

Abstract
Colon cancer is the fourth most common cancer globally with 639,000 deaths reported annually. Typical chemotherapy is provided by injection route to reduce tumor growth and metastasis. Recent research investigates the oral delivery profiles of chemotherapeutic agents. In comparison to injection, oral administration of drugs in the form of a colon-specific delivery system is expected to increase drug bioavailability at target site, reduce drug dose and systemic adverse effects. Pectin is suitable for use as colon-specific drug delivery vehicle as it is selectively digested by colonic microflora to release drug with minimal degradation in upper gastrointestinal tract. The present review examines the physicochemical attributes of formulation needed to retard drug release of pectin matrix prior to its arrival at colon, and evaluate the therapeutic value of pectin matrix in association with colon cancer. The review suggests that multi-particulate calcium pectinate matrix is an ideal carrier to orally deliver drugs for site-specific treatment of colon cancer as (1) crosslinking of pectin by calcium ions in a matrix negates drug release in upper gastrointestinal tract, (2) multi-particulate carrier has a slower transit and a higher contact time for drug action in colon than single-unit dosage form, and (3) both pectin and calcium have an indication to reduce the severity of colon cancer from the implication of diet and molecular biology studies. Pectin matrix demonstrates dual advantages as drug carrier and therapeutic for use in treatment of colon cancer.
Key words: colon cancer, oral drug delivery, pectin
COLON ANATOMY AND PHYSIOLOGY
The term colon refers to the lower part of gastrointestinal tract, extending from ileocecal junction to anus (Fig. 1). It is divided into several sections, i.e., cecum, ascending colon, transverse colon, descending colon, sigmoid colon, and rectum with characteristic lengths of 6–7, 20, 45, 30, 40 and 12 cm, respectively, and an average internal diameter of 6 cm (1,2). The main role of colon is consolidation of intestinal content into feces by absorption of water and electrolytes. The colon has a large absorptive capacity of which 2000 ml of fluid entering the colon each day will be absorbed by more than 90% (1). In healthy human subjects, sodium and chloride ions are usually absorbed and potassium and bicarbonate ions are usually secreted. Fluid and electrolyte absorption is assisted by segmental contraction which circulates chyme across colonic mucosa.
Fig. 1.

Gastrointestinal tract and its distribution probability of colon cancer
The colon epithelium is covered and protected by mucus of which the major glycoprotein constituents are mucins (3). Two structurally and functionally distinct classes of mucins have been identified: secreted gel-forming mucins (MUC2, MUC5AC, MUC5B, and MUC6) and transmembrane mucins (MUC1, MUC3A, MUC3B, MUC4, MUC12, and MUC17). The mucins contain a high content of carbohydrates which are present in the form of clustered oligosaccharides linked to tandem repeat peptides rich in threonine, serine, and proline. They possess a high molecular weight, are resistant to degradation in intestinal tract and recognizable by a number of antibodies and lectins.
The colon contains an almost neutral and reducing medium. In healthy human subjects, the part of gastrointestinal tract which exhibits the highest pH level is terminal ileum (7.5 ± 0.5) (1). On the contrary in the colon, pH is reduced to 6.4 ± 0.6, 6.6 ± 0.8, and 7.0 ± 0.7 at proximal, middle, and distal colon regions, respectively (1,2,4). Under normal physiological condition of healthy human subjects, the colon exhibits irregular alternation of quiescence, non-propagating, segmental contractions and far less-frequent propagated contractions (5). While segmental and propagated contractions predominate in mid-region and distal colon, respectively, retrograde movements have been demonstrated at proximal colon and contribute to increase mass retention in ascending colon and cecum thereby promoting fermentation, electrolyte and water resorption (1).
A dosage form that does not undergo disintegration/dissolution in the upper gastrointestinal tract could require 4 to 12 h to reach colon following oral administration (1). In comparison to upper gastrointestinal tract, the transit of chyme through colon is slower and the total transit time is highly variable and affected by several factors. Under normal condition, the mean total colonic transit time is 35 h as indicated by radiopaque marker technique. The mean segmental transit times of right (ascending and part of transverse), left (part of transverse and descending), and rectosigmoid colon are 11.3, 11.4 and 12.4 h, respectively. The total colonic transit time is shorter in male than female. Smaller dosage forms such as microparticulate systems (granules, pellets, beads), travel at a slower rate in colon than larger units (tablets) unlike gastric emptying profile of the same dosage forms (1–4). The transit rate of colonic content does not exceed the doubling time of bacteria (6). The bacteria can proliferate and ferment the chyme along its colonic passage.
COLONIC MICROFLORA
The human colon consists of over 400 distinct species of bacteria with a population of 1011 to 1012 CFU/ml and a small number of fungi (1,2,5,6). A breakdown of microbial count with respect to aerobic or facultative bacteria, fungi, and anaerobic bacteria is summarized in a review by Vandamme et al. (2). In a healthy adult, the microbial community consists of autochthonous and allochthonous microbes (6). The main species include Bacteroides, Bifidobacterium, Eubacterium, and Lactobacillus (2,5–7). These bacteria produce reductive and hydrolytic enzymes which are active in carbohydrate and protein fermentation, bile acid, and steroid transformation and metabolism of xenobiotic substances (5). The proximal colon has the highest rate of microbial growth as it receives the highest concentration of energy source (1). The primary source of nutrition for these anaerobic bacteria is carbohydrates such as non-starch polysaccharides from the intestinal chyme (5). The non-starch polysaccharides are fermented during transit through colon via enzymatic action of α-L-arabinofunosidase, β-D-fucosidase, β-D-galactosidase, β-D-glucosidase and β-xylosidase (5,6). Polysaccharide enzymatic breakdown is greater in proximal colon than distal colon, while is negligible in stomach and small intestine (1,5).
The end products of fermentation include short-chain fatty acid, carbon dioxide, hydrogen, methane, and hydrogen sulfide (1). The highest level of short-chain fatty acid is produced at right colon (127 mM) with a molar ratio distribution of 57:22:21 for acetate, propionate, and butyrate, respectively (2). The total short-chain fatty acid concentrations at transverse and left colon are 117 and 90 mM, respectively, with corresponding molar ratio distributions of 55:22:23 and 57:21:22 for acetate, propionate and butyrate. The short-chain fatty acids are responsible for the pH lowering observed in colon, nonetheless they are rapidly absorbed from large intestine stimulating at the same time sodium and water absorption (6). The fatty acids are utilized as energy source of intestinal epithelium. The butyrate in addition regulates nucleic acid metabolism of epithelial cells and maintains the health of epithelium. In human, the composition of colonic bacteria population and enzymes levels are influenced by age, diet, geographic prevalent, intestinal pH, motility, and food content (2,5). Diseases, drugs and bacterial metabolites can greatly affect host microflora profiles.
COLON CANCER
Colon is liable to a number of pathological conditions, namely infections, inflammatory bowel diseases (Crohn’s disease and ulcerative colitis), irritable bowel syndrome and cancer. Colon cancer, commonly known as colorectal cancer or large bowel cancer refers to cancerous growth in colon, rectum, or cecum. A total of 639,000 deaths related to colon cancer are reported worldwide each year (8). Colon cancer is the fourth most common form of cancer globally. Colon cancer is reckoned to be a disease of affluence, as it occurs most frequently in North America, Australia, New Zealand, Japan, and Western Europe (9–12).
It is generally accepted that colon cancer is mainly attributable to diet in one way or another (9,11,13). Several additional risk factors have been related to colon cancer, including gender and ethnicity with a higher risk in male than female and black than white, respectively (9–11,14), old age (10,11,13), presence of adenomatous polyps (10,11,13), previous history of ovary, uterus or breast cancer (15), smoking and alcohol drinking habits (9,10,16), physical inactivity (9,11), contraction of specific strains of human papilloma viral infection (15), Streptococcus bovis bacteremia and inflammatory bowel diseases (10,11,13), particularly ulcerative colitis. Administration of exogenous hormones such as estrogen in hormone replacement therapy, as well as, regular use of non-steroidal anti-inflammatory drugs is reported to exert some protective effects against colon cancer (9–11,17,18).
Sixty to 80% of colon cancer cases do not appear to be attributed to inherited symptoms or familial tendency, and are known as sporadic cancer (19). The risk of developing sporadic colon cancer is significantly modulated by environmental factors. The majority of sporadic colon cancer manifests via tumor suppressor/chromosomal instability pathway which could involve the mutation/loss of APC (adenomatous polyposis coli), p53, DCC (deleted in colonic cancer), SMAD 4 and K-ras genes, as well as, activation of telomerase (Fig. 2) (10,18–21). Ten to 15% of sporadic colon cancer express via microsatellite pathway which involves mutation of MMR (mismatch repair) genes (10,19). Ninety-five percent of all malignant tumors of colon are carcinomas (22). Early colon carcinomas are limited to submucosa as polypoid lesion, pedunculated, semipedunculated, or sessile (14,22). Flat lesion with no or slight elevation of not more than twice the mucosa height can exist possibly with slight central depression. Advanced carcinoma invades beyond submucosa and is frequently found ulcerated with sharply demarcated margin or to a lesser extent as polypoid. Adenocarcinoma and mucinous adenocarcinoma represent 90% to 95% of carcinomas. Some adenocarcinomas produce abundant mucus. These carcinomas remain as adenocarcinomas unless mucus contributes to more than 50% of tumor bulk. The severity of carcinoma is indicated by histopathological grading G1 to G4 which ranges from well-differentiated carcinoma with cellular features closely resemble to normal epithelium to undifferentiated carcinoma with no glandular and squamous differentiation at all. The anatomical extent of colon carcinoma is described by Dukes classification where A, B, and C denote tumor invading submucosa, muscularis and lymph node respectively, and D denotes distant metastasis or TNM (tumor node metastasis) system with T grades the level of tumor invasiveness, N grades the involvement of regional lymph node and M grades the level of distant metastasis (11,13,21).
