

Abstract
Background & Aims
Ablation of Notch signaling within the intestinal epithelium results in loss of proliferating crypt progenitors, due to their conversion into post-mitotic secretory cells. We aimed to confirm that Notch was active in stem cells (SC), investigate consequences of loss of Notch signaling within the intestinal SC compartment, and identify the physiological ligands of Notch in mouse intestine. Furthermore, we investigated whether the induction of goblet cell differentiation that results from loss of Notch requires the transcription factor Krüppel-like factor 4 (Klf4).
Methods
Trasgenic mice that carried a reporter of Notch1 activation were used for lineage tracing experiments. The in vivo functions of the Notch ligands Jagged1 (Jag1), Delta-like1 (Dll1), Delta-like4 (Dll4), and the transcription factor Klf4 were assessed in mice with inducible, gut-specific gene targeting (Vil-Cre-ERT2).
Results
Notch1 signaling was found to be activated in intestinal SC. Although deletion of Jag1 or Dll4 did not perturb the intestinal epithelium, inactivation of Dll1 resulted in a moderate increase in number of goblet cells without noticeable effects of progenitor proliferation. However, simultaneous inactivation of Dll1 and Dll4 resulted in the complete conversion of proliferating progenitors into post-mitotic goblet cells, concomitant with loss of SC (Olfm4+, Lgr5+ and Ascl2+). Klf4 inactivation did not interfere with goblet cell differentiation in adult wild-type or in Notch pathway-deficient gut.
Conclusions
Notch signaling in SC and progenitors is activated by Dll1 and Dll4 ligands and is required for maintenance of intestinal progenitor and SC. Klf4 is dispensable for goblet cell differentiation in intestines of adult Notch-deficient mice.
Keywords: GI development, knockout mice, intestinal stem cells, gene regulation
Introduction
The epithelium of the small intestine consists of four principal cell types: absorptive enterocytes, mucus secreting goblet cells, antimicrobial Paneth cells, and hormone secreting enteroendocrine cells. While most differentiated cell types reside within the villus, Paneth cells are found at the bottom of the crypt. They are in close contact with the leucine-rich repeat-containing G-protein-coupled receptor 5 (Lgr5) and olfactomedin-4 (Olfm4) expressing intestinal stem cells (SC)1, 2. These SC, also known as crypt base columnar (CBC) cells, proliferate constantly1 and give rise to transient amplifying (TA) cells, which localize within the crypt compartment just above the Paneth cells. TA cells migrate upwards and differentiate at the crypt-villus boundary into one of the 4 known cell types of the small intestine. These regeneration and differentiation processes have to be under stringent control to ensure proper homeostasis.
The Notch cascade mediates cell-to-cell signaling and has been shown to be essential for the maintenance of the proliferative crypt compartment, as well as for the formation of adsorptive enterocytes (reviewed in3). The mammalian Notch receptor-ligand family consists of four single trans-membrane Notch receptors (Notch1-4) and five single trans-membrane ligands (Delta-like1, 3, 4, and Jagged1, 2; hereafter Dll1, 3, 4 and Jag1, 2)4. Ligand-receptor interactions between neighboring cells lead to sequential proteolytic cleavages of the receptor liberating its intracellular domain (NICD). Once released, NICD translocates to the nucleus and heterodimerizes with the transcription factor CSL (also known as RBP-J in the mouse) inducing transcription of downstream target genes. Inhibition of Notch signaling in the intestinal epithelium using conditional gene targeting of RBP-J or by pharmacological γ-secretase inhibitors, which block the release of NICD, results in the loss of the proliferative crypt compartment and conversion of crypt progenitors into secretory cells5. We recently showed that Notch1 and Notch2 receptors act redundantly in mediating Notch signaling in the intestine6. Only simultaneous conditional gene inactivation or antibody-mediated inhibition of both receptors resulted in the complete conversion of the crypt progenitors into postmitotic goblet cells6, 7. Taken together, these results suggest that Notch signaling is not only a gatekeeper for proliferating crypt progenitors but is also involved in controlling the balance between secretory and absorptive cell types. However, whether the role Notch plays in SC maintenance reflects direct activation of Notch signaling in intestinal SC is currently unknown.
