

Abstract
The aim of this study was to develop a drug-specific absorption model for gliclazide (GLK) using mechanistic gastrointestinal simulation technology (GIST) implemented in GastroPlusTM software package. A range of experimentally determined, in silico predicted or literature data were used as input parameters. Experimentally determined pH-solubility profile was used for all simulations. The human jejunum effective permeability (Peff) value was estimated on the basis of in vitro measured Caco-2 permeability (literature data). The required PK inputs were taken from the literature. The results of the simulations were compared with actual clinical data and revealed that the GIST-model gave accurate prediction of gliclazide oral absorption. The generated absorption model provided the target in vivo dissolution profile for in vitro–in vivo correlation and identification of biorelevant dissolution specification for GLK immediate-release (IR) tablets. A set of virtual in vitro data was used for correlation purposes. The obtained results suggest that dissolution specification of more than 85% GLK dissolved in 60 min may be considered as “biorelevant” dissolution acceptance criteria for GLK IR tablets.
Key words: biorelevant dissolution specification, gliclazide, in vitro–in vivo correlation (IVIVC), mechanistic absorption simulation
INTRODUCTION
Gliclazide (GLK) is a second-generation sulfonylurea derivative, widely used for the treatment of type II diabetes mellitus. GLK is available as oral tablets (30 and 80 mg strength) with the recommended dosage between 40 and 320 mg/day. Reports from the in vivo studies show that, after oral administration, gliclazide is almost completely absorbed (1,2). However, due to its low and pH-dependent aqueous solubility (3–5), GLK absorption rate appears to be slow and variable (3,6–8), and therefore, its absorption profile is difficult to decipher.
Drug absorption and sufficient and reproducible bioavailability are recognized as some of the major issues considering drug delivery from solid oral dosage forms. In order to establish the relationship between drug physicochemical data and its clinical performance, a mechanistic approach to oral drug absorption based on the Biopharmaceutics Classification System (BCS) was introduced (9). According to the BCS concept, drug dose solubility and dissolution rate from pharmaceutical preparations along with intestinal permeability are major determinants of its absorption. Oral absorption of ionizable poorly water-soluble drugs (BCS class II weak electrolytes) is influenced by both their physicochemical properties and physiological conditions in the gastrointestinal (GI) tract (e.g., pH and the presence of surfactants). Although characterized by low aqueous solubility, these drugs often show high bioavailability after oral administration.
Oral drug absorption is usually estimated from the in vivo plasma concentration (Cp)–time data following intra- and extravascular drug inputs by means of conventional pharmacokinetic (PK) methods (numerical deconvolution, Wagner Nelson, Loo Riegelman). However, if intravenous data are lacking due to poor drug solubility and/or if the drug shows nonlinear kinetics, this might present a limitation for conventional pharmacokinetic analysis. Development of the in silico prediction tools capable of forecasting in vivo absorption solely on the basis of drug physicochemical and PK properties has therefore received widespread attention over the past few years (10–14). Based on the theory of the BCS and GI physiology, a semiphysiological absorption model named Advanced Compartmental Absorption and Transit model (ACAT; commercially available as GastroPlusTM software package) for in silico prediction of oral drug absorption was developed. The form of ACAT model implemented in GastroPlus is modeled by a system of coupled linear and nonlinear rate equations used to simulate the effect of physiological conditions on drug absorption as it transits through successive GI compartments. The equations include the consideration of six states (unreleased, undissolved, dissolved, degraded, metabolized, and absorbed), 18 compartments (stomach, seven compartments for the small intestine, colon, and nine enterocyte compartments), three states of excreted material (unreleased, undissolved, and dissolved), and the amount of drug in up to three PK compartments (when PK parameters are available). The total amount of absorbed material is summed over the integrated amounts being absorbed/exsorbed from each absorption/transit compartment (12). Besides physiological parameters, this model requires certain input parameters regarding drug physicochemical and PK data along with some product characteristics. Such parameters should adequately reflect drug biopharmaceutical properties.