Fig. 2.

Development of colon cancer by chromosomal and microsatellite instability pathways
Surgery remains the primary mode of colon cancer treatment with chemotherapy and/or radiotherapy recommended upon the nature and extent of severity of disease. Chemotherapy can be provided after surgery as adjuvant, before surgery as neo-adjuvant, or as primary therapy to reduce tumor size and growth as well as the likelihood or propensity of metastasis. Chemotherapy is given as adjuvant when the cancer has spread to lymph nodes. Typical regimen of adjuvant chemotherapy is based on the combination infusion of 5-fluorouracil, leucovorin, and oxaliplatin (11). In case of metastasis, the chemotherapy is given as a primary treatment and is represented by a combination of infusional 5-fluorouracil, leucovorin, and oxaliplatin with bevacizumab or 5-fluorouracil, leucovorin, and irinotecan with bevacizumab (15,23,24). The majority of metastases arising from colon cancer are confined to liver (11). Regional delivery of chemotherapy can be performed via hepatic artery, as this has been known for years to supply blood to micrometastasis (25).
Alternatively, the chemotherapeutic agents can be administered via the oral route. Many oral chemotherapeutic agents, mainly fluorinated pyrimidines, have been developed and evaluated for the treatment of advanced and curatively resected colon cancer (26). In comparison to injection, oral administration of anti-cancer agents is expected to improve the quality of life of patients and increase the cost-effectiveness of treatment through reducing the duration of hospitalization. Possible examples of these anti-cancer agents are 5-fluorouracil, hexycarbamoyl-5-fluorouracil (carmofur), uracil/tegafur, uracil/tegafur/leucovorin, and N4-pentoxylcarbonyl-5′-deoxy-5-fluorocytidine (capecitabine) (11,24,26). Oral administration of uracil-tegafur with leucovorin or capecitabine demonstrates approximately equal efficacy and similar median survival time, but less mucositis, neutropenia, and alopecia than intra-venous 5-fluorouracil (11,24). Various positive clinical outcomes on usage of oral chemotherapeutic agents for treatment of colon cancer have been reviewed by Sakamoto et al. (26). The transformation of injectable to oral route is deemed to be a feasible approach in drug administration. Indeed, the orally administered capecitabine has been recommended clinically as an equivalent alternative to intravenously administered 5-fluorouracil-leucovorin (27).
COLON-SPECIFIC ORAL DRUG DELIVERY SYSTEM
With respect to the treatment of colon cancer by orally administered chemotherapeutic agents, it has been suggested by several authors that if the drug is released from the dosage form specifically in the colonic region, it can achieve maximal pharmacological effect. Indeed, colon-targeted drug delivery can increase drug bioavailability at target site, possibly allowing for the reduction of the administered dose and decrease of systemic side effects (4,6). Delivering the drug specifically to the colon is ideal for the exploitation of new biotechnology drugs, such as peptides or protein drugs, due to minimized acidic and enzymatic drug degradation otherwise predominant in the upper gastrointestinal tract. The degree of drug absorption can be enhanced by exploiting the greater colon susceptibility to absorption enhancer effects and prolonged dosage form transit time in comparison to upper gastrointestinal tract (28). However, to succeed in colon targeting, it is imperative to deliver drugs at adequate local concentration and without premature drug release/loss in the upper gastrointestinal tract.
Broadly, five colon-specific drug delivery technologies have been developed and evaluated for their suitability over the years. They all have a common key concept of exploiting unique physiological features of the gastrointestinal tract to obtain system activation and drug release only upon reaching the colonic region. They are prodrug/azo-polymer systems, time-dependent systems, pH-dependent systems, pressure-dependent systems and microbial triggered systems (1,2,4,6,29,30).
Azo-polymers are relatively stable in upper gastrointestinal tract. The azo bonds can resist chemical and enzymatic actions in stomach and small intestine. Nevertheless, the hydrophobic azo-polymers undergo slow degradation by enterobacteria making the intended drug release at specific site delayed (31). However, a risk has been evidenced that azo-polymeric matrices degradation results in the formation of toxic by-products, making such systems unsuitable for long-term treatments. The prodrug approach, on the other hand, is restricted by the availability of functional groups on drug moiety for chemical linkage and new drug entity requiring lengthy evaluation from a regulatory point of view prior to its use in the market.
pH-dependent systems are commonly formulated using enteric coating techniques (32). However, the small intestine and large intestine do not have a markedly different pH environment and, as a consequence, the reproducibility of drug release at a well-defined site from a pH-dependent system can be poor. In other words, the use of enteric coated dosage form may be accompanied by premature drug release in the small intestine. This depends also on the profile of gastrointestinal motility which varies among individuals and disease/dietary conditions which lead to variations in pH of intestine.
In a similar way, changes in motility, pH, and microflora activity of the gastrointestinal tract affect the site specificity of time-dependent systems. Time-dependent systems exploit gastrointestinal transit time as a tool to tune drug delivery to the colon by means of a suitably delayed release profile (33). It is practically difficult to utilize transit time as a measure to target drug delivery of these systems at colon. The suitability of pressure-dependent systems is a function of the reproducibility in pressure and duration of peristaltic waves as well as of disease condition (34). The high-pressure phases in colon are known to be infrequent and follow circadian rhythm with maximum frequency after waking, meals, and with defecation. The success rate of drug release from a pressure-dependent system in colon is therefore low or unpredictable.
Colon-specific drug delivery by means of prodrugs, azo-polymers, time-, pH- and pressure-sensitive approaches has respective pros and cons. Each of them encounters limitations in degree of site specificity, toxicity, ease of preparation, and reproducibility of performance. In comparison to these approaches, drug release triggered by colonic microflora is envisaged to have a higher degree of site selectivity. Polysaccharides can be selectively degraded by colonic enzymes and their use as drug vehicle can trigger drug release to start in the colon. The degradation of polysaccharides by microflora exhibits a relatively consistent pattern across diverse human population (35), thus assuring the reproducibility of drug release performance of polysaccharide-based drug carriers. Using naturally occurring dietary polysaccharides, the issues of toxicity and safety are minimized. The main saccharolytic species in colon are Bacteroides and Bifidobacterium (6). The success of polysaccharides as drug carriers also lies in their ability to hydrate and swell creating a diffusion barrier during its gastrointestinal transit. Upon arrival at the colon, the hydrated vehicle permits the access of colonic bacteria/enzymes to degrade the polysaccharide network and release the drug at the intended site. Examples of polysaccharides which are potentially useful in the formulation of colon-specific drug delivery systems include alginate, amylose, arabinogalactan, arabinoxylan, cellulose, chitosan, chondroitin sulfate, dextran, guar gum, locust bean gum, inulin, karaya gum, laminarin, pectin, starch, tragacanth gum, xanthan gum, and xylan (1,2,4,6,29,36).
PECTIN AND COLON CANCER
Pectin is a natural heteropolysaccharide with at least 65% by weight of galacturonic acid units (37). It is made of 1,4 linked α-D-galactosyluronic acid residues and a range of neutral sugars such as rhamnose, galactose, arabinose and lesser amounts of others (37,38). Pectin is available in the form of free acid, simple salt such as sodium, potassium, and calcium salts, methyl ester, acetylester, feruloylester, or amidated polysaccharide (36,37,39,40). Pectin is polymolecular and polydisperse. It contains a few hundreds to about 1,000 saccharide units in a chain-like configuration, corresponding to an average molecular weight between 50,000 and 150,000 Da (41).
Pectin is suitable for use as colon-specific drug delivery vehicle in treatment of colon cancer and other colon diseases as it is selectively digested by microflora in colon and exhibits potential to prevent colon cancer from the implication of diet (42). Being a soluble dietary fiber, pectin is reported to increase the transit time through gastrointestinal tract, fecal bulk, bile acid excretion and short-chain fatty acid production. Unlike insoluble fiber such as cellulose, it undergoes almost 100% fermentation in colon (43). Fermentation of pectin leads to the generation of short-chain fatty acids (42,43). The short-chain fatty acids are the primary energy source of colonocyte (12,43,44). Moreover, these fatty acids lower the colonic pH, which is protective against colon cancer (42,43,45). In fact, epidemiology studies indicated that colon cancer subjects have a fecal pH of 7, whereas normal subjects have a fecal pH of 6.5. 7-α-Dehydroxylase is the bacterial enzyme which converts primary bile acids into secondary bile acids such as lithocholic acid and deoxycholic acid, known as promoter of colon cancer. Though debatable, the lowering of pH obtained by pectin fermentation can reduce the risk of colon cancer by diminishing the production of secondary bile salts, via inhibiting the activity of 7-α-dehydroxylase at a pH below 6.5 and by simultaneously decreasing free bile acid solubility (43,45). The level of bile acids can be further lowered by pectin via increasing their excretion in feces (42,46).
In addition to colonic pH lowering effects, short-chain fatty acids decrease colon hyperproliferation induced by deoxycholate in association with reduced expression of c-Fos and c-Jun (42). They have shown to promote apoptosis in a variety of colon tumor cell lines and are able to depress proliferation of tumor cells in vivo (12,42,43). The butyrate induces apoptosis in tumor cells which lack a functional p53 pathway via a caspase-dependent process (12). It downregulates bcl-2 oncogene thereby removes the barrier to apoptotic cell death (16). The butyrate induces histone hyperacetylation via the inhibition of histone deacetylase (12,18,43). Acetylation of histone favors an open, transcriptionally active chromatin configuration which in turn facilitates the expression of pro-apoptotic genes.
