H). HREIMS Calcd C17H23N5 (M+) m/z 297.3980, found 297.1958. Anal. Calcd for : C17H23N5•13 H2O; C, 68.05; H, 7.67, N, 23.35. Found: C, 68.12; H, 7.82; N, 23.36
4.3.23. 2-(4-Methylpiperidin-1-yl)-N-phenethylpyrimidin-4-amine (8e)
The product was obtained after coupling 8 with 4-methylpiperidine (0.13 mL, 1.11 mmol) to afford a light brown solid (0.20 g, 79%). mp: 65–67 °C. IR (film, CH2CL2): 3439 cm−1(NH); 1H-NMR (300 MHz, CDCl3): δ 7.84 (d, J = 6.0 Hz, 1H), δ 7.18–7.33 (m, 5H), δ 5.57 (d, J = 6.0 Hz, 1H), δ 4.64–4.69 (m, 2H), δ 4.53 (br s, 1H), δ 3.52–3.56 (m, 2H), δ 2.86–2.90 (m, 2H), δ 2.73–2.82 (m, 2H), δ 1.63–1.68 (m, 3H), δ 1.33–1.40 (m, 1H), δ 1.08–1.16 (m, 3H), δ 0.92 (d, J = 6.0 Hz, 3H). HREIMS Calcd C18H24N4 (M+) m/z 296.4100, found 296.2380. C18H24N4; C, 72.94; H, 8.16, N, 18.90. Found: C, 72.38; H, 7.99; N, 18.34.
4.3.24. 1-[4-(4-(Phenethylamino)pyrimidin-2-yl)piperazin-1-yl]ethanone (8f)
The product was obtained after coupling 8 with acetylpiperazine (0.14 g, 1.11 mmol) to afford a yellow solid (0.24 g, 86%). mp: 150–152 °C. IR (film, CH2Cl2): 3437 cm−1 (NH); 1HNMR (300 MHz, CDCl3): δ 7.85 (d, J = 6.0 Hz, 1H), δ 7.19–7.33 (m, 5H), δ 5.66 (d, J = 6.0 Hz, 1H), δ 4.61 (br s, 1H), δ 3.73–3.80 (m, 4H), δ 3.65 (t, J = 6.0 Hz, 2H), δ 3.55 (t, J = 6.0 Hz, 2H), δ 3.46–3.50 (m, 2H), δ 2.86–2.90 (m, 2H), δ 2.12 (s, 3H). HREIMS Calcd C18H23N5O (M+) m/z 325.4081, found 325.1913. C18H23N5•0.6 EtOAc; C, 64.78; H, 7.41, N, 18.52. Found: C, 64.66; H, 7.43; N, 18.30.
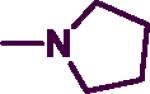
4.3.25. N-(Naphth-1-ylmethyl)-2-(pyrrolidine)pyrimidin-4-amines (9a)
The product was obtained after coupling 9 with pyrrolidine to afford a light brown solid (0.16 g, 70%). mp: 105–107 °C. IR (film, CH2CL2): 3433 cm−1 (NH); 1H-NMR (300 MHz, CDCl3) δ 8.05 (d, J = 6.0 Hz, 1H), δ 7.78–7.91 (m, 3H), δ 7.38–7.51 (m, 4H), δ 5.64 (d, J = 6.0 Hz, 1H), δ 4.96 (d, J = 6.0 Hz, 2H), δ 4.79 (br s, 1H), δ 3.53–3.57 (m, 4H), δ 1.91–1.95 (m, 4H). HREIMS Calcd C19H20N4 (M+) m/z 304.3889, found 304.2086. C19H20N4•0.2 H2O; C, 74.02; H, 6.49, N, 18.18. Found: C, 74.09; C, 6.68; N, 18.19.
4.3.26. 2-Morpholino-N-(naphth-1-ylmethyl)pyrimidin-4-amine (9b)
The product was obtained after coupling 9 with morpholine (0.08 mL, 0.96 mmol) to afford a light yellow solid (0.18 g, 75%). mp: 170–172 °C. IR (film, CH2CL2): 3437 cm−1 (NH); 1H-NMR (300 MHz, CDCl3): δ 8.01 (d, J = 6.0 Hz, 1H), δ 7.79–7.90 (m, 3H), δ 7.39–7.52 (m, 4H), δ 5.71 (d, J = 6.0 Hz, 1H), δ 4.92 (d, J = 6.0 Hz, 2H), δ 4.77 (br s, 1H), δ 3.69–3.77 (m, 8H). HREIMS Calcd C19H20N4O (M+) m/z 320.3883, found 320.1825. C19H20N4O; C, 71.23; H, 6.29, N, 17.49. Found: C, 71.28; H, 6.37; N, 17.08.