Cell fate decisions and differentiation processes within the intestinal epithelium involve the Notch target gene Hes1, which represses the bHLH transcription factor Math18. The intestine of Hes1 deficient mice contain increased Paneth, goblet and enteroendocrine cell numbers and have a decrease in absorptive cells8, 9, while Math1 mutant animals have lost most secretory cells and are only colonized by enterocytes10, 11. These results suggest that Hes1-mediated repression of Math1 contributes to the regulation of the secretory versus adsorptive lineage decision. Another important transcription factor shown to be repressed via Notch mediated Hes1 expression is the zinc finger transcription factor Krüppel-like factor 4 (Klf4)12-14. Klf4 deficient mice (p1) showed a 90% decrease of goblet cell numbers within the colon indicating that Klf4 is important for goblet cell differentiation during fetal life15, 16. The fact that blocking Notch signaling correlates with increased Klf4 mRNA expression and results in goblet cell metaplasia, combined with in vitro studies showing that Notch or Hes1 can repress Klf4 promoter driven transcription, resulted in a model in which Notch signaling suppresses the goblet cell fate through Hes1 mediated repression of Klf412-14.
Here, we identify the physiological Notch ligands of the murine intestine, and demonstrate the importance of the Notch pathway for proper intestinal SC homeostasis. Furthermore, our results show that Klf4 is dispensable for goblet cell differentiation in adult intestines of Notch mutant mice, complementing recent observations that Math1 acts downstream of Notch in driving the goblet cell fate17.
Material and methods
Animals
All animal work was conducted according to Swiss national guidelines. This study has been reviewed and approved by the Service Vétérinaire Cantonal of Etat de Vaud. All NIP1::CreERT2 study related animals were housed in the Washington University mouse facility and were approved by the Animal Studies Committee of Washington University.
The Cre-ERT2 recombinase activity was induced by injecting 2-3 week old mice with tamoxifen (10mg/kg body weight in Corn oil; Sigma) intraperitoneally for 5 consecutive days. The γ-secretase inhibitor DBZ (20 μM/kg body weight in 0.5% HPMC, 0.1% w/v NP40 in water; Calbiochem) was injected intraperitoneally for 4 consecutive days. Mice were analyzed 2 weeks after the last injection, or 3-5 days after the last injection in case of lethal phenotypes (Dll1-Dll4vil-Cre-ERT2, RBP-Jvil-Cre-ERT2, RBPJ-Klf4vil-Cre-ERT2 and DBZ treated mice).
The targeting and screening of NIP1::CreERT2 ES cells were described18 and detailed analysis of the animals will be described elsewhere. The NIP1::CreERT2 reporter was activated by injecting 1 month old mice with tamoxifen (220mg/kg) for three consecutive days. Mice were analyzed 8 months after the last injection.
Tissue sample preparation, immunohistochemistry, in situ hybridization, Southern blot and qRT-PCR are described in supplementary material.
Results
Notch1 activation occurs in adult intestinal stem cells
Notch signaling in the intestine plays an important role as a gatekeeper of progenitor cells and also as a regulator of the absorptive/secretory lineage5, 6. To gain further insight whether Notch signaling might also occur at the level of adult intestinal SC we generated a tamoxifen-inducible Notch1 activation-dependent reporter knock-in mouse line, NIP1::CreERT2 (Figure 1A)18. The intracellular domain of the targeted Notch1 allele was replaced with a cDNA encoding a 6x myc tagged CreERT2 (6mtCreERT2). Binding of Notch ligands to the NIP1::CreERT2 will trigger the release of the 6mtCreERT2 from the membrane, but only in the presence of tamoxifen can CreERT2 enter the nucleus. Within the nucleus CreERT2 will mediate the excision of a floxed stop cassette in reporter strains such as RosaR26R or the Rosa26-EYFP mice and permanently label the cells and all their offspring with LacZ or EYFP, respectively.
Figure1. Notch1 is activated in intestinal stem cells.