While low methoxyl pectin, high methoxyl pectin and citrus pectin are found to increase the risk of colon tumor (47,48), apple pectin and citrus pectin have been reported lately to decrease the number and incidence of colon tumor (49,50). Pectins and pecticoligosaccharides are found to increase the apoptosis frequency of colonic adenocarcinoma cell line HT29 (51). Dietary supplementation of pectin indicates that pectin can suppress colon cancer significantly and exert anti-proliferative effect in mouse distal colon (52,53). Pectin can negate colon cancer via inhibiting galectin-3 biological functions. Galectin-3 is a chimeric gene product with a monomer subunit of approximately 30,000 Da which undergoes noncovalent homodimerization (3). It is a β-galactoside-binding protein which can be found in the cytoplasm and nucleus, on the cell surface, and is secreted by tumor cells. Galectin-3 mediates cell–cell adhesion to reduce apoptosis incurred by anoikis, and to induce initial adhesion of cancer cells to endothelium and subsequent tumor cell homotypic aggregation (Fig. 3) (3,54–56). It promotes tumor cells to interact with extracellular matrix proteins in development of a secondary tumor growth, clonogenic survival of cancer cells, and stimulates endothelial cell morphogenesis and angiogenesis necessary for tumor growth. Specific gene expression and anti-apoptotic activity are regulated by nucleus and cytoplasmic galectin-3, respectively, whereas extracellular galectin-3 mediates cell migration, cell adhesion and cell–cell interaction through its carbohydrate-binding property. Galectin is expressed at higher levels in colon cancer than normal colon. It binds to colonic MUC 2 mucin with altered carbohydrate structure. Carbohydrate or polysaccharide-mediated interference with galectin-3 binding to mucin can lead to therapeutic or prevention advantages in colon cancer. Modified citrus pectin, low methoxyl pectin and high methoxyl pectin are reported as antagonists to galectin-3 (3,54–58). Modified citrus pectin has shown to reduce the growth and metastasis of colon tumor (3,56–58). Orally administered modified citrus pectin at 0.8 and 1.6 mg/ml for 20 days to balb-c mice implanted with colon-25 tumors exhibits a dose-dependent reduction in tumor size (57). The modified citrus pectin has a potential to raise the apoptotic response of tumor cells to chemotherapy (56). The summative observation indicates the likelihood of pectin being utilized as drug, matrix vehicle, and therapeutic sensitizer of anti-cancer agents.
Fig. 3.

Modulation of galectin-3-mediated colon cancer development by pectin
Citrus pectin modified by heat treatment is reported to lead to significant levels of tumor cell apoptosis comparable to fractionated pectin powder (56). pH treatment of modified citrus pectin is critical to reduce cell–cell adhesion in the development of tumor though it is reported that mild base treatment removes ester linkages of pectin and destroys its apoptotic activity. Pectin exhibits a higher strength of mucoadhesion on large intestinal mucosa than small intestinal mucosa (59). The mucoadhesion property of pectin is promoted by high degree of esterification and large molecular weight attributes of pectin. High esterified pectin tends to form gel network with endogenous mucin on the surfaces of colonic mucosa (60). The strong tendency of pectin to interact with large intestinal mucosa is expected to interrupt the biochemical processes mediating cell–cell adhesion in tumor growth. Modification or processing of pectin by means of heat or pH treatment shall avoid excessive molecular chain breakdown or de-esterification of pectin which can translate to a loss of its mucoadhesiveness and therapeutic function.
PECTIN AS COLON-SPECIFIC ORAL DRUG DELIVERY VEHICLE
Pectin is completely fermented in colon by microflora with low esterified pectin being fermented faster than high esterified pectin (61). It appears that only a partial degradation is possible at the pH 2 to 4 conditions of stomach via side chain hydrolysis and at pH 5 to 6 conditions of small intestine via β-elimination of main chain or de-esterification (61,62). Nonetheless, pectin is an aqueous soluble polymer. The matrix made of pectin is prone to swelling as well as erosion in aqueous medium leading to premature drug release at upper gastrointestinal tract and thereby defeating its ability as colon-specific drug delivery vehicle.
The pectin-based matrix is available as single-unit dosage form such as tablet or multiple-unit dosage form, namely beads, pellets, and microparticles. In designing colon-specific drug delivery systems, numerous formulation approaches have been taken in the past years to prevent pectin-based matrix from undergoing early drug release in the upper gastrointestinal tract (Fig. 4). The pectin in matrix has been subjected to crosslinking by di- or multi-valent cations, coacervation with an oppositely charged polyelectrolyte, mixing with viscous polymer and/or calcium salt, or coating with viscous, pH resistant or poorly water-soluble polymer, coacervate and/or complex to reduce its drug dissolution propensity in aqueous medium prior to reaching the colon (Table I). Pectin, in combination with a crosslinking agent or a polymer, may also be employed itself as a delayed release coat to be applied onto a drug core via film or compression-coating technique. In particular, multiple coat layers can be applied onto the drug core and these coats can vary in their chemical composition in order to retard/modulate drug release at different sites of upper gastrointestinal tract. Pectin has also been used to prepare prodrugs which will release the free drug upon arrival at colon. Table I summarizes several types of pectin-based colon-specific dosage forms and their associated formulation mechanisms. In addition to the anti-cancer agents 5-fluorouracil and methotrexate, anti-inflammatory drugs, antibiotics, anti-nociceptive agents, and enzymes have been formulated in the form of pectin-based matrix for colon delivery. A schematic flowchart in relation to processing technology employed in crosslinking, coacervation, compression, and coating of various formulations is shown in Fig. 5.
Fig. 4.
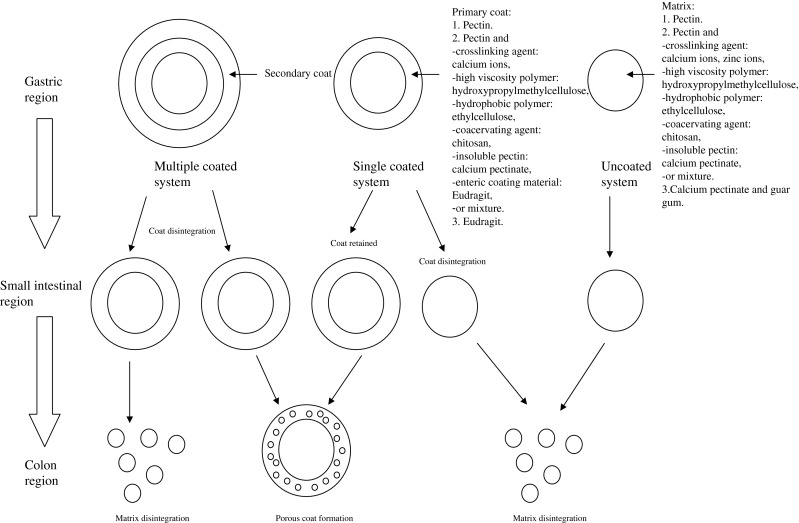
Formulation and design of microflora-triggered pectin-based colon-specific oral drug delivery system
Table I.