4.3.27. N-(Naphth-1-ylmethyl)-2-thiomorpholinopyrimidin-4-amine (9c)
The product was obtained after coupling 9 thiomorpholine (0.10 mL, 0.96 mmol) to afford a yellowish brown solid (0.19 g, 76%). mp: 105–107 °C. IR (film, CH2Cl2 ): 3439 cm−1 (NH); 1H-NMR (300 MHz, CDCl3): δ 8.01 (d, J = 6.0 Hz, 1H), δ 7.73–7.87 (m, 3H), δ 7.39–7.51 (m, 4H), δ 5.68 (d, J = 6.0 Hz, 1H), δ 4.93 (d, J = 6.0 Hz, 2H), δ 4.82 (br s, 1H), δ 4.05–4.12 (m, 4H), δ 2.59–2.65 (m, 4H). HREIMS Calcd C19H20N4S (M+) m/z 336.4539, found 336.2171. C19H20N4S; C, 67.83; H, 5.99, N, 16.65. Found: C, 67.78; H, 5.86; N, 16.50.
4.3.28. 2-(4-Methylpiperazin-1-yl)-N-(naphth-1-ylmethyl)pyrimidin-4-amine (9d)
The product was obtained after coupling 9 with methylpiperazine (0.11 mL, 0.96 mmol) to afford a yellow solid (0.23 g, 80%). mp: 118–120 °C. IR (film, CH2CL2): 3436 cm−1 (NH); 1HNMR (300 MHz, CDCl3): δ 8.03 (d, J = 6.0 Hz, 1H), δ 7.78–7.90 (m, 3H), δ 7.39–7.52 (m, 4H), δ 5.67 (d, J = 6.0 Hz, 1H), δ 4.94 (d, J = 6.0 Hz, 2H), δ 4.80 (br s, 1H), δ 3.81–3.85 (m, 4H), δ 2.41–2.45 (m, 4H), δ 2.32 (s, 3H). HREIMS Calcd C20H23N5 (M+) m/z 333.4301, found 333.1961. C20H23N5•0.5 EtOAc; C, 70.01; H, 7.21, N, 18.55. Found: C, 69.72; H, 7.25; N, 18.21.
4.3.29. 2-(4-Methylpiperidin-1-yl)-N-(naphth-1-ylmethyl)pyrimidin-4-amine (9e)
The product was obtained after coupling 9 with 4-methylpiperidine (0.11 mL, 0.96 mmol). The residue was purified using 3:1 ether: hexanes column to afford an off-white/light yellow semi-solid (0.14 g, 55%). IR (film, CH2CL2): 3439 cm−1 (NH); 1H-NMR (300 MHz, CDCl3): δ 8.03 (d, J = 6.0 Hz, 1H), δ 7.78–7.91 (m, 3H), δ 7.38–7.53 (m, 4H), δ 5.62 (d, J = 6.0 Hz, 1H), δ 4.94 (d, J = 6.0 Hz, 2H), δ 4.75 (br s, 1H), δ 4.68–4.72 (m, 3H), δ 2.76–2.84 (m, 2H), δ 1.64–1.68 (m, 2H), 1.57–1.60 (m, 1H), 1.10–1.14 (m, 4H), 0.92 (d, J = 6.0 Hz, 3H). HREIMS Calcd C21H24N4 (M+) m/z 332.4421, found 332.2246. C21H24N4•0.5 EtOAc; C, 73.38; H, 7.50, N, 14.88. Found: C, 73.07; H, 7.52; N, 14.64.