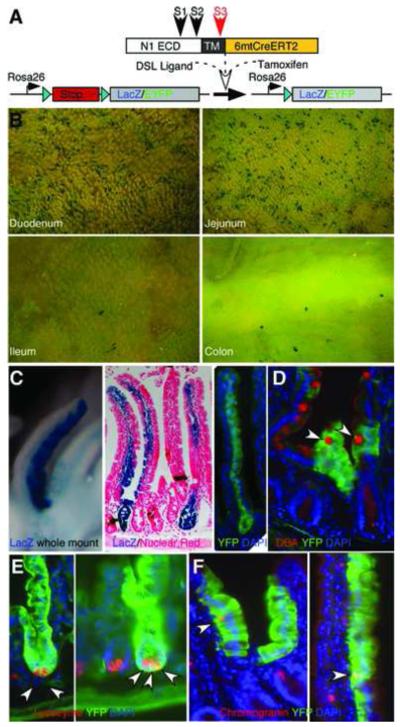
(A) Schematic illustration of NIP1::CreERT2 knock-in reporter mice (see text for details). N1 ECD, Notch1 extracellular domain; TM, Notch1 trans-membrane domain. (B-C) LacZ whole mount staining and EYFP expression in different intestinal segments eight months after reporter activation. Note fully labeled crypt-villus unit from both the Rosa LacZ and EYFP reporters. (D-F) Double staining of sections derived from the small intestine of NIP1::CreERT2 Rosa EYFP reporter mice for the goblet cell marker DBA (D), Paneth cell marker lysozyme (E) and enteroendocrine marker chromogranin (F). Double positive cells are highlighted with arrowheads.
To ascertain that adult intestinal SC were labeled at the time of activation, we examined the intestine of NIP1::CreERT2 Rosa LacZ or EYFP mice eight months after tamoxifen administration for three consecutive days. Labeled villi were maintained along the entire intestine, with more labeled villi visible in the duodenum and jejunum (Figure 1B) similar to NIP::CRE mice19. Entire crypt-villus units were clearly labeled (Figure 1C), and double immunofluorescence for EYFP and cell type specific markers identified all the differentiated intestinal cell types: goblets cells (EYFP+, DBA+ (Dolichos Biflorus Agglutinin); Figure 1D), Paneth cells (EYFP+, lysozyme+; Figure 1E) and enteroendocrine cells (EYFP+, chromogranin+; Figure 1F). The in vivo lineage tracing demonstrates that Notch1 activation occurs within the adult intestinal SC under physiological condition, however, the physiological ligands triggering Notch signaling within the intestine remain to be identified.
Dll1 deficiency induces increased goblet cell numbers
In situ hybridization based expression studies as well as LacZ-knock-in alleles for Dll1 and Dll4 point to Jag1, Dll1 and Dll4 as potential candidates for the physiological Notch1 and Notch2 ligands in the intestine20-22. We investigated the role of each ligand in intestinal homeostasis using an inducible loss of function approach. Specifically, mice carrying floxed alleles for the Dll123, Dll424 and Jag125 genes were intercrossed with transgenic mice expressing Cre-ERT2 under the control of the villin promoter (vil-Cre-ERT2)26. Two to three week old Dll1flox/flox vil-Cre-ERT2 (Dll1vil-Cre-ERT2), Dll4flox/flox vil-Cre-ERT2 (Dll4vil-Cre-ERT2) and Jag1flox/flox vil-Cre-ERT2 (Jag1vil-Cre-ERT2), as well as corresponding littermate controls lacking the vil-Cre-ERT2 transgene (control) were injected with tamoxifen on 5 consecutive days and analyzed 2 weeks after the last injection. Successful gene ablation in here and subsequent experiments was confirmed by Southern blot analysis of genomic DNA derived from epithelial cells of the small intestine and colon (Figure S1), and/or combined with qRT-PCR for mRNA expression of the targeted ligands (Figure 2C, 3D).
Figure2. Increased goblet cell differentiation in Dll1 deficient intestine.
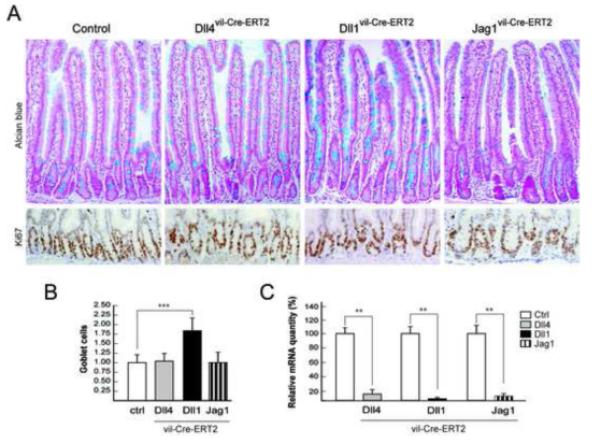
(A) Alcian blue (top panel) and Ki67 (bottom panel) staining of representative sections from the proximal small intestine of control, Dll4vil-Cre-ERT2, Dll1vil-Cre-ERT2 and Jag1vil-Cre-ERT2 mice. (B) Quantification of goblet cells in control and the indicated mutant mice. The number of Alcian blue-positive goblet cells per crypt-villus unit was counted and expressed as relative values to littermate controls (1.00). Dll1 mutant mice show a 1.83 fold increase (P<.0001) in goblet cells number. (C) Relative mRNA quantification of Dll4, Dll1 and Jag1, derived from control and single mutant mice (P< .01).