The most commonly used drug physicochemical property to assess its in vivo performance is in vitro dissolution of a drug product. It is, therefore, important to define drug release methodology that would be predictive of its clinical performance and to establish quantitative in vitro–in vivo correlation (IVIVC). Validated IVIVC model, and consequently, identification of biorelevant dissolution method can aid in reducing the number of human in vivo studies during the development of generic formulations, their approval by the regulatory agencies, and certain post-approval changes.
The purpose of this study was: (1) to develop a drug-specific absorption model for gliclazide using gastrointestinal simulation technology (GIST), (2) to use the generated absorption model to provide the target in vivo dissolution profile for IVIVC, and (3) to identify biorelevant dissolution specifications for GLK IR tablets based on a set of virtual in vitro data. Level A IVIVC based on deconvolution and convolution approaches were applied to assess the relationship between the in vitro and in vivo data. Dissolution acceptance criteria were discussed in terms of the extent in which the differences in drug release kinetics observed in vitro were reflected on the simulated in vivo dissolution profile.
MATERIALS AND METHODS
In Vivo Data
The pharmacokinetic inputs required for the simulation, calculated on the basis of the actual clinical data, were taken from the literature (7). Published data from gliclazide bioequivalence (BE) studies (Diamicron® 80 mg IR tablets) (2) were used to evaluate the resultant gliclazide absorption model. These data were also used for in vitro–in vivo correlation purposes.
In Vitro Studies
Solubility Determination
Equilibrium solubility was determined by a “shake-flask” method using various buffer media in the pH range 1.1–9.0. Excess amount of gliclazide powder (Zhejiang Jiuzhou Pharmaceutical Co. Ltd., China) was placed into the vials containing 40 mL of each tested media and shaken at 250 rpm for 48 h at 37 ± 0.5°C. Samples withdrawn were filtered, properly diluted, and assayed for gliclazide UV spectrophotometrically (Evolution 300, Thermo Fisher Scientific, UK) at the wavelength of the relative maximum absorption. All measurements were performed in triplicate.
Dissolution Studies
Dissolution studies of 80-mg generic gliclazide IR tablets commercially available on Serbian market (Glioral, Galenika a.d., Serbia; Glikosan, Slaviamed, Serbia; and Diprian, Hemofarm a.d., Serbia) were carried out in a rotating paddle apparatus (Erweka DT 70, Germany) at 37 ± 0.5°C and rotational speed of 100 rpm, using 900 ml of various dissolution media (media pH 1.2, 4.0, 4.5, 6.8, 7.2, and 7.4). Withdrawn samples were filtered and after appropriate dilution, assayed for gliclazide UV spectrophotometrically at the wavelength of the relative maximum absorbance.
Experimental in vitro data for 80-mg generic GLK tablets and literature in vitro data for Diamicron® 80-mg GLK tablets (3) were used to construct a set of virtual in vitro data used for GastroPlusTM simulation. The investigated in vitro profiles were generated to reflect the situation where less than 85% of the drug is dissolved (incomplete dissolution because of limited GLK solubility in the pH range 3.0–4.2; profile a); more than 85% of the drug is dissolved in 15 (“very rapid” dissolution criteria; profile e), 30 (“rapid” dissolution criteria; profile d), 45 (profile c), or 60 min (profile b).
Mechanistic Simulations
GastroPlusTM (version 6.1.0003, Simulations Plus, Inc., Lancaster, CA, USA) was used to simulate the in vivo absorption profile of gliclazide. The program has three input tabs, namely Compound, Physiology, and Pharmacokinetics, comprising three sets of factors influencing oral drug absorption. The required input parameters related to gliclazide physicochemical and pharmacokinetic properties were experimentally determined, in silico predicted and/or taken from the literature. Summary of the input parameters used is given in Table I. Considering controversial literature data regarding gliclazide solubility (3,5), experimentally determined pH-solubility profile was employed for all simulations. For drug absorption simulation based on GLK physicochemical and PK data, the “IR tablet mode” was specified; when in vitro dissolution profiles of IR GLK tablets were used as an input function in GastroPlusTM, the “tabulated in vitro dissolution data” function together with the “CRU-dispersed” dosage form was selected.