Types of Pectin-Based Colon-specific Oral Drug Delivery System
| No. | Type of dosage form | Type of drug | Type of pectin | Formulation mechanism | Reference |
|---|---|---|---|---|---|
| Single-unit | |||||
| 1. | Calcium pectinate tablet | Sodium fluorescein (model drug), indomethacin | Low methoxyl and high methoxyl pectins | Mixing of pectin with varying contents of calcium ions in tablet. | (63,64) |
| 2. | Calcium pectinate tablet | Indomethacin | Low esterified pectin | Use of calcium pectinate as matrix substance of tablet. | (65) |
| 3. | Pectin-HPMC-calcium tablet | Indomethacin | High methoxyl pectin | Mixture of pectin, HPMC and calcium salt as matrix substance of tablet. | (66) |
| 4. | Zinc pectinate tablet | Ketoprofen | Amidated low methoxyl pectin | Ionotropic gelation of pectin with zinc ions into microparticles followed by compression of dry microparticles into tablet with dextran. | (67) |
| 5. | Eudragit L coated calcium pectinate-pectin tablet | Samaium oxide 152 | – | Tablet made of calcium pectinate and pectin is enteric coated by Eudragit L to target isotope release at ascending colon. | (68) |
| 6. | Eudragit L coated calcium pectinate-guar gum tablet | Samaium oxide 152 | – | Tablet made of calcium pectinate and guar gum is enteric coated by Eudragit L to target isotope release at ascending and transverse colon. | (68) |
| 7. | Eudragit L100-coated ethylcellulose-pectin tablet | Ropivacaine | Amidated pectin and high methoxyl pectin | Drug core consists of pectin and ethylcellulose and is coated with Eudragit L100. | (69) |
| 8. | Pectin-coated tablet | Naphthol green B (model drug) | Pectin USP | Pectin as compression coat onto drug core. | (70) |
| 9. | Pectin-HPMC-coated tablet | 5-aminosalicylic acid, nisin | Pectin USP | Mixture of pectin and high molecular weight hydroxypropylmethylcellulose (HPMC) as compression coat onto drug core. | (28,71) |
| 10. | Pectin-chitosan-HPMC-coated tablet | Paracetamol | High methoxyl pectin USP | Mixture of pectin, chitosan and HPMC as film coat onto drug core. | (72–74) |
| 11. | Ethylcellulose-pectin-coated tablet | Paracetamol | Pectin USP | Mixing of pectin with hydrophobic ethylcellulose as film coat of tablet. | (75) |
| 12. | Pectin–chitosan-coated tablet | Indomethacin, paracetamol | High methoxyl pectin USP | Pectin-chitosan as inter-polymer complex coat onto drug core. | (76) |
| Multiple-unit | |||||
| 1. | Zinc pectinate beads | Ketoprofen | Amidated low methoxyl pectin | Ionotropic gelation of pectin with zinc ions in low pH aqueous medium to form beads. Low pH reaction medium promotes protonation of carboxylate moiety of pectin, suppresses intermolecular charge–charge repulsion, reduces polymer chain solubility and facilitates polymer conformational ordering. Beads are subsequently introduced in enteric coated capsule. | (77,78) |
| 2. | Zinc pectinate microparticles | Ketoprofen | Amidated low methoxyl pectin | Ionotropic gelation of pectin with zinc ions to form microparticles. | (67) |
| 3. | Calcium pectinate beads | Penicillinase (β-lactamase) | Low methoxyl amidated and non-amidated pectins | Ionotropic gelation of pectin with calcium ions into beads. | (79) |
| 4. | Calcium pectinate microparticles | Methotrexate | – | Ionotropic gelation of pectin with calcium ions into microparticles. | (80) |
| 5. | Pectinate-chitosan beads | Indomethacin, sulphamethoxazole | Amidated low methoxyl pectin | Coacervation/crosslinking of pectin with chitosan and calcium ions in a single pot process. | (81) |
| 6. | Pectin-chitosan composite particles | Bovine serum albumin | Low methoxyl pectin | Coacervation/crosslinking of pectin with chitosan and calcium ions in a single pot process. | (82) |
| 7. | Chitosan/pectin-coated pellets | Riboflavin | Low methoxyl pectin | Immersion coating of calcium acetate pellets with pectin followed by chitosan. | (83) |
| 8. | Pectin-ethylcellulose-coated pellets | 5-fluorouracil | Low methoxyl pectin | Mixing of pectin with hydrophobic ethylcellulose as film coat of pellets. | (84,85) |
| 9. | Pectin-Eudragit RL30D-coated pellets | Theophylline | High methoxyl pectin | Pectin-Eudragit RL30D as inter-polymer complex coat onto drug core. | (86) |
| 10. | Aquacoat ECD30, Surelease and Eudragit NE30D, RS30D/HM or LM calcium pectinate-coated pellets | Theophylline | High methoxyl (HM) and low methoxyl (LM) pectins | Coating of pellets with Aquacoat ECD30, Surelease and Eudragit NE30D, RS30D containing high methoxyl pectin or low methoxyl calcium pectinate. | (87) |
| 11. | Eudragit S100-coated calcium pectinate microparticles | Theophylline | Amidated and non-amidated low methoxyl pectins | Crosslinking of pectin with calcium ions into microparticles followed by Eudragit S100 coating onto microparticles by immersion-solvent evaporation technique. | (88) |
| 12. | Eudragit S100-coated calcium pectinate beads | Theophylline, 5-fluorouracil | Low methoxyl pectin | Crosslinking of pectin with calcium ions into beads followed by Eudragit S100 coating onto beads. | (89,90) |
| 13. | Eudragit S100-coated pectin microparticles | 5-Fluorouracil | – | Pectin microparticles are formed by emulsion dehydration technique and subsequently coated by Eudragit S100 via oil-in-oil solvent evaporation method. | (91) |
| 14. | HM pectin-coated LM pectinate beads | Coomassie Brilliant Blue G 250 (model drug) | High methoxyl and low methoxyl pectins | HM pectin as coat onto LM pectinate core. | (92) |
| Prodrug | |||||
| 1. | Pectin-ketoprofen prodrug | Ketoprofen | High esterified pectin | Chemical linking between pectin and drug. | (93) |
| 2. | Pectin-5-fluorouracil conjugate | 5-Fluorouracil | High esterified pectin | Chemical linking between pectin and drug. | (94) |
Fig. 5.

Schematic flow of processing technology employed in design of pectin-based colon-specific oral drug delivery system
The proposed formulation strategies to prevent premature drug release from pectin-based colon-specific dosage form exploit a number of mechanisms as follow:
enhancement of hydrophobicity
reduction of aqueous solubility
enhancement of physical strength
enhancement of viscosity
reduction of swelling capacity
reduction of erosion capacity
of the pectin-based matrix or coat. A functional colon-specific dosage form must not exhibit premature drug release at upper gastrointestinal tract. It must nevertheless be able to hydrate, swell and absorb enzyme- or microflora-rich fluids in colon. The pectinolytic enzymes would then digest the pectin components of matrix or coat of a dosage form thereby initiating drug release.
High methoxyl pectin has been advocated as the polymer of choice because it is inherently less water soluble than low methoxyl pectin (75,76,86). However, it is reported that low methoxyl pectinate beads coated with high methoxyl pectin exhibit a higher propensity of drug release than uncoated beads (92). This is ascribed to prolonged exposure of drug core to moist high methoxyl pectin layer during coating process. It results in drug migration to the surface and leads to a burst release effect upon erosion and dissolution of the high methoxyl pectin coat. Low methoxyl pectin is prepared by hydrolytic demethylation of high methoxyl pectin (83). This reaction is known to reduce the molecular weight and chain length of pectin. Shorter chain length of pectin is characterized by its higher aqueous solubility, unstable polymeric network and faster drug release. In conjunction with colon-specific dosage form prepared using chitosan and/or multi-valent cationic salt, low methoxyl pectin is nevertheless preferable as it is able to interact with chitosan or cationic salt to a greater extent than high methoxyl pectin due to the availability of a higher fraction of free carboxylic acid groups.
Crosslinking of pectin with calcium ion or zinc ions, as well as, coacervation/complexation with polymers such as chitosan and/or Eudragit™ decreases the solubility, swelling and erosion extent of the polymer, but increases the mechanical strength and hydrophobicity of the dosage form. An increase in hydrophobicity of the dosage form due to charge neutralization reduces its tendency to interact with dissolution medium thereby lowering the rate and extent of drug release. A reduction in solubility, swelling and erosion and an increase in mechanical strength, following complex or crosslink formation, minimize the breakdown of dosage form, the generation of surface pore or cracks and similarly reduce the abrupt release of drug from dosage form to surrounding dissolution medium. The hydrophobicity and drug release retardation capacity of colon-specific dosage form can be further promoted by coating the matrix with ethylcellulose, a water-insoluble polymer. The mechanical strength of dosage form can be modulated and enhanced through blending of pectin with hydroxypropylmethylcellulose or dextran. The combination with hydroxypropylmethylcellulose or dextran can increase the gel viscosity of the hydrated dosage form. It reduces the erosion propensity of matrix thereby lowering drug release.
The chemical composition of the matrix and/or coat polymer is the key parameter affecting the drug release pattern of colon-specific dosage form. Sustaining drug release will be less possible when calcium pectinate is used instead of pectin to complex with Eudragit™ (95). The calcium pectinate has a lower fraction of carboxylate moiety than pectin as some of its active sites are occupied by calcium ions. The level of calcium pectinate-Eudragit™ complexation is lower than that of pectin-Eudragit and the formed complexes are less able to retain drug within the matrix. The lowest drug release can be obtained when a coat is made of a high content of pectin complexed to chitosan, using low methoxyl pectin and highly deacetylated chitosan which provide a substantial fraction of carboxylate and amino moieties respectively for coacervation reaction (96). In any case, coacervation of pectin with chitosan however does not negate the drug release of pectin-based dosage form in response to pectinolytic enzymes (76,81).
In drug delivery system with coat composed of pectin and hydrophobic polymer, the digestion of pectin results in the formation of pores for drug release (72,73,75,85,95). The fraction of hydrophobic polymer in the coat must not be excessively high in order for pectin to hydrate for enzymatic digestion to take place at colon. It must not be too low as this will bring about premature drug release. The optimal pectin content has been reported to constitute less than 20% in the composite (36). In coat made of complex of water-soluble polymers, the action of pectinolytic enzymes on the coat complex gives rise to pectin leaching and drug release in colon (86). The hydrophilic polymer counterpart of pectin, once released from complex, similarly becomes freely solvated, swells, and leads to distortions in coat thereby further facilitating drug release. Enzymatic degradation of pectin components forms the primary mechanism of colon-specific drug release. Nevertheless, the leaching of pectin components from coat may suppress the formation of hydrated pectin channels (87,92). The undigested coat on matrix can be reconstructed via distension, plugging up pores, reducing free volume between polymer chains and preventing drug release. The restructuring of coat is possible when glass transition temperature of coat is close to temperature of dissolution medium and it can be lowered when polymers are placed in a liquid environment. In accentuation of drug release from the core to the surrounding colonic medium, a disintegrant such as crosslinked polyvinylpyrrolidone can be added to such matrix (72). The digestion of pectin by pectinolytic enzymes is accompanied by water influx into core and build-up of pressure by crosslinked polyvinylpyrrolidone onto surrounding coat/matrix substances, thus accelerating the release of drug in colon.
Calcium ions are commonly used as crosslinking agent in preparation of pectin-based colon-specific dosage form. The incorporation of a calcium salt into a pectin matrix enhances the susceptibility of pectin to enzymatic digestion in human colon since many pectinases have shown to be stimulated by or have an absolute requirement for calcium ions for their activity (36,63,64). The calcium pectinate matrix is found to release the major part of drug in intestinal medium (67,69,77). Generally, the use of a higher calcium ion content can reduce drug release while the matrix travels along the gastrointestinal tract towards colon and it could provide sigmoidal drug release pattern (63,64).