4.4. Preparation of 4-[4-(benzylamino)pyrimidin-2-yl]thiomorpholine-1-oxide (7s)
To a mixture of N-benzyl-2-thiomorpholinopyrimidin-4-amine (7c) (0.20 g, 0.70 mmol) in 4 mL of 1,4-dioxane, kept at 0 °C (ice-bath), mCPBA (0.19 g, 1.12 mmol) was added dropwise. The reaction is allowed to stir on the ice-bath for 5 minutes and then was kept at r.t for 3 hours. DCM was added to the mixture to aid in the 1,4-dioxane in vacuo evaporation. The residue was re-dissolved in a solvent mixture of EtOAc and DCM in ~ 3:1 ratio and successfully washed with a concentrated NaHCO3 and NaCl solution (1 × 15 mL). Aqueous layer was washed with EtOAc (3 × 15 mL) and the combined organic layer was dried over anhydrous MgSO4 and filtered. The organic layer was evaporated in vacuo and the resulting solid residue was further purified by silica gel column chromatography using 3:1 EtOAc: hexanes to afford a light yellow solid (0.16 g, 75%). mp: 78–80 °C. IR (film, CH2Cl2): 3439 cm−1 (NH); 1H-NMR (300 MHz, CDCl3): δ 7.87 (d, J = 6.0 Hz, 1H), δ 7.24–7.35 (m, 5H), δ 5.74 (d, J = 6.0 Hz, 1H), δ 4.99 (br s, 1H), δ 4.49 (d, J = 6.0 Hz, 2H), δ 4.37 (t, J = 6.0 Hz, 1H), δ 4.32 (t, J = 6.0 Hz, 1H), 4.10–4.19 (m, 2H), 2.69–2.73 (m, 4H). HREIMS Calcd C15H18N4OS (M+) m/z 302.3946, found 302.1259. C15H18N4OS•0.3 DCM; C, 56.05; H, 5.72, N, 17.09. Found: C, 56.38; H, 5.74; N, 17.22.
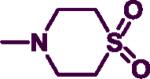
4.5. Preparation of 4-[4-(benzylamino)pyrimidin-2-yl]thiomorpholine-1,1-dioxide(7t)
To a mixture of N-benzyl-2-thiomorpholinopyrimidin-4-amine (7c) (0.20 g, 0.70 mmol) in 5 mL of MeOH, kept at 0 °C (ice-bath), potassium peroxymonosulphate (0.23 g, 1.54 mmol) in H2O was added dropwise. The reaction was allowed to stir on the ice-bath for 5 minutes and then was kept at 70–75 °C for 1 hour. 4 mL of 1,4-dioxane was added to the mixture and the reaction vessel was moved to r.t for 4 hours. MeOH and 1,4-dioxane were evaporated in vacuo and the residue was re-dissolved in 3:1 EtOAc: DCM and successfully washed with a concentrated NaHCO3 and NaCl solution (1 × 15 mL). Aqueous layer was washed with EtOAc (3 × 15 mL) and the combined organic layer was dried over anhydrous MgSO4 and filtered. The organic layer was evaporated in vacuo and the resulting solid residue was further purified by silica gel column chromatography using 3:1 EtOAc: hexanes to afford a light orange solid (0.17 g, 75%). Mp: 65–67 °C. IR (film, CH2Cl2): 3464 cm−1 (NH); 1H-NMR (300 MHz, CDCl3): δ 7.87 (d, J = 6.0 Hz, 1H), δ 7.25–7.35 (m, 5H), δ 5.79 (d, J = 6.0 Hz, 1H), δ 5.14 (br s, 1H), δ 4.49 (d, J = 6.0 Hz, 2H), δ 4.21–4.25 (m, 4H), δ 2.88–2.92 (m, 4H), 4.10–4.19 (m, 2H), 2.69–2.73 (m, 4H). HREIMS Calcd C15H10N4O2S (M+) m/z 318.3946, found 318.1310. C15H18N4OS2•0.2 DCM; C, 51.95; H, 5.28, N, 15.94. Found: C, 52.04; H, 5.34; N, 15.71.