Table I.
Summary of the GLK Input Parameters Employed for Gastrointestinal Simulation
| Parameter | Value |
|---|---|
| Molecular weight | 323.4 g/mol |
| log P | 1.448a |
| pKa | 2.9; 5.8; 9.6b |
| Human jejunal permeability | 3.683 × 10−4 cm/sc |
| Dose | 80 mg |
| Dose volume | 250 ml |
| Solubility (pH 4.37) | 0.025 mg/mld |
| Mean precipitation time | 900 se |
| Diffusion coefficient | 0.782 × 10−5 cm2/sa |
| Drug particle density | 1.2 g/mle |
| Effective particle radius | 25 μme |
| Body weight | 74 kg |
| FPE (liver) | 30%f |
| Blood/plasma conc. ratio | 1e |
| Unbound percent in plasma | 4.7%f |
| CL | 0.012 L/h/kgf |
| V c | 0.23 L/kgf |
| Elimination half-life, t 1/2 | 13.29 h |
| Simulation time | 48 h |
GLK gliclazide, FPE first-pass extraction (liver), CL clearance, V c volume of distribution
a In silico predicted (ADMETPredictor™ module)
bEstimated by GastroPlus™ on the basis of experimentally determined pH-solubility profile
cValue calculated on the basis of in vitro measured permeability (Caco-2 cell line; 24) using permeability converter utility integrated in GastroPlus™ software
dExperimental values
eDefault GastroPlus™
fLiterature value taken from (7)
In the Physiology tab, the Opt logD Model SA/V 6.1 was used to estimate the changes in permeability as drug travels along the GI tract. The absorption gradient coefficients C1–C4 were adjusted (using the Optimization module) to best match the resultant model to the experimental data. These coefficients were used to calculate the absorption scale factors (ASF) which scale the effective permeability to account for variations in absorption-rate-determining effects (e.g., pH effects, the presence of influx and efflux transporters) that differ from one compartment to another (12). All other parameters were fixed at default values that represent human fasted physiology.
Parameter sensitivity analysis (PSA) was used to assess the effect of input PK parameters (systemic clearance, volume of distribution, first-pass effect) on the predicted rate and extent of GLK absorption.
In Vitro–In Vivo Correlation
Numerical Convolution
A set of virtual in vitro data representing different dissolution scenarios was used as the input function in GastroPlusTM software to estimate the expected GLK in vivo absorption profiles. The profiles obtained were compared with the mean plasma concentration profile of gliclazide observed after oral administration of 80-mg tablets (2).
Numerical Deconvolution
In the deconvolution approach, the percent of drug absorbed at the specified time points estimated by GastroPlusTM was plotted against the percent dissolved in vitro at the same time points. Time scaling factor was taken into account considering the time discrepancies between the in vitro and in vivo release profiles.
Linear regression analysis was used to evaluate the obtained correlation plots.
RESULTS AND DISCUSSION
Solubility
The experimentally obtained solubility data indicate that gliclazide is an ampholyte with pH-dependent solubility in the GI pH range (Fig. 1).
Fig. 1.

pH-dependent solubility of gliclazide at 37 ± 0.5°C and calculated dose numbers (D 0) for 80 mg gliclazide dose. Dotted lines represent the critical dose numbers for GLK (conservative upper limit of D 0 = 1, and the calculated value of D 0 = 14.5)
According to the BCS (15), GLK meets the criteria of a low solubility drug. Regarding the conservative criterion for the upper limit of dose number (D0 = 1), solubility is expected to be the limiting factor for GLK absorption up to pH 6.9. In order to determine the solubility-limited region for GLK in a more accurate way, the critical dose number value for GLK was calculated using the equation (Eq. 1) that relates the extent of drug absorption to its dose number, assuming the drug is highly permeable (9):
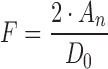 |
1 |
where F is the fraction of drug absorbed, and An is the absorption number calculated by GastroPlusTM (An = 7.27). The critical value of D0 was calculated under the assumption that the drug is completely absorbed (F = 1).