Lately, zinc ions have been adopted as crosslinking agent of interest. The zinc ion-crosslinked pectin matrix releases drug at a lower rate than the calcium ion-crosslinked sample (67,77,78). In comparison with calcium ions, zinc ions form a more extensive crosslink with pectin and reduce the degree of rehydration and molecular porosity of matrix to a greater extent. Rapid drug release from calcium ion-crosslinked matrix is presumably due to a greater level of solvent penetration into the calcium pectinate network followed by a greater propensity for ion exchange of calcium with sodium and potassium ions. The monovalent ions displace calcium ions from the gelled structure and transform insoluble calcium pectinate into soluble pectin which has less drug release retardation capacity.
PECTIN-BASED COLON-SPECIFIC ORAL DRUG DELIVERY SYSTEM AND COLON CANCER
Over the past 5 years, in vivo assessments indicate that colon-specific pectin-based oral dosage forms can be designed primarily through coating of drug matrix with pectin-ethylcellulose or Eudragit film (85,91). The early drug release in gastrointestinal tract is negated by coat hydrophobicity and reduced matrix solubility. The majority of drug is released or accumulated at the colonic region of intestinal tract. Pharmacokinetically, the propensity of drug absorption at colon can be low due to reduced absorption surface area and low drug permeability across colonic barrier. Some of the released drug may still be located in the colon lumen 24 h after oral administration of matrix due to slow transit (85). Low drug absorption may be undesirable for systemically acting drugs. In colon cancer chemotherapy, low plasma drug concentration can nevertheless increase the safety margin of the treatment by lowering peak plasma drug levels and sustaining tumor exposure to the drug. Low plasma drug concentrations coupled to prolonged exposure to high local colonic drug concentration could facilitate DNA-directed action of the drug, which contributes to the anti-cancer effect.
5-Fluorouracil has been loaded into pectin microspheres by means of emulsion dehydration technique (91). Briefly, a w/o emulsion is first obtained by mixing the solution mixture of drug and pectin with isooctane. This is followed by dehydration of dispersed phase by acetone to produce the micropsheres. The formed microspheres, having a mean diameter between 25 and 30 μm, are then coated with Eudragit S100 by an oil-in-oil solvent evaporation method. The coated microspheres are able to prevent the release of 5-fluorouracil at pH 1.2 and 4.5, otherwise observed for uncoated matrix. These microspheres can efficiently deliver drug to the colon after 6 to 8 h of administration in rats. In another work reported by Wei et al. (85), 5-fluorouracil is encapsulated in pellets by means of extrusion–spheronization technique using microcrystalline cellulose as processing aid. The formed pellets are subsequently coated with pectin and ethylcellulose using the fluid-bed coating technique. In vivo bioavailability analysis of coated pellets indicates that a Tmax of 14 h is obtainable when compared to uncoated samples which are characterized by a short Tmax of 0.75 h. Formulation of 5-fluorouracil in these coated structures can delay the release of drug in gastrointestinal tract.
Recent reports on pectin-based colonic delivery system containing 5-fluorouracil and methotrexate indicate that sustained-release pellets and microparticles are preferred as oral dosage form to deliver chemotherapeutic agents in colon cancer (80,84,85,91). Unlike gastric transit, small dosage forms show a slower colon transit than large dosage forms (1). Prolonged colonic retention of dosage form and/or consistent local colonic exposure to anti-cancer drug is more easily attainable by using multiple-unit particulate system such as beads, pellets and microparticles as drug delivery systems instead of single-unit tablets (often large because of high drug doses).
Crosslinking agents have been part of the formulation for many pectin-based multi-particulate colon-specific drug delivery systems. In spite of recent reports favoring the use of zinc ions, calcium ions will possibly remain as the mainstream crosslinking agent as they promote pectinolytic enzymatic degradation of pectin and exhibits biological properties which aid to counteract the development of colon cancer. In a recent work of Chaurasia et al. (80), calcium pectinate beads in the size range of 20 to 30 μm are produced by modified emulsification method using calcium chloride as crosslinking agent. These beads have a good encapsulation efficiency of methotrexate (up to 74%) and release only 8% of loaded drug when they are incubated for 5 h in simulated gastric fluid followed by enzymeless simulated intestinal fluid. The majority of drug release only when pectinase or cecal rat content is present in the simulated intestinal fluid, demonstrating that the pectinate matrix crosslinked by calcium ions is still an efficient carrier to target the release of anti-cancer drug at colon through exploiting the physiological features of colonic environment.
The role of calcium in colon cancer has been evaluated in animals, epidemiologic studies and human intervention studies (97). Though there is no clear relationship between calcium intake and colon cancer development, dietary supplementation with calcium has been found to reduce adenoma size and number (18). High calcium and vitamin intake increases apoptosis of colonic epithelium and may reduce risk of colon cancer (16–18,98). The capability of calcium to combat colon cancer is likely ascribed by its binding affinity to potentially carcinogenic free bile acids in colon.
Both pectin and calcium have indicated their biological significance in combating the development of colon cancer. The present review hitherto suggests that multi-particulate drug delivery system made of calcium pectinate can be an ideal carrier to orally administer and deliver chemotherapeutic agents to the colon for treatment of colon cancer, upon modification to prevent early drug release in the upper part of gastrointestinal tract. Both pectin and calcium should be the main excipients used in the formulation of the delivery system. These excipients can be used as matrix/coat substances. The success of chemotherapy is dependent on the ability of the delivery system to hinder premature drug release in the upper gastrointestinal tract, and be digested and released the loaded drug, possibly together with pectin and calcium at the site of disease. It is envisaged that the reproducibility of matrix/coat digestion and drug release at colon is an interplay effect of colonic pH and microflora environment with the physicochemical properties of drug, pectin and other excipients of the dosage form. The composition of every individual microflora can fluctuate under illness and to a lesser extent be induced by dietary intervention (99). High risk of colon cancer is associated with the presence of Bacteroides vulgatus and Bacteroides stercosis in human. However, the exact complications brought about by microflora changes in presence of colon cancer on the performance of pectin-based drug delivery systems are not known and likely to remain elusive. One main reason for this is that the intestinal bacteria are adapted to an anaerobic environment (100). They are difficult or impossible to culture extraintestinally for study. With the advent of genetic technology, only 1,822 of more than 20,000 rRNA gene sequences in GenBank are in fact documented as derived from human gastrointestinal tract, and 1,689 are uncultured bacteria. Another issue is related to pH differences. The colonic mucosa pH is alkaline (pH = 8) and is similar on both sides of colon in healthy and colon cancer patients (101,102). Both healthy right colostomates and cancer patients have the same luminal pH at 7.6, but the fecal pH of healthy right colostomates is lower than that of colon cancer patients (pH = 6.6) (101). Variations in rate and extent of drug release from pectin-based dosage forms are deemed to be induced by different fecal pH environments at the proximal colon in diseased and healing states, and such variations are expected to be complicated by and/or be a resultant effect of concomitant changes of microflora composition during the treatment of colon cancer.
FUTURE ISSUES
Continuing efforts to design pectin-based delayed-release dosage form for use in colonic drug delivery are undertaken by many researchers. Gelatin, alginate, and xyloglucan are some polymers explored in formulation of pectin-based drug delivery system (103–106). The pectin-based dosage form has been recently subjected to treatment by microwave to evaluate the capacity of radiation to further reduce the drug release propensity of matrix at upper gastrointestinal tract (107,108). Ongoing research is conducted to produce water-resistant pectin derivatives without compromising their susceptibility to microflora fermentation in the colon (36). Pectin receives an overwhelming attention on its practical applications and implications in drug delivery. Nonetheless, one shall highlight that there is no clear picture on the exact complex structure of pectin and the physicochemical attributes of pectin vary with its source and manufacturing process (37,56). Further work shall be conducted to characterize the physicochemical attributes of pectin and evaluate its structure-activity relationship with reference to colon-specific drug delivery. Pectin has shown an indication to be able to suppress colon cancer growth lately. The collective effectiveness of pectin as drug delivery vehicle, drug and/or therapeutic enhancer in colon-specific delivery of anti-cancer drugs requires verification. The distribution probability of cancer varies from one site to another site of colon and 99% of carcinomas occur singly (Fig. 1) (21). The possibility of pectin-based colon-specific dosage form to perform solely and precisely at a cancer site of an abnormal colon is a fascinating challenge for future pharmaceutists. Polymers, other than pectin, require further investigation on their potential use as concurrent drug carrier and chemotherapeutic agent through a multi-disciplinary approach.
Acknowledgments
The authors wish to thank the Ministry of Higher Education, Malaysia for facility support (code: 0141903).
References
- 1.Watts PJ, Illum L. Colonic drug delivery. Drug Dev Ind Pharm. 1997;23(9):893–913. [Google Scholar]
- 2.Vandamme ThF, Lenourry A, Charrueau C, Chaumeil J-C. The use of polysaccharides to target drugs to the colon. Carbohydr Polym. 2002;48:219–231. [Google Scholar]
- 3.Byrd JC, Bresalier RS. Mucins and mucin binding proteins in colorectal cancer. Cancer Metastasis Rev. 2004;23:77–99. doi: 10.1023/a:1025815113599. [DOI] [PubMed] [Google Scholar]
- 4.Singh BN. Modified release solid formulations for colonic delivery. Recent Pat Drug Deliv Formul. 2007;1:53–63. doi: 10.2174/187221107779814122. [DOI] [PubMed] [Google Scholar]
- 5.Yang L. Biorelevant dissolution testing of colon-specific delivery systems activated by colonic microflora. J Control Release. 2008;125:77–86. doi: 10.1016/j.jconrel.2007.10.026. [DOI] [PubMed] [Google Scholar]
- 6.Sinha VR, Kumria R. Microbially triggered drug delivery to the colon. Eur J Pharm Sci. 2003;18:3–18. doi: 10.1016/s0928-0987(02)00221-x. [DOI] [PubMed] [Google Scholar]
- 7.Guarner F. Enteric flora in health and disease. Digestion. 2006;73(suppl 1):5–12. doi: 10.1159/000089775. [DOI] [PubMed] [Google Scholar]
- 8.World Health Organization: Health topics. http://www.who.int/topics/cancer/en (2009). Accessed 25 Feb 2009.