4.6. Preparation of N-benzyl-2-(piperazin-1-yl)pyrimidin-4-amine (7u)
To a mixture of tert-butyl 4-(4-(benzyl amino)pyrimidin-2-yl)piperazine-1-carboxylate (7g) (0.20 g, 0.54 mmol) in 4 mL of DCM, kept at 0 °C (ice-bath), TFA (4 mL, 53.83 mmol) was added dropwise. The reaction was allowed to stir on the ice-bath for 5 minutes and then was kept at r.t for 2 hours. DCM is evaporated in vacuo and the residue was re-dissolved in 3:1 EtOAc: DCM and successfully washed with a concentrated NaHCO3 and NaCl solution (1 × 15 mL). Aqueous layer was washed with EtOAc (3 × 15 mL) and the combined organic layer was dried over anhydrous MgSO4 and filtered. The organic layer was evaporated in vacuo to afford a light yellow solid (0.11 g, 75%). mp: 70–72 °C. IR (film, CH2Cl2): 3438 cm−1 (NH); 1H-NMR (300 MHz, CDCl3): δ 7.86 (d, J = 6.0 Hz, 1H), δ 7.24–7.34 (m, 5H), δ 5.66 (d, J = 6.0 Hz, 1H), δ 4.91 (br s, 1H), δ 4.49 (d, J = 6.0 Hz, 2H), δ 3.73–3.76 (m, 4H), 3.09 (s, 1H), δ 2.88–2.91 (m, 4H). HREIMS Calcd C15H19N (M+ m/z 269.3449, found 269.1953.
4.7. Cholinesterase Inhibition Assay
The ability of the test compounds (7a–u, 8a–f and 9a–e) to inhibit human AChE (product number C3389, Sigma-Aldrich, St. Louis, MO) and equine serum BuChE (product number C1057, Sigma-Aldrich, St. Louis, MO) was determined using Ellman's method (IC50 values, μM) using appropriate reference agents tacrine hydrochloride (item number 70240, Cayman Chemical, Ann Arbor, MI), bis(7)-tacrine (item number 10005836, Cayman Chemical, Ann Arbor, MI), and galanthamine hydrobromide (product number G1660, Sigma, St. Louis, MO). Stock solutions of test compounds were dissolved in a minimum volume of DMSO (1%) and were diluted using the buffer solution (50 mM Tris–HCl, pH 8.0, 0.1 M NaCl, 0.02 M MgCl2.6H2O). In 96-well plates, 160 μL 5,5'-dithiobis(2-nitrobenzoic acid) (1.5 mM DTNB), 50 μLof AChE (0.22 U/mL prepared in 50 mM Tris-HCl, pH 8.0, 0.1% w/v bovine serum albumin, BSA) or 50 μL of BuChE (0.06 U/mL prepared in 50 mM Tris–HCl, pH 8.0, 0.1% w/v BSA) were incubated with 10 μL of various concentrations of test compounds (0.001–100 μM) at room temperature for 5 min followed by the addition of the substrates (30 μL) acetylthiocholine iodide (15 mM ATCl) or S-butyrylthiocholine iodide (15 mM BTCI) and the absorbance was measured at different time intervals (0, 60, 120 and 180 s) at a wavelength of 405 nm BioTek ELx800 microplate reader. Percent inhibition was calculated by the comparison of compound treated to various control incubations that included 1% DMSO. The concentration of the test compound causing 50% inhibition (IC50, μM) was calculated from the concentration-inhibition response curve on logarithmic scale (duplicate to quadruplicate determinations).
4.8. hAChE-induced Aβ Aggregation Inhibition Studies
Thioflavin T (ThT) is a benzothiazole salt frequently used in the detection of amyloid plaque formation. As Aβ peptides start to aggregate into oligomers and fibrils, ThT binds to the beta sheets formed as a result of the aggregation and the detectable change in its emission spectrum is used to quantify the degree of aggregation.32–34 Aβ1–40 was purchased from GL Biochem Ltd. Human recombinant AChE lyophilized powder was purchased from Sigma Aldrich (product number C1682) and ThT was purchased from Fisher Scientific. Aβ1–40 was prepared as previously described.32 Lyophilized Aβ1–40 was dissolved in DMSO to obtain a 2 mM solution and then diluted into 500 μM solution with 0.215 M sodium phosphate buffer (pH 8.0). Aliquots (2 μL) of Aβ1–40 were incubated with 16 μL of hAChE, which was dissolved in 0.215 M sodium phosphate buffer (pH 8.0), to give a final concentration of 50 μM of Aβ1–40 and 230 μM of hAChE. For co-incubation experiments, 2 μL of the test compounds in 0.215 M sodium phosphate buffer pH 8.0 solution (final concentration 100 μM) were added into aliquots (2 μL) of Aβ1–40 (final concentration 50 μM) along with 16 μL of hAChE (final concentration 230 μM). The reaction mixtures were incubated at room temperature for 24 h and 100 μL of ThT (20 μM) in 50 mM glycine-NaOH buffer (pH 8.5) was added. Fluorescence was monitored at 442 nm and emission at 490 nm using a Proton Technology International (PTI, Birmingham, NJ) spectrofluorometer. Each assay was run in triplicates along with donepezil (item number 13245, Caymen Chemical, Ann Arbor, MI) and propidium iodide (product number C4170, Sigma-Aldrich, St. Louis, MO) as reference agents. The fluorescence intensities in the presence and absence of inhibitors were compared using appropriate controls containing 1% DMSO and the percentage of inhibition was calculated using the equation: 100-(IFi/IFo×100) where IFi and IFo are the fluorescence intensities obtained for Aβ1–40 + hAChE in the presence and absence of inhibitor, respectively.