Regarding the obtained D0 value (D0 > 14.5), pH 3.0–4.2 can be considered as a solubility-limited region of GLK absorption. Above the pH 4.2 boundary, gliclazide solubility markedly increased, indicating that solubility is not the limiting factor for the absorption throughout the intestine. Therefore, GLK might be classified as intermediate solubility drug with non-solubility-limited absorption after oral administration.
Mechanistic Simulation–Model Validation
Gastrointestinal simulation for GLK IR tablets based on the input parameters presented in Table I was performed using the GastroPlusTM Single Simulation Mode. The simulated GLK absorption profile is presented in Fig. 2, together with the mean plasma profile taken from the literature (2).
Fig. 2.

GastroPlusTM predicted (line) and observed (2) (open square) mean GLK plasma C p–time profiles following administration of a single 80-mg gliclazide IR tablet
The predicted fraction of drug absorbed (Fa) was 99.94% which is in accordance with the literature-reported almost 100% bioavailability of gliclazide after oral administration (1,2). The predicted and in vivo observed pharmacokinetic parameters following oral administration of 80-mg GLK IR tablets are shown in Table II. The percent prediction error values were less than 10% for Cmax and area-under-the-curve values indicating that the generated absorption model gave good prediction of gliclazide oral absorption. Although the percent prediction error for tmax was relatively high (18.22%), it has to be considered that this value was calculated in comparison to the mean tmax value estimated for a particular in vivo observed data set (2). Considering the variable gliclazide in vivo kinetics (reported mean tmax values after oral administration of IR tablets varied between 2.3 and 4.5 h (2,6,16), the simulated value of 3.68 h can be considered as a reasonable estimate.
Table II.
Comparison of Pharmacokinetic Parameters Between Simulated and In Vivo Observed Data for GLK After Oral Administration of 80-mg IR Tablets
| Parameter | Observed | Simulated | PE (%) |
|---|---|---|---|
| C max (μg/ml) | 2.98 | 2.77 | 7.05 |
| t max (h) | 4.50 | 3.68 | 18.22 |
| AUC0→∞ ( μg h/mL) | 67.15 | 63.03 | 6.13 |
| AUC0→t ( μg h/mL) | 52.40 | 57.07 | −8.92 |
GLK gliclazide, IR immediate-release, PE prediction error values, AUC area under the curve
The resultant ASF values, adjusted to best fit the observed plasma concentration–time data for GLK IR tablets, were lower than GastroPlusTM generated values, indicating possible influence of efflux transporters on gliclazide absorption through the small intestine. This assumption is supported by the results of Al-Salami and associates revealing that gliclazide is a substrate of the ileal efflux drug transporters Mrp2 and Mrp3 (17,18). The role of transporters in oral drug absorption have been emphasized by the introduction of the Biopharmaceutics Drug Disposition Classification System (BDCCS) that enables predictability of in vivo drug transport, absorption, and disposition on the basis of drug solubility and the extent of metabolism (19,20). Regarding experimentally obtained pH-solubility data and literature-reported extensive metabolism in liver (2), gliclazide would be assigned as BDCCS class II drug wherein efflux transporters are expected to affect the extent and rate of the oral drug absorption.
Parameter sensitivity analysis (data not shown) revealed that the percent of GLK absorbed (Fa) is rather insensitive to the variations in PK parameters tested. The PSA also showed that broad variations in the selected PK parameters affect the predicted Cmax and tmax, but there were no significant differences observed when these PK inputs were varied in a range covering literature-reported values (1,6).
GastroPlusTM generated regional absorption distribution demonstrated that majority of GLK is absorbed in duodenum and jejunum (69.9%), while the rest of the dose is absorbed in mid and distal GI regions (Fig. 3). This is in accordance with solubility results indicating that GLK solubility in proximal parts of the intestine might not be enough for drug absorption to be completed in these compartments. However, in mid and distal GI regions (ileum, caecum), GLK solubility is high enough for the total amount of remaining undissolved drug to completely dissolve and, consequently, pass into the systemic circulation.
Fig. 3.

Compartmental absorption of gliclazide
In Vitro–In Vivo Correlation
Numerical Convolution
Different virtual dissolution profiles used as inputs for gastrointestinal simulations are presented in Fig. 4a. The corresponding Cp–time profiles (Fig. 4b), estimated on the basis of the generated GLK-specific absorption model, were plotted against the in vivo observed data in order to develop a level A IVIVC model (Fig. 5). The obtained correlation coefficients and slopes of the regression lines are given in Table III.
Fig. 4.

Virtual gliclazide dissolution profiles (a) and the corresponding simulated in vivo profiles, along with the actual in vivo data (2). The simulated profiles b, c, and d overlap (b)
Fig. 5.

IVIVC plot for GLK IR tablets (convolution approach)
Table III.
Statistical Parameters of the Obtained IVIVC (Convolution Approach)
| In vitro inputs | a Value | r Value |
|---|---|---|
| Profile a | 0.440 | 0.382 |
| Profile b | 0.894 | 0.897 |
| Profile c | 0.896 | 0.910 |
| Profile d | 0.898 | 0.923 |
| Profile e | 0.867 | 0.947 |
a slope of the regression line, r coefficient of correlation
The results obtained indicated that variations in drug input kinetics were well reflected on the simulated in vivo profiles. However, it is evident that differences observed in vitro were less pronounced in the predicted PK profiles (the simulated profiles b, c, and d overlap). The highest degree of deviation from the in vivo observed profile was demonstrated for profile a, representing scenario in which less than 85% of the drug is dissolved. On the other hand, values of the slope close to unity as well as high coefficients of correlation indicated the presence of level A correlation for the profiles b, c, e, and d.
Numerical Deconvolution
In an attempt to establish IVIVC using deconvolution approach, hypothetical in vivo absorption profile estimated by GastroPlusTM from the in vivo Cp–time curve (2) was compared with the virtual in vitro dissolution profiles. In order to produce a meaningful correlation, it was assumed that these profiles must not differ in their morphology. In the case of GLK IR tablets, the in vitro dissolution profile ran ahead the in vivo dissolution, so rescaling of the time axis when progressing from in vitro to in vivo was necessary. IVIVC plot of the percentage dissolved in vitro versus the percentage absorbed in vivo is presented in Fig. 6.
Fig. 6.

IVIVC plot for GLK IR tablets (deconvolution approach)
The outcomes of deconvolution approach (Table IV) are in accordance with those obtained by convolution approach. It was shown that the in vitro profile e (stretched by 12-fold linear rescaling of the time axis) has the same general shape (morphology) as the estimated hypothetical in vivo dissolution profile. However, good correlation was also achieved for the in vitro profiles b, c, and d (as illustrated by the estimated statistical parameters).
Table IV.
Statistical Parameters of the Obtained IVIVC (Deconvolution Approach)
| In vitro inputs | a Value | r Value |
|---|---|---|
| Profile a | 2.289 | 0.875 |
| Profile b | 1.031 | 0.894 |
| Profile c | 1.056 | 0.929 |
| Profile d | 0.946 | 0.896 |
| Profile e | 1.189 | 0.999 |
a slope of the regression line, r coefficient of correlation
Dissolution Test Requirements
British Pharmacopoeia (BP) gives recommendation for dissolution test conditions for GLK IR tablets (900 ml of media pH 7.4, paddle apparatus at 100 rpm), but no dissolution test requirements have been appointed so far in BP or other regulatory documents. Regarding the pH-solubility profile of GLK, the dissolution process at pH 7.4 is expected to be fast and complete (complying with the hypothetical profile representing “very rapid” dissolution scenario). Our results suggest that dissolution specification of >85% GLK dissolved in 60 min may be considered as biorelevant dissolution acceptance criteria for GLK IR tablets.
Biowaiver Considerations
The most common type of biowaiver adopted by the regulatory authorities include the application of the BCS-based scheme (similar or rapid/very rapid dissolution profiles of the test and reference product in pH 1.2, 4.5, and 6.8 media) or the application of IVIVC. In the case of IR dosage forms, biowaivers may be requested solely for highly soluble and highly permeable substances (BCS class I) when its drug product is (very) rapidly dissolving and exhibits similar dissolution profile to the reference product, while the IVIVC-based approach has been narrowed down to applications for extended-release products (15,21,22). Recent suggestions point out that IVIVC-based biowaiver concept could be extended to some BCS class II drugs under the assumption that the drug dissolves completely during the GI passage (23). In the case of highly soluble (BCS class I) drugs, it is generally accepted that complete dissolution in vivo complies with very rapid/rapid in vitro dissolution kinetics (15,21). However, in the case of BCS class II drugs, complete in vivo dissolution might occur at later time points, allowing wider biorelevant in vitro dissolution specification to be set. This assumption is supported by the results of the present study demonstrating that differences in GLK in vitro dissolution kinetics such as 85% drug dissolved within the 15–60 min time frame would not be reflected on in vivo pharmacokinetic profile.
In order to assess whether dissolution is a rate-limited factor for gliclazide absorption, dissolution behavior of different GLK IR tablets was studied in various pH media (pH 1.2, 4.0, and 7.2). Experimental data for generic tablets commercially available on Serbian market, together with the literature data for Diamicron tablets (3) are shown in Fig. 7. In medium pH 4.0, dissolution was slow and incomplete for all the investigated formulations, presumably due to the low solubility of gliclazide at pH 3.0–4.2. Although gliclazide exhibits comparable solubility at pH 1.2 and 7.2, slower release rate at pH 1.2 (less than 85% of GLK dissolved in 60 min from Glikosan and Diamicron tablets) indicated that solubility is not a major factor that governs gliclazide dissolution rate in this medium. According to Hong et al. (3), slow dissolution at pH 1.2 could be explained by slow water penetration into the dosage form at this pH. On the other hand, the release process in pH 7.2 medium was fast and complete, except in the case of Diprian tablets. Less than 10% gliclazide released into the pH 7.2 buffer within 60 min indicate that Diprian tablets might correspond to a modified-release (MR) formulation. However, further in vivo studies are needed in order to investigate whether the resultant plasma concentration–time profile following oral administration of Diprian tablets resemble the one resulting from typical MR gliclazide formulation. It should be noted that, regardless of the favorable dissolution test conditions (high solubility at pH 7.2, intensive rotational speed of 100 rpm), this medium was still able to discriminate between different gliclazide formulations (drug release rates from Glioral and Glikosan tablets were “very rapid”, while Diamicron tablets complied with “rapidly” dissolving formulation). In the absence of in vivo bioequivalence data (gliclazide plasma levels after administration of Glioral and Glikosan tablets in comparison to Diamicron tablets were not available), there is not enough information to conclude whether pH 7.2 media at 100 rpm would provide biorelevant and discriminatory test conditions for in vitro evaluation of GLK IR tablets. However, the present results indicate that GLK solubility and dissolution from IR tablets are not expected to be the rate-limiting factors for gliclazide in vivo absorption, and since this is a highly permeable drug, there is a rationale to postulate that biowaiver extension might be applicable in the case of GLK IR tablets. Additionally, other factors such as the potential impact of intestinal efflux transporters on GLK absorption should be thoroughly investigated and understood when considering biowaiver extension.
Fig. 7.

Dissolution profiles of generic 80-mg gliclazide tablets commercially available on Serbian market (experimental data), together with the literature data for Diamicron 80-mg gliclazide tablets (3) in different media: pH 1.2 (a), 4.0 (b), and 7.2 (c)
CONCLUSION
The presented data demonstrate that gastrointestinal simulation technology can be successfully used to predict GLK absorption profile. In the present case, both convolution and deconvolution approaches were successful in establishing a level A IVIVC. Based on the results obtained by IVIVC in conjunction with GIST, dissolution specification of >85% GLK dissolved in 60 min could be suggested as biorelevant dissolution acceptance criteria for GLK IR tablets. The dissolution criteria depicted in this study can be used to develop in vitro methodology that would be predictive of drug products in vivo behavior and that might eventually serve as surrogate for clinical BE studies (biowaiver) for GLK IR tablets.
Acknowledgments
This work was done under the project Biopharmaceutical Characterization of the Selected BCS Class II and III Drugs: In Vitro and In Silico Methods Evaluation (TR-23015) supported by the Ministry of Science and Technological Development, Republic of Serbia.
References
- 1.Delrat P, Paraire M, Jochemsen R. Complete bioavailability and lack of food-effect on pharmacokinetics of gliclazide 30 mg modified release in healthy volunteers. Biopharm Drug Dispos. 2002;23:151–157. doi: 10.1002/bdd.303. [DOI] [PubMed] [Google Scholar]
- 2.Najib N, Idkaidek N, Beshtawi M, Bader M, Admour I, Mahmood Alam S, et al. Bioequivalence evaluation of two brands of gliclazide 80 mg tablets (Glyzide® & Diamicron®)—in healthy human volunteers. Biopharm Drug Dispos. 2002;23:197–202. doi: 10.1002/bdd.310. [DOI] [PubMed] [Google Scholar]
- 3.Hong SS, Lee SH, Lee YJ, Chung SJ, Lee MH, Shim CK. Accelerated oral absorption of gliclazide in human subjects from a soft gelatin capsule containing a PEG 400 suspension of gliclazide. J Control Release. 1998;51:185–192. doi: 10.1016/S0168-3659(97)00167-3. [DOI] [PubMed] [Google Scholar]
- 4.Özkan Y, Atay T, Dikmen N, Işimer A, Aboul-Enein HY. Improvement of water solubility and in vitro dissolution rate of gliclazide by complexation with β-cyclodextrin. Pharm Acta Helv. 2000;74:365–370. doi: 10.1016/S0031-6865(99)00063-1. [DOI] [PubMed] [Google Scholar]
- 5.Shewale BD, Fursule RA, Sapkal NP. Effect of pH and hydroxylpropyl-β-cyclodextrin on solubility and stability of gliclazide. Int J Health Res. 2008;1:95–99. [Google Scholar]
- 6.Kobayashi K, Kimura M, Sakoguchi T, Kitani Y, Hata M, Matsuoka A. Influence of blood proteins on biomedical analysis. III. Pharmacokinetics and protein binding of gliclazide. J Pharm Dyn. 1981;4:436–442. doi: 10.1248/bpb1978.4.436. [DOI] [PubMed] [Google Scholar]
- 7.Davis TME, Daly F, Walsh JP, Ilett KF, Beilby JP, Dusci LJ, et al. Pharmacokinetics and pharmacodynamics of gliclazide in Caucasians and Australian Aborigines with type 2 diabetes. Br J Clin Pharmacol. 2000;49:223–230. doi: 10.1046/j.1365-2125.2000.00162.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rana MKZ. Gliclazide and glibenclamide interactions with antacids and H2-antagonists. Ph.D Thesis, Department of Chemistry, University of Karachi, Pakistan. 2003. http://prr.hec.gov.pk/Chapters/973-1.pdf. Accessed 23 Jun 2010.
- 9.Amidon GL, Lennernas H, Shah VP, Crison JR. A theoretical basis for a biopharmaceutic drug classification: the correlation of in vitro drug product dissolution and in vivo bioavailability. Pharm Res. 1995;12:413–420. doi: 10.1023/A:1016212804288. [DOI] [PubMed] [Google Scholar]
- 10.Yu LX, Lipka E, Crison JR, Amidon G. Transport approaches to the biopharmaceutical design of oral drug delivery systems: prediction of intestinal absorption. Adv Drug Deliv Rev. 1996;19:359–376. doi: 10.1016/0169-409X(96)00009-9. [DOI] [PubMed] [Google Scholar]
- 11.Norris DA, Leesman GD, Sinko PJ, Grass GM. Development of predictive pharmacokinetic simulation models for drug discovery. J Control Release. 2000;65:55–62. doi: 10.1016/S0168-3659(99)00232-1. [DOI] [PubMed] [Google Scholar]
- 12.Agoram B, Woltosz WS, Bolger MB. Predicting the impact of physiological and biochemical processes on oral drug bioavailability. Adv Drug Deliv Rev. 2001;50(Suppl 1):S41–67. doi: 10.1016/S0169-409X(01)00179-X. [DOI] [PubMed] [Google Scholar]
- 13.Yokoe J, Iwasaki N, Haruta S, Kadono K, Ogawara K, Higaki K, et al. Analysis and prediction of absorption behavior of colon-targeted prodrug in rats by GI-transit-absorption model. J Control Release. 2003;86:305–313. doi: 10.1016/S0168-3659(02)00424-8. [DOI] [PubMed] [Google Scholar]
- 14.Okumu A, DiMaso M, Löbenberg R. Computer simulations using GastroPlusTM to justify a biowaiver for etoricoxib solid oral drug products. Eur J Pharm Biopharm. 2009;72:91–98. doi: 10.1016/j.ejpb.2008.10.019. [DOI] [PubMed] [Google Scholar]
- 15.Food and Drug Administration/Center for Drug Evaluation and Research. Guidance for industry: waiver of in vivo bioavailability and bioequivalence studies for immediate-release solid oral dosage forms based on a biopharmaceutics classification system. 2000. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm070246.pdf. Accessed 23 Jun 2010.
- 16.Głowka FK, Hermann TW, Zabel M. Bioavailability of gliclazide from some formulation tablets. Int J Pharm. 1998;172:71–77. doi: 10.1016/S0378-5173(98)00167-7. [DOI] [Google Scholar]
- 17.Al-Salami H, Butt G, Tucker I, Fawcett P, Golocorbin-Kon S, Mikov I, Mikov M. Gliclazide reduces MKC intestinal transport in healthy but not diabetic rats. Eur J Drug Metabol Pharmacokinet. 2009;34:43–50. doi: 10.1007/BF03191383. [DOI] [PubMed] [Google Scholar]
- 18.Al-Salami H, Butt G, Tucker I, Mikov M. Influence of the semisynthetic bile acid (MKC) on the ileal permeation of gliclazide in healthy and diabetic rats. Pharmacol Rep. 2008;60:532–541. [PubMed] [Google Scholar]
- 19.Wu CY, Benet L. Predicting drug disposition via application of BCS: transport/absorption/elimination interplay and development of a biopharmaceutics drug disposition classification system. Pharm Res. 2005;22:11–23. doi: 10.1007/s11095-004-9004-4. [DOI] [PubMed] [Google Scholar]
- 20.Benet L, Amidon G, Barends D, Lennernäs H, Polli J, Shah V, Stavchansky S, Yu L. The use of BDDCS in classifying the permeability of marketed drugs. Pharm Res. 2007;52:483–488. doi: 10.1007/s11095-007-9523-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.European Medicines Agency, Committee For Medicinal Products For Human Use (CHMP), Guideline On The Investigation Of Bioequivalence. 2010. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2010/01/WC500070039.pdf. Accessed 5 Nov 2010
- 22.Food and Drug Administration, Center for Drug Evaluation and Research (CDER). Guidance for industry Extended Release Oral Dosage Forms: Development, Evaluation, and Application of In Vitro/In Vivo Correlations. 1997. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm070239.pdf. Accessed 26 Oct 2010
- 23.Yu LX, Amidon GL, Polli JE, Zhao H, Mehta MU, Conner DP, et al. Biopharmaceutics classification system: the scientific basis for biowaiver extensions. Pharm Res. 2002;19:921–925. doi: 10.1023/A:1016473601633. [DOI] [PubMed] [Google Scholar]
- 24.Stetinova V, Polaskova A, Smetanova L, Kholova D, Herout V, Kvetina J. Toxicological studies, membrane transport and pharmacodynamic effect of gliclazide in rats. Toxicol Lett. 2008;180(Suppl 1):S58–59. doi: 10.1016/j.toxlet.2008.06.639. [DOI] [Google Scholar]