- 9.Boyle P, Leon ME. Recent developments in the epidemiology of colorectal cancer. In: Bleiberg H, Kemeny N, Rougier P, Wilke H, editors. Colorectal cancer— A clinical guide to therapy, chapter 2. UK: Taylor and Francis; 2002. pp. 11–29. [Google Scholar]
- 10.Lieberman D. Colorectal cancer screening and surveillance. In: Ginsberg GG, Kochman ML, Norton I, Gostout CJ, editors. Clinical gastrointestinal endoscopy, Chapter 37. UK: Elservier; 2005. pp. 537–547. [Google Scholar]
- 11.Midgley RS, Merrie A, Kerr DJ, Mortensen N. Colorectal cancer: a multidisciplinary approach. In: Weinstein WM, Hawkey CJ, Bosch J, editors. Clinical gastroenterology and hepatology, Chapter 60D. Spain: Elsevier; 2005. pp. 421–430. [Google Scholar]
- 12.Johnson IT. New approaches to the role of diet in the prevention of cancers of the alimentary tract. Mutat Res. 2004;551:9–28. doi: 10.1016/j.mrfmmm.2004.02.017. [DOI] [PubMed] [Google Scholar]
- 13.Yen EF. Colon neoplasms. In: Shiels A, Lin TL, editors. The Washington ManualTM gastroenterology subspecialty consult, Chapter 17. USA: Lippincott Williams & Wilkins; 2004. pp. 93–100. [Google Scholar]
- 14.Soetikno R, Friedland S, Matsuda T, Gotoda T. Colonoscopic polypectomy and endoscopic mucosal resection. In: Ginsberg GG, Kochman ML, Norton I, Gostout CJ, editors. Clinical gastrointestinal endoscopy, Chapter 38. UK: Elservier; 2005. pp. 549–568. [Google Scholar]
- 15.Wikipedia. Colorectal cancer. Wikipedia: the free Encyclopedia. Accessed 17 Apr 2009.
- 16.Greenwald P, Clifford CK, Milner JA. Diet and cancer prevention. Eur J Cancer. 2001;37:948–965. doi: 10.1016/s0959-8049(01)00070-3. [DOI] [PubMed] [Google Scholar]
- 17.Benamouzig R. Chemoprevention of colorectal cancer. In: Bleiberg H, Kemeny N, Rougier P, Wilke H, editors. Colorectal cancer— A clinical guide to therapy, chapter 5. UK: Taylor and Francis; 2002. pp. 47–52. [Google Scholar]
- 18.Watson AJM. An overview of apoptosis and the prevention of colorectal cancer. Crit Rev Oncol Hematol. 2006;57:107–121. doi: 10.1016/j.critrevonc.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 19.Strate L, Pieramici E, Ahnen D. Biology and genetics of colorectal cancer. In: Weinstein WM, Hawkey CJ, Bosch J, editors. Clinical gastroenterology and hepatology, Chapter 60A. Spain: Elsevier; 2005. pp. 397–406. [Google Scholar]
- 20.Abramowicz MJ. Biology of colorectal cancer: an overview of genetic factors. In: Bleiberg H, Kemeny N, Rougier P, Wilke H, editors. Colorectal cancer—a clinical guide to therapy, chapter 1. UK: Taylor and Francis; 2002. pp. 3–9. [Google Scholar]
- 21.Liu C, Crawford JM. The gastrointestinal tract. In: Kumar V, Abbas AK, Fausto N, editors. Pathologic basis of disease, Chapter 17. USA: Elsevier; 2005. pp. 797–875. [Google Scholar]
- 22.Hermanek P. Pathology of colorectal cancer. In: Bleiberg H, Kemeny N, Rougier P, Wilke H, editors. Colorectal cancer— A clinical guide to therapy. UK: Taylor and Francis; 2002. pp. 55–72. [Google Scholar]
- 23.Davies JM, Goldberg RM. First-line therapeutic strategies in metastatic colorectal cancer. Oncol Huntingt. 2008;22:1470–1479. [PubMed] [Google Scholar]
- 24.Midgley R. Systemic adjuvant therapy of colon cancer. In: Bleiberg H, Kemeny N, Rougier P, Wilke H, editors. Colorectal cancer—a clinical guide to therapy, Chapter 12. UK: Taylor and Francis; 2002. pp. 127–134. [Google Scholar]
- 25.Ammori JB, Kemeny NE. Regional hepatic chemotherapies in treatment of colorectal cancer metastases to the liver. Semin Oncol. 2010;37:139–148. doi: 10.1053/j.seminoncol.2010.03.003. [DOI] [PubMed] [Google Scholar]
- 26.Sakamoto J, Oba K, Matsui T, Kobayashi M. Efficacy of oral anticancer agents for colorectal cancer. Dis Colon Rectum. 2006;49:S82–S91. doi: 10.1007/s10350-006-0601-7. [DOI] [PubMed] [Google Scholar]
- 27.National Cancer Institute, US National Institute of Health. Colon cancer treatment (PDQ®). http://www.cancer.gov/cancertopics/pdq/treatment/colon/HealthProfessional/69.cdr. Accessed 27 July 2010.
- 28.Ugurlu T, Turkoglu M, Gurer US, Akarsu BG. Colonic delivery of compression coated nisin tablets using pectin/HPMC polymer mixture. Eur J Pharm Biopharm. 2007;67:202–210. doi: 10.1016/j.ejpb.2007.01.016. [DOI] [PubMed] [Google Scholar]
- 29.Sinha VR, Kumria R. Polysaccharides in colon specific drug delivery. Int J Pharm. 2001;224:19–38. doi: 10.1016/s0378-5173(01)00720-7. [DOI] [PubMed] [Google Scholar]
- 30.Yang L, Chu JS, Fix JA. Colon-specific drug delivery, new approaches and in vitro/in vivo evaluation. Int J Pharm. 2002;235:1–15. doi: 10.1016/s0378-5173(02)00004-2. [DOI] [PubMed] [Google Scholar]
- 31.Van den Mooter G, Maris B, Samyn C, Augustijns P, Kinget R. Use of azopolymers for colon specific drug delivery. J Pharm Sci. 1997;86:1321–1327. doi: 10.1021/js9702630. [DOI] [PubMed] [Google Scholar]
- 32.Sinha VR, Kumria R. Coating polymers for colon specific drug delivery: a comparative in vitro evaluation. Acta Pharm. 2003;53:41–47. [PubMed] [Google Scholar]
- 33.Gazzaniga A, Maroni A, Sangalli ME, Zema L. Time-controlled oral delivery systems for colon targeting. Expert Opin Drug Deliv. 2006;3:583–597. doi: 10.1517/17425247.3.5.583. [DOI] [PubMed] [Google Scholar]
- 34.Shibata N, Ohno T, Shimokawa T, Hu Z, Yoshikawa Y, Koga K, Murakami M, Takada K. Application of pressure-controlled colon delivery capsule to oral administration of glycyrrhizin in dogs. J Pharm Pharmacol. 2001;53:441–447. doi: 10.1211/0022357011775730. [DOI] [PubMed] [Google Scholar]
- 35.Liu CS, Fishman ML, Hicks KB. Pectin in controlled drug delivery—a review. Cellulose. 2007;14:15–24. [Google Scholar]
- 36.Liu LS, Fishman ML, Kost J, Hicks KB. Pectin-based systems for colon-specific drug delivery via oral route. Biomaterials. 2003;24:3333–3343. doi: 10.1016/s0142-9612(03)00213-8. [DOI] [PubMed] [Google Scholar]
- 37.May CD. Pectins. In: Phillips GO, Williams PA, editors. Handbook of hydrocolloids, Chapter 10. UK: Woodhead Publishing Limited; 2003. pp. 169–188. [Google Scholar]
- 38.Ridley BL, O’Neill MA, Mohnen D. Pectins: structure, biosynthesis, and oligogalacturonide-related signaling. Phytochem. 2001;57:929–967. doi: 10.1016/s0031-9422(01)00113-3. [DOI] [PubMed] [Google Scholar]
- 39.Denev P. Kratchanov Chr. Influence of some cations on the reaction of apple pectin with ammonia in homogeneous media. In: Visser J, Voragen AGJ, editors. Pectins and pectinases. Amsterdam: Elsevier; 1996. pp. 527–540. [Google Scholar]
- 40.Rinaudo M. Physicochemical properties of pectins in solution and gel states. In: Visser J, Voragen AGJ, editors. Pectins and pectinases. Amsterdam: Elsevier; 1996. pp. 21–33. [Google Scholar]
- 41.Sriamornsak P. Chemistry of pectin and its pharmaceutical uses: a review. Silpakorn Uni Int J. 2003;3(1–2):206–228. [Google Scholar]
- 42.Moore MA, Park CB, Tsuda H. Soluble and insoluble fiber influences on cancer development. Crit Rev Oncol Hematol. 1998;27:229–242. doi: 10.1016/s1040-8428(98)00006-7. [DOI] [PubMed] [Google Scholar]
- 43.Lupton JR. Is fiber protective against colon cancer? Where the research is leading us. Nutrition. 2000;16(7/8):558–561. doi: 10.1016/s0899-9007(00)00350-6. [DOI] [PubMed] [Google Scholar]
- 44.Wong JM, de Souza R, Kendall CW, Emam A, Jenkins DJ. Colonic health: fermentation and short chain fatty acids. J Clin Gastroenterol. 2006;40:235–243. doi: 10.1097/00004836-200603000-00015. [DOI] [PubMed] [Google Scholar]
- 45.Ferguson LR, Harris PJ. Studies on the role of specific dietary fibres in protection against colorectal cancer. Mutat Res. 1996;350:173–184. doi: 10.1016/0027-5107(95)00105-0. [DOI] [PubMed] [Google Scholar]