4.9. SH-SY5Y Neuroblastoma Cell Toxicity Studies
The cellular reduction of 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide, MTT (Product No: 30-1010K, from American Type Culture Collection, Manassas, VA) was measured by detecting a purple formazan intermediate at 570 nm. By incubating the cells with varying concentrations of test compounds, their toxicity attributes can be determined directly by determining the % reduction of MTT or indirectly by reporting the % viability of the cell line used.35,36 The SH-SY5Y neuroblastoma cells were plated at a density of 50,000 cells per well in 100 μ-L of complete media consisting of a 1:1 mixture of Eagle's Minimum Essential Medium (EMEM) and Ham's F12, supplemented with 10% Fetal Bovine Serum (FBS). The cells were incubated overnight before treatment with 100 μL of test sample solutions and select controls (tacrine hydrochloride, item number 70240, Caymen Chemical, Ann Arbor, MI and galanthamine hydrobromide, product number G1660 Sigma-Aldrich, St. Louis, MO) at various concentrations (0–160 μM) in 2% DMSO for 24 hours at 37 °C (n = 4). A 20 μL of the MTT reagent solution was added and the cells were incubated for an additional 3 h. The cells were subsequently solubilized with 80 μL of MTT reagent solution and incubated at room temperature overnight. Cell viability was determined by measuring the absorbance at 570 nm using a Molecular Devices Spectramax 190 microplate reader. All results were expressed as a percent reduction of MTT relative to untreated controls that included 2% DMSO (defined as 100% viability) and the average absorbance value for each treatment was subtracted with the absorbance reading of wells containing only media, MTT reagent, and detergent reagent.
5.0. Molecular Modeling (docking) studies
Docking experiments were performed using Discovery Studio Client v2.5.0.9164 (2005–09), Accelrys Software Inc. running on a HP xw4600 workstation (Processor x86 family 6 model 23 stepping 10 GenuineIntel 2999 ~Mhz). The coordinates for the X-ray crystal structure of the enzyme hAChE and hBuChE were obtained from the RCSB Protein Data Bank and hydrogens were added. The ligand molecules were constructed using the Build Fragment tool and energy minimized for 1000 iterations reaching a convergence of 0.01 kcal/mol Å. The docking experiment on hAChE was carried out by superimposing the energy minimized ligand in the PDB file 1B41 after which fasciculin was deleted. The coordinates for hBuChE was obtained from PDB file 1P0I and the energy minimized ligand was superimposed and the resulting ligand–enzyme complex was subjected to docking using the Libdock command in the receptor–ligand interactions protocol of Discovery Studio after defining subsets of the enzyme within 10 Å sphere radius of the ligand. The force field, Chemistry at HARvard Macromolecular Mechanics (CHARMM) was employed for all docking purposes. The ligand–enzyme assembly was then subjected to a molecular dynamics (MD) simulation using Simulation protocol at a constant temperature of 300 K with a 100 step equilibration for over 1000 iterations and a time step of 1 fs using a distance dependent dielectric constant 4r. The optimal binding orientation of the ligand–enzyme assembly obtained a013;fter docking was further minimized for 1000 iterations using the conjugate gradient method until a convergence of 0.001 kcal/mol Å was reached after which Eintermolecular (kcal/mol) of the ligand–enzyme assembly was evaluated.
Supplementary Material
Figure 4.

Superimposition of the binding modes of N-benzyl-2-(4-methylpiperidin-1-yl)pyrimidin-4-amine (7e, red) and 2-(4-methylpiperidin-1-yl)-N-(naphthalene-1-ylmethyl)pyrimidin-4-amine (9e, blue) within the active site of hBuChE.