Figure3. Complete conversion of crypt progenitors into post-mitotic goblet cells in Dll1-Dll4 double mutant mice.
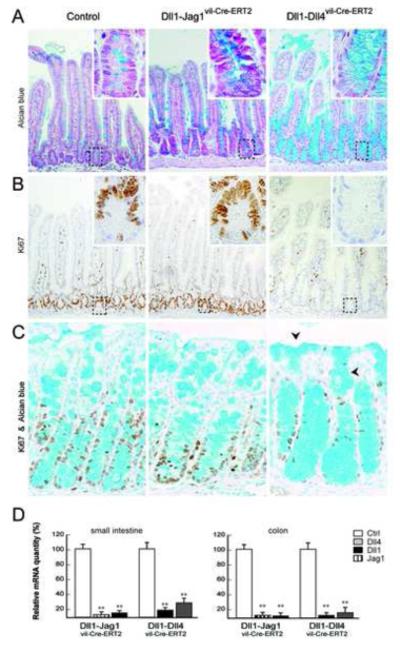
(A) Alcian blue and (B) Ki67 antibody staining of representative sections from the small intestine of control, Dll1-Jag1vil-Cre-ERT2 and Dll1-Dll4vil-Cre-ERT2 mice. The Dll1-Jag1 double mutant mice show an increase in goblet cell numbers similar to single Dll1 mutant mice (1.88 P< .0001) while the proliferative crypt compartment of Dll1-Dll4vil-Cre-ERT2 is completely converted into post-mitotic goblet cells. Insets in panels show high magnification (400x) of the outlined crypt regions. (C) Alcian blue and Ki67 staining of colonic crypts revealed a similar phenotype. Not only the crypt but also the inter crypt epithelium is converted into goblet cells (black triangles). (D) Quantitative mRNA analysis from epithelial cells of the small intestine and colon confirmed efficient gene inactivation (P< .01).
Loss of Notch signaling within the small intestine resulted in goblet cell metaplasia accompanied by a loss of proliferating progenitors5, 6 (see also Figure 6A). Therefore, we performed histological analysis combined with Alcian blue and Ki67 staining of individual mice gene-targeted for each ligand. The small intestine and colon of Dll4 and Jag1 mutant mice revealed no significant differences in morphology and goblet cell number (Figure 2A-B and S2). Moreover, the proliferative crypt compartment appeared to be intact (Figure2A). Lysozyme and synaptophysin antibody staining of intestinal sections derived from Dll4, Dll1 and Jag1 mutant mice identified no change within the Paneth and enteroendocrine cell compartments relative to controls (Figure S3). In contrast, inactivation of Dll1 within the small intestine resulted in an average increase of 83% of goblet cells (P<.0001) (Figure 2B), reminiscent of Notch1 loss7, 19. Despite the increase of goblet cell numbers in Dll1vil-Cre-ERT2 animals there was no significant change of Ki67+ crypt progenitor cells compared to control mice.
Figure6. Klf4 is dispensable for goblet cell metaplasia induced by loss of Notch signaling.
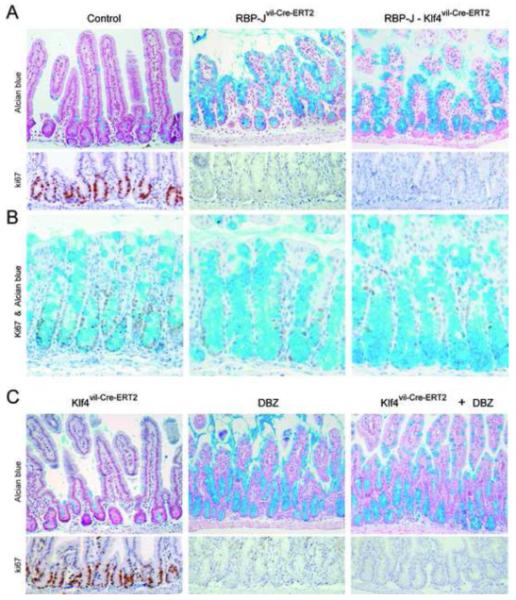
(A-B) Alcian blue (top panels) and Ki67 (bottom panels) staining on sections derived from the proximal intestine (A) and colon (B) of the indicated mice. Note the complete loss of the proliferative crypt compartment and the complete conversion into goblet cells in RBP-J and RBP-J Klf4 mutant intestine. (C) Pharmaceutical inhibition of Notch signaling using the γ-secretase inhibitor DBZ after gut specific inactivation of Klf4 results in the loss of proliferating crypt progenitors due to their conversion into postmitotic goblet cells.