- 46.Park HS, Choi JS, Kim KH. Docosahexaenoic acid-rich fish oil and pectin have a hypolipidemic effect, but pectin increases risk factor for colon cancer in rats. Nutr Res. 2000;20(12):1783–1794. [Google Scholar]
- 47.Bauer HG, Asp NG, Oste R. Effect of dietary fiber on the induction of colorectal tumors and fecal β-glucuronidase activity in the rat. Cancer Res. 1979;39(9):3752–3756. [PubMed] [Google Scholar]
- 48.Bauer HG, Asp NG, Dahigvist A. Effect of two kinds of pectin and guar gum on 1, 2-dimethylhydrazine initiation of colon tumors and on fecal β-glucuronidase activity in the rat. Cancer Res. 1981;41(6):2518–2523. [PubMed] [Google Scholar]
- 49.Ohkami H, Tazawa K, Yamashita I, Shimizu T, Murai K, Kobashi K, Fujimaki M. Effects of apple pectin on fecal bacterial enzymes in azoxymethane-induced rat colon carcinogenesis. Jpn J Cancer Res. 1995;86(6):523–529. doi: 10.1111/j.1349-7006.1995.tb02429.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tazawa K, Okami H, Yamashita I, Ohnishi Y, Kobashi K, Fujimaki M. Anticarcinogenic action of apple pectin on fecal enzyme activities and mucosal or portal prostaglandin E2 levels in experimental rat colon carcinogenesis. J Exp Clin Cancer Res. 1997;16(1):33–38. [PubMed] [Google Scholar]
- 51.Olano-Martin E, Rimbach GH, Gibson GR, Rastall RA. Pectin and pecticoligosaccharides induce apoptosis in in vitro human colonic adenocarcinoma cells. Anticancer Res. 2003;23(1A):341–346. [PubMed] [Google Scholar]
- 52.Heitman DW, Hardman WE, Cameron IL. Dietary supplementation with pectin and guar gum on 1, 2-dimethylhydrazine-induced colon carcinogenesis in rats. Carcinogenesis. 1992;13(5):815–818. doi: 10.1093/carcin/13.5.815. [DOI] [PubMed] [Google Scholar]
- 53.Umar S, Morris AP, Kourouma F, Sellin JH. Dietary pectin and calcium inhibit colonic proliferation in vivo by differing mechanisms. Cell Prolif. 2003;36(6):361–375. doi: 10.1046/j.1365-2184.2003.00291.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nakahara S, Raz A. Regulation of cancer-related gene expression by galectin-3 and the molecular mechanism of its nuclear import pathway. Cancer Metastasis Rev. 2007;26:605–610. doi: 10.1007/s10555-007-9095-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jacobasch G, Dongowski G, Florian S, Müller-Schmehl K, Raab B, Schmiedl D. Pectin does not inhibit intestinal carcinogenesis in APC-deficient Min/+ mice. J Agri Food Chem. 2008;56(4):1501–1510. doi: 10.1021/jf070872l. [DOI] [PubMed] [Google Scholar]
- 56.Glinsky VV, Raz A. Modified citrus pectin anti-metastatic properties: one bullet, multiple targets. Carbohydr Res. 2009;344:1788–1791. doi: 10.1016/j.carres.2008.08.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hayashi A, Gillen AC, Lott JR. Effects of daily oral administration of quercetin chalcone and modified citrus pectin on implanted colon-25 tumour growth in balb-c mice. Altern Med Rev. 2000;5(6):546–552. [PubMed] [Google Scholar]
- 58.Nangia-Makker P, Hogan V, Honjo Y, Baccarini S, Tait L, Bresalier R, Raz A. Inhibition of human cancer cell growth and metastasis in nude mice by oral intake of modified citrus pectin. J Natl Cancer Inst. 2002;94(24):1854–1862. doi: 10.1093/jnci/94.24.1854. [DOI] [PubMed] [Google Scholar]
- 59.Thirawong N, Nunthanid J, Puttipipatkhachorn S, Sriamornsak P. Mucoadhesive properties of various pectins on gastrointestinal mucosa: an in vitro evaluation using texture analyzer. Eur J Pharm Biopharm. 2007;67:132–140. doi: 10.1016/j.ejpb.2007.01.010. [DOI] [PubMed] [Google Scholar]
- 60.Liu LS, Fishman ML, Hicks KB, Kende M. Intercation of various pectin formulations with porcine colonic tissues. Biomaterials. 2005;26:5907–5916. doi: 10.1016/j.biomaterials.2005.03.005. [DOI] [PubMed] [Google Scholar]
- 61.Dongowski G, Anger H. Metabolism of pectin in the gastrointestinal tract. In: Visser J, Voragen AGJ, editors. Pectins and pectinases. Amsterdam: Elsevier; 1996. pp. 659–666. [Google Scholar]
- 62.Saito D, Nakaji S, Fukuda S, Shimoyama T, Sakamoto J, Sugawara K. Comparison of the amount of pectin in the human terminal ileum with the amount of orally administered pectin. Nutrition. 2005;21:914–919. doi: 10.1016/j.nut.2005.01.005. [DOI] [PubMed] [Google Scholar]
- 63.Ashford M, Fell J, Attwood D, Sharma H, Woodhead P. Studies on pectin formulations for colonic drug delivery. J Control Release. 1994;30:225–232. [Google Scholar]
- 64.Wei X, Sun N, Wu B, Yin C, Wu W. Sigmoidal release of indomethacin from pectin matrix tablets: Effect of in situ crosslinking by calcium cations. Int J Pharm. 2006;318:132–138. doi: 10.1016/j.ijpharm.2006.03.024. [DOI] [PubMed] [Google Scholar]
- 65.Rubinstein A, Radai R, Ezra M, Pathak S, Rokem JS. In vitro evaluation of calcium pectinate: a potential colon-specific drug delivery carrier. Pharm Res. 1993;10(2):258–263. doi: 10.1023/a:1018995029167. [DOI] [PubMed] [Google Scholar]
- 66.Wu B, Deng D, Lu Y, Wu W. Biphasic release of indomethacin from HPMC/pectin/calcium matrix tablet: II. Influencing variables, stability and pharmacokinetics in dogs. Eur J Pharm Biopharm. 2008;69:294–302. doi: 10.1016/j.ejpb.2007.10.001. [DOI] [PubMed] [Google Scholar]
- 67.El-Gibaly I. Oral delayed-release system based on Zn-pectinate gel (ZPG) microparticles as an alternative carrier to calcium pectinate beads for colonic drug delivery. Int J Pharm. 2002;232:199–211. doi: 10.1016/s0378-5173(01)00903-6. [DOI] [PubMed] [Google Scholar]
- 68.Adkin DA, Kenyon CJ. Itzhack Lerner E, Landau I, Strauss E, Caron D, Penhasi A, Rubinstein A, Wilding IR. The use of scintigraphy to provide “proof of concept” for novel polysaccharide preparations designed for colonic drug delivery. Pharm Res. 1997;14(1):103–107. doi: 10.1023/a:1012019820603. [DOI] [PubMed] [Google Scholar]
- 69.Ahrabi SF, Madsen G, Dyrstad K, Sande SA, Graffner C. Development of pectin matrix tablets for colonic delivery of model drug ropivacaine. Eur J Pharm Sci. 2000;10:43–52. doi: 10.1016/s0928-0987(99)00087-1. [DOI] [PubMed] [Google Scholar]
- 70.Wakerly Z, Fell JT, Attwood D, Parkins DA. In vitro evaluation of pectin-based colonic drug delivery systems. Int J Pharm. 1996;129:73–77. [Google Scholar]
- 71.Turkoglu M, Ugurlu T. In vitro evaluation of pectin-HPMC compression coated 5-aminosalicylic acid tablets for colonic delivery. Eur J Pharm Biopharm. 2002;53:65–73. doi: 10.1016/s0939-6411(01)00225-9. [DOI] [PubMed] [Google Scholar]
- 72.Macleod GS, Fell JT, Collett JH. An in vitro investigation into the potential for bimodal drug release from pectin/chitosan/HPMC-coated tablets. Int J Pharm. 1999;188:11–18. doi: 10.1016/s0378-5173(99)00197-0. [DOI] [PubMed] [Google Scholar]
- 73.Ofori-Kwakye K, Fell JT. Biphasic drug release from film-coated tablets. Int J Pharm. 2003;250:431–440. doi: 10.1016/s0378-5173(02)00578-1. [DOI] [PubMed] [Google Scholar]
- 74.Ofori-Kwakye K, Fell JT, Sharma HL, Smith A-M. Gamma scintigraphic evaluation of film-coated tablets intended for colonic or biphasic release. Int J Pharm. 2004;270:307–313. doi: 10.1016/j.ijpharm.2003.11.009. [DOI] [PubMed] [Google Scholar]
- 75.Wakerly Z, Fell JT, Attwood D, Parkins D. Studies on drug release from pectin/ethylcellulose film-coated tablets: a potential colonic delivery system. Int J Pharm. 1997;153:219–224. [Google Scholar]
- 76.Fernández-Hervás MJ, Fell JT. Pectin/chitosan mixtures as coatings for colon-specific drug delivery: an in vitro evaluation. Int J Pharm. 1998;169:115–119. [Google Scholar]
- 77.Chambin O, Dupuis G, Champion D, Voilley A, Pourcelot Y. Colon-specific drug delivery: influence of solution reticulation properties upon pectin beads performance. Int J Pharm. 2006;321:86–93. doi: 10.1016/j.ijpharm.2006.05.015. [DOI] [PubMed] [Google Scholar]
- 78.Dupuis G, Chambin O, Génelot C, Champion D, Pourcelot Y. Colonic drug delivery: influence of crosslinking agent on pectin beads properties and role of the shell capsule type. Drug Dev Ind Pharm. 2006;32(7):847–855. doi: 10.1080/03639040500536718. [DOI] [PubMed] [Google Scholar]
- 79.Bourgeois S, Gernet M, Pradeau D, Andremont A, Fattal E. Evaluation of critical formulation parameters influencing the bioactivity of β-lactamases entrapped in pectin beads. Int J Pharm. 2006;324:2–9. doi: 10.1016/j.ijpharm.2006.05.059. [DOI] [PubMed] [Google Scholar]
- 80.Chaurasia M, Chourasia MK, Jain NK, Jain A, Soni V, Gupta Y, Jain SK. Methotrexate bearing calcium pectinate microspheres: a platform to achieve colon-specific drug release. Curr Drug Deliv. 2008;5(3):215–219. doi: 10.2174/156720108784911668. [DOI] [PubMed] [Google Scholar]
- 81.Munjeri O, Collett JH, Fell JT. Hydrogel beads based on amidated pectins for colon-specific drug delivery: the role of chitosan in modifying drug release. J Control Release. 1997;46:273–278. [Google Scholar]
- 82.Chang KLB, Lin J. Swelling behavior and the release of protein from chitosan-pectin composite particles. Carbohydr Polym. 2000;43:163–169. [Google Scholar]
- 83.Hiorth M, Versland T, Heikkilä J, Tho I, Sande SA. Immersion coating of pellets with calcium pectinate and chitosan. Int J Pharm. 2006;308:25–32. doi: 10.1016/j.ijpharm.2005.10.012. [DOI] [PubMed] [Google Scholar]
- 84.Wei H, Qing D, De-Ying C, Bai X, Li-Fang F. Pectin/ethylcellulose as film coatings for colon-specific drug delivery: Preparation and in vitro evaluation using 5-fluorouracil pellets. PDA J Pharm Sci Technol. 2007;61(2):121–130. [PubMed] [Google Scholar]
- 85.Wei H, Qing D, De-Ying C, Bai X, Li-Fang F. Study on colon-specific pectin/ethylcellulose film-coated 5-fluorouracil pellets in rats. Int J Pharm. 2008;348:35–45. doi: 10.1016/j.ijpharm.2007.07.005. [DOI] [PubMed] [Google Scholar]
- 86.Semdé R, Amighi K, Devleeschouwer MJ, Moës AJ. Studies of pectin HM/Eudragit® RL/Eudragit® NE film-coating formulations intended for colonic drug delivery. Int J Pharm. 2000;197:181–192. doi: 10.1016/s0378-5173(99)00467-6. [DOI] [PubMed] [Google Scholar]
- 87.Semdé R, Amighi K, Devleeschouwer MJ, Moës AJ. Effect of pectinolytic enzymes on the theophylline release from pellets coated with water insoluble polymers containing pectin HM or calcium pectinate. Int J Pharm. 2000;197:169–179. doi: 10.1016/s0378-5173(99)00465-2. [DOI] [PubMed] [Google Scholar]
- 88.Maestrelli F, Cirri M, Corti G, Mennini N, Mura P. Development of enteric-coated calcium pectinate microspheres intended for colonic drug delivery. Eur J Pharm Biopharm. 2008;69:508–518. doi: 10.1016/j.ejpb.2007.12.004. [DOI] [PubMed] [Google Scholar]
- 89.Chan WA, Boswell CD, Zhang Z. Comparison of the release profiles of a water soluble drug carried by Eudragit-coated capsules in different in-vitro dissolution liquids. Powder Technol. 2001;119:26–32. [Google Scholar]
- 90.Jain A, Gupta Y, Jain SK. Potential of calcium pectinate beads for target specific drug release to colon. J Drug Targeting. 2007;15(4):285–294. doi: 10.1080/10611860601146134. [DOI] [PubMed] [Google Scholar]
- 91.Paharia A, Yadav AK, Rai G, Jain SK, Pancholi SS, Agrawal GP. Eudragit-coated pectin microspheres of 5-fluorouracil for colon targeting. AAPS Pharm Sci Tech. 2007;8(1):Article 12 E1– E7. [DOI] [PMC free article] [PubMed]
- 92.Atyabi F, Majzoob S, Iman M, Salehi M, Dorkoosh F. In vitro evaluation and modification of pectinate gel beads containing trimethyl chitosan, as a multi-particulate system for delivery of water-soluble macromolecules to colon. Carbohydr Polym. 2005;61(1):39–51. [Google Scholar]
- 93.Xi MM, Zhang SQ, Wang XY, Fang KQ, Gu Y. Study on the characteristics of pectin-ketoprofen for colon targeting in rats. Int J Pharm. 2005;298:91–97. doi: 10.1016/j.ijpharm.2005.04.012. [DOI] [PubMed] [Google Scholar]
- 94.Wang Q-W, Liu X-Y, Liu L, Feng J, Li Y-H, Guo Z-J, Mei Q-B. Synthesis and evaluation of the 5-fluorouracil-pectin conjugate targeted at the colon. Med Chem Res. 2007;16:370–379. [Google Scholar]
- 95.Semdé R, Amighi K, Pierre D, Devleeschouwer MJ, Moës AJ. Leaching of pectin from mixed pectin/insoluble polymer films intended for colonic drug delivery. Int J Pharm. 1998;174:233–241. [Google Scholar]
- 96.Hiorth M, Tho I, Sande SA. The formation and permeability of drugs across free pectin and chitosan films prepared by a spraying method. Eur J Pharm Biopharm. 2003;56:175–182. doi: 10.1016/s0939-6411(03)00065-1. [DOI] [PubMed] [Google Scholar]
- 97.Shike M. Diet and lifestyle in the prevention of colorectal cancer: an overview. Excerpta Med. 1999;106(1A):11S–15S. doi: 10.1016/s0002-9343(98)00340-4. [DOI] [PubMed] [Google Scholar]
- 98.Alabaster O, Tang Z, Shivapurkar N. Dietary fiber and the chemopreventive modelation of colon carcinogenesis. Mutat Res. 1996;350:185–197. doi: 10.1016/0027-5107(95)00114-x. [DOI] [PubMed] [Google Scholar]
- 99.Guarner F, Malagelada J-R. Gut flora in health and disease. Lancet. 2003;361(9356):512–519. doi: 10.1016/S0140-6736(03)12489-0. [DOI] [PubMed] [Google Scholar]
- 100.Oscar C, Thompson-Chagoyán OC, Maldonado J, Gil A. Colonization and impact of disease and other factors of intestinal microbiota. Dig Dis Sci. 2007;52:2069–2077. doi: 10.1007/s10620-006-9285-z. [DOI] [PubMed] [Google Scholar]
- 101.Charalambides D, Segal I. Colonic pH: a comparison between patients with colostomies due to trauma and colorectal cancer. Am J Gastroenterol. 1992;87(1):74–78. [PubMed] [Google Scholar]
- 102.McDougall CJ, Wong R, Scudera P, Lesser M, DeCosse JJ. Colonic mucosal pH in humans. Dig Dis Sci. 1993;38(3):542–545. doi: 10.1007/BF01316512. [DOI] [PubMed] [Google Scholar]
- 103.Pillay V, Fassihi R. In vitro release modulation from crosslinked pellets for site specific drug delivery to the gastrointestinal tract I. Comparison of pH-responsive drug release and associated kinetics. J Control Release. 1999;59:229–242. doi: 10.1016/s0168-3659(98)00196-5. [DOI] [PubMed] [Google Scholar]
- 104.Pillay V, Fassihi R. In vitro release modulation from crosslinked pellets for site specific drug delivery to the gastrointestinal tract II. Physicochemical characterization of calcium-alginate, calcium pectinate and calcium-alginate-pectinate pellets. J Control Release. 1999;59:243–256. doi: 10.1016/s0168-3659(98)00197-7. [DOI] [PubMed] [Google Scholar]
- 105.Joseph I, Venkataram S. Indomethacin sustained release from alginate-gelatin or pectin-gelatin coacervates. Int J Pharm. 1995;126:161–168. [Google Scholar]
- 106.Itoh K, Yahaba M, Takahashi A, Tsuruya R, Miyazaki S, Dairaku M, Togashi M, Mikami R, Attwood D. In situ gelling xyloglucan/pectin formulations for oral sustained drug delivery. Int J Pharm. 2008;356:95–101. doi: 10.1016/j.ijpharm.2007.12.049. [DOI] [PubMed] [Google Scholar]
- 107.Nurjaya S, Wong TW. Effects of microwave on drug release properties of matrices of pectin. Carbohydr Polym. 2005;62(3):245–257. [Google Scholar]
- 108.Wong TW, Nurjaya S. Drug release property of chitosan-pectinate beads and its changes under the influence of microwave. Eur J Pharm Biopharm. 2008;69:176–188. doi: 10.1016/j.ejpb.2007.09.015. [DOI] [PubMed] [Google Scholar]