Acknowledgments
The authors would like to thank the Department of Biology and the School of Pharmacy at the University of Waterloo for financial support of this research project. This work was also partially supported by the Alzheimer's Association (NIRG-08-91651) and the Alzheimer's Disease Research Center (NIH 3P50 AG005131). The NSF is also acknowledged for a CAREER Award to JY (CHE-0847530).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supporting Information Available A table of microanalytical data is presented for representative compounds along with their molecular volume (Å3).
References and notes
- 1.Holden M, Kelly C. Adv. Psychiatr. Treat. 2002;8:89. [Google Scholar]
- 2.Klafki H, Staufenbiel M, Kornhuber J, Wiltfang J. Brain. 2006;129:2840. doi: 10.1093/brain/awl280. [DOI] [PubMed] [Google Scholar]
- 3.Edwards PD, Albert JS, Sylvester M, Aharony D, Andisik D, Callaghan O, Campbell JB, Carr RA, Chessari G, Congreve M, Frederickson M, Folmer RHA, Geschwindner S, Koether G, Kolmodin K, Krumrine J, Mauger RC, Murray CW, Olsson L, Patel S, Spear N, Tian G. J. Med. Chem. 2007;50:5912. doi: 10.1021/jm070829p. [DOI] [PubMed] [Google Scholar]
- 4.Selkoe DJ. Science. 2002;298:789. doi: 10.1126/science.1074069. [DOI] [PubMed] [Google Scholar]
- 5.Aguzzi A, O'Conner T. Nat. Rev. Drug Disc. 2010;9:237. doi: 10.1038/nrd3050. [DOI] [PubMed] [Google Scholar]
- 6.Melnikova I. Nat. Rev. Drug Disc. 2007;6:341. doi: 10.1038/nrd2314. [DOI] [PubMed] [Google Scholar]
- 7.Suh WH, Suslick KS, Suh Y. Curr. Med. Chem. 2005;5:259. [Google Scholar]
- 8.Shen T, Tai K, Henchman RH, McCammon JA. Acc. Chem. Res. 2002;35:340. doi: 10.1021/ar010025i. [DOI] [PubMed] [Google Scholar]
- 9.Darvesh S, Hopkins DA, Geula C. Nature Rev. Neurosci. 2003;4:131. doi: 10.1038/nrn1035. [DOI] [PubMed] [Google Scholar]
- 10.Green KD, Fridman M, Garneau-Tsodikova S. ChemBioChem. 2009;10:2191. doi: 10.1002/cbic.200900309. [DOI] [PubMed] [Google Scholar]
- 11.Soreq H, Seidman S. Nat. Rev. Neurosci. 2001;2:294. doi: 10.1038/35067589. [DOI] [PubMed] [Google Scholar]
- 12.Villalobos A, Blake JF, Biggers CK, Butler TW, Chapin DS, Chen YL, Ives JL, Jones SB, Liston DR, Nagel AA, Nason DM, Neilson JA, Shalaby IA, White WF. J. Med. Chem. 1994;37:2721. doi: 10.1021/jm00043a012. [DOI] [PubMed] [Google Scholar]
- 13.Kamal MA, Qu X, Yu Q, Tweedie D, Holloway HW, Li Y, Tan Y, Greig NH. J. Neural. Transm. 2008;115:889. doi: 10.1007/s00702-008-0022-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Campiani G, Fattorusso C, Butini S, Gaeta A, Agnusdei M, Gemma S, Persico M, Catalanotti B, Savini L, Nacci V, Novellino E, Holloway HW, Greig NH, Belinskaya T, Fedorko JM, Saxena A. J. Med. Chem. 2005;48:1919. doi: 10.1021/jm049510k. [DOI] [PubMed] [Google Scholar]
- 15.Gibbs ME, Maksel D, Gibbs Z, Hou X, Summers RJ, Small DH. Neurobiol. Aging. 2010;31:614. doi: 10.1016/j.neurobiolaging.2008.05.018. [DOI] [PubMed] [Google Scholar]