Taken together, these results suggest that inactivation of individual ligands is not sufficient to recapitulate the phenotypes observed in mice in which Notch signaling was completely abrogated5, 6 (see also Figure 6A). Nevertheless, inactivation of the Dll1 gene showed a moderate increase in goblet cell numbers indicating that Dll1 might be one of several physiological Notch ligands in the crypt compartment.
Dll1 and Dll4 cooperate to maintain the crypt progenitor compartment
To investigate whether functional redundancy existed at the level of the ligands as well as the receptors, we generated compound conditional knockout mice of Dll1 with other ligands (Dll1-Jag1vil-Cre-ERT2 and Dll1-Dll4vil-Cre-ERT2) to assess functional redundancy. Simultaneous inactivation of both Jag1 and Dll1 within the small intestine did not differ from loss of Dll1 alone (compare Figure 3A-B with Figure 2A). Furthermore, the large intestine of these double mutant mice was comparable to control animals (Figure 3C). Thus, under physiological conditions Jag1 does not contribute to Notch activation in the gut.
In contrast, Dll1-Dll4 double KO mice showed a rapid and progressive loss of weight, accompanied by cachexia and poor grooming, leading to death within 4-6 days after the last tamoxifen injection. Histological examination of the small intestine revealed a complete conversion of crypt progenitors into goblet cells as well as loss of Ki67+ cells, indicating loss of the proliferative crypt compartment (Figure 3A-B). Similarly, in the colon we observed reduced numbers of Ki67+ cells and a corresponding increase of goblet cell numbers reaching the apical epithelium (Figure 3C). Active Notch1 signaling within the crypt compartment of the small intestine was investigated by NICD1 immunostaining, Hes1 in situ hybridization and qRT-PCR for Math1. Dll1-Dll4 double deficient and RBP-J mutant intestinal crypts did not show any NICD1 staining and were also negative for Hes1 mRNA expression (Figure 4A-B). In contrast, Math1 mRNA expression, which is normally repressed by Hes1 in the crypt compartment, was strongly upregulated in both Notch mutant animals compared to control mice (Figure 4C). Taken together these data demonstrate that Notch signaling is abrogated in Dll1-Dll4 double mutant mice and that these animals phenocopy RBP-J and Notch1-Notch2 mutant mice5, 6.
Figure4. Notch signaling is disrupted in the small intestine of Dll1-Dll4 and RBP-J mutant mice.
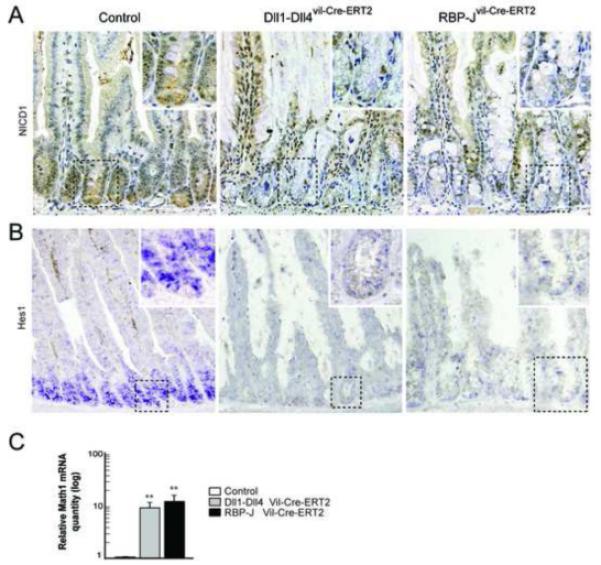
(A) Active Notch signaling was visualized with anti-NICD1 antibody staining on representative sections from control, Dll1-Dll4 and RBP-J deficient intestines. Disruption of Notch signaling was confirmed by the absence of nuclear staining in the crypt compartment of Dll1-Dll4 and RBP-J deficient mice. (B) In situ hybridization of the Notch target gene Hes1 on sections from indicated mice, and (C) quantification of Math1 mRNA level derived from epithelial cells of control, Dll1-Dll4 and RBP-J mutant mice (P< .01).
Similarly to RBP-J deficient mice the Paneth and enteroendocrine cell compartments were not significantly affected in the Dll1-Dll4 deficient intestine (Figure S3). This may be explained in terms of the relatively long lifetimes of these secretory cell types: given that the intestinal tissues of this mice were fixed only 3-5 days after the final injection of tamoxifen, there will not have been sufficient time for the adjustment of Paneth and enteroendocrine cell numbers according to their production rate27. These results identified Dll1 and Dll4 as the physiological ligands mediating Notch signaling in the crypt compartment of the small intestine and colon. Thus the presence of both Dll1 and Dll4 ligands is essential for maintenance of the proliferative crypt compartment.
Loss of Lgr5+ intestinal stem cells in Notch mutant mice
Earlier we showed that Notch1 was active in intestinal SC. To address whether loss of Notch signaling affect intestinal SC in addition to crypt progenitor cells, we investigated the consequences of inactivating different combinations of Notch ligands in the SC compartment of the small intestine. Lineage tracing experiments for both Bmi1 and Lgr5 identified SC that are capable of replenishing all cell types of the small intestine1, 28. Bmi1 expression marks the so called +4 position which was proposed to be the location of the SC due to their ability to retain DNA labels29. In contrast Lgr5+ cells are cycling and although they occasionally localize to the +4 position it is currently not clear whether Bmi1+ and Lgr5+ cells represent distinct, overlapping or identical SC populations within the small intestine. Olfm4 was recently identified as being a specific and robust marker of mouse and human Lgr5-type SC in the small intestine3. We therefore analyzed control, Dll4, Dll1, Jag1, Dll1-Jag1 and Dll1-Dll4 mutant mice for the presence or absence of Lgr5-type SC using Olfm4 in situ hybridization. As expected, the intestine of control, Dll4, Dll1, Jag1 and Dll1-Jag1 mutant mice showed the presence of normal numbers of Olfm4+ intestinal SC at the bottom of the crypt (Figure S4). In contrast, the intestine of Dll1-Dll4 and RBP-J deficient mice lacked Olfm4+ SC in crypts where gene inactivation was complete (Figure 5). We further confirmed that loss of Notch signaling is not only affecting Olfm4 expression on SC, but the SC compartment itself by analyzing two Wnt targets/SC markers, Lgr51 and Ascl230. Expression of both genes was ablated in Dll1-Dll4 and RBP-J deficient small intestine (Figure 5). The concomitant loss of Olfm4, Lgr5, and Ascl2 expression, the labeling of SC with NIP1::CreERT2, and the complete loss of Ki67+ cells within the crypt (Figure 3B) indicates that cycling Lgr5+ SC convert into postmitotic goblet cells when Notch signaling is lost. Taken together, these results indicate that Notch signaling is directly involved in the maintenance of SC within the small intestine.
Figure5. Loss of intestinal stem cell markers in Notch signaling deficient Dll1-Dll4 and RBP-J mutant mice.
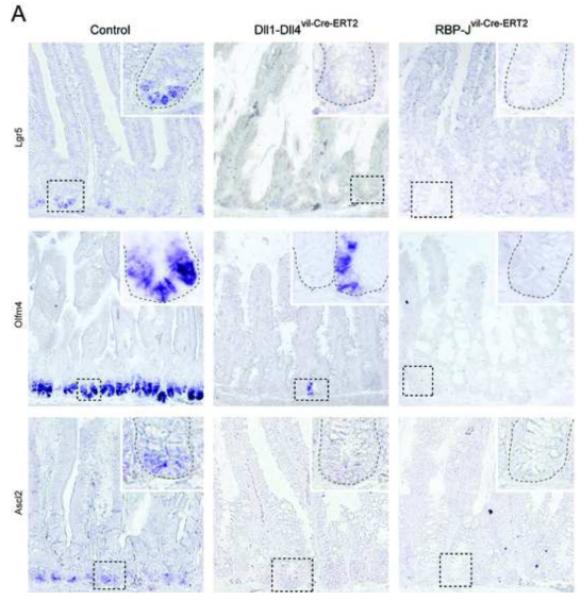
(A) In situ hybridization analysis for SC markers Lgr5, Olfm4 and Ascl2 