- 16.Dinamarca MC, Sagal JP, Quintanilla RA, Godoy JA, Arrazola MS, Inestrosa NC. Mol. Neurodegener. 2010;5:2. doi: 10.1186/1750-1326-5-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rosini M, Simoni E, Bartolini M, Cavalli A, Ceccarini L, Pascu N, McClymont DW, Tarozzi A, Bolognesi ML, Minarini A, Tumiatti V, Andrisano V, Mellor IR, Melchiorre C. J. Med. Chem. 2008;51:4381. doi: 10.1021/jm800577j. [DOI] [PubMed] [Google Scholar]
- 18.Pakaski M, Kalman J. Neurochem. Int. 2008;53:103. doi: 10.1016/j.neuint.2008.06.005. [DOI] [PubMed] [Google Scholar]
- 19.Racchi M, Mazzucchelli M, Lenzken SC, Porrello E, Lanni C, Govoni S. Chem. Biol. Interact. 2005;157–158:335. doi: 10.1016/j.cbi.2005.10.099. [DOI] [PubMed] [Google Scholar]
- 20.Inestrosa NC, Dinamarca MC, Alvarez A. FEBS J. 2008;275:625. doi: 10.1111/j.1742-4658.2007.06238.x. [DOI] [PubMed] [Google Scholar]
- 21.Inestrosa NC, Alvarez A, Perez CA, Moreno RD, Vincete M, Linker C, Casanueva OI, Soto C, Garrido J. Neuron. 1996;16:881. doi: 10.1016/s0896-6273(00)80108-7. [DOI] [PubMed] [Google Scholar]
- 22.Belluti F, Rampa A, Piazzi L, Bisi A, Gobbi S, Bartolini M, Andrisano V, Cavalli A, Recanatini M, Valenti P. J. Med. Chem. 2005;48:4444. doi: 10.1021/jm049515h. [DOI] [PubMed] [Google Scholar]
- 23.Harel M, Schalk I, Ehret-Sabatier L, Bouet F, Goeldner M, Hirth C, Axelsen PH, Silman I, Sussman JL. Proc. Natl. Acad. Sci. USA. 1993;90:9031. doi: 10.1073/pnas.90.19.9031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pang P, Quiram P, Jelacic T, Hong F, Brimijoin S. J. Biol. Chem. 1996;271:23646. doi: 10.1074/jbc.271.39.23646. [DOI] [PubMed] [Google Scholar]
- 25.Bolognesi ML, Andrisano V, Bartolini M, Banzi R, Melchiorre C. J. Med. Chem. 2005;48:24. doi: 10.1021/jm049156q. [DOI] [PubMed] [Google Scholar]
- 26.Greenblatt HM, Silman I, Sussman JL. Drug Dev. Res. 2000;50:573. [Google Scholar]
- 27.Lee LL, Ha H, Chang Y, Delisa MP. Protein Sci. 2009;18:277. doi: 10.1002/pro.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mohamed T, Rao PPN. Bioorg. Med. Chem. Lett. 2010;20:3606. doi: 10.1016/j.bmcl.2010.04.108. [DOI] [PubMed] [Google Scholar]
- 29.Nugiel DA, Cornelius LAM, Corbett JW. J. Org. Chem. 1997;62:201. doi: 10.1021/jo961909w. [DOI] [PubMed] [Google Scholar]
- 30.Fiorini MT, Abell C. Tetrahedron Lett. 1998;39:1827. [Google Scholar]
- 31.Ellman GL, Courtney KD, Andres V. Biochem. Pharmacol. 1961;7:88. doi: 10.1016/0006-2952(61)90145-9. [DOI] [PubMed] [Google Scholar]
- 32.Zhao X, Yang J. ACS Chem. Neurosci. 2010;1:655. doi: 10.1021/cn100067e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Khurana R, Coleman C, Ionescu-Zanetti C, Carter SA, Krishna V, Grover RK, Roy R, Singh S. J. Struc. Biol. 2005;151:229. doi: 10.1016/j.jsb.2005.06.006. [DOI] [PubMed] [Google Scholar]
- 34.Eubanks LM, Rogers CJ, Beuscher AE, IV, Koob GF, Olson AJ, Dickerson TJ, Janda KD. Mol. Pharmaceut. 2006;3:773. doi: 10.1021/mp060066m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mosmann T. J. Immunol. Methods. 1983;65:55. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- 36.Vellonen K, Honkakoski P, Urtti A. Eur. J. Pharm. Sci. 2004;23:181. doi: 10.1016/j.ejps.2004.07.006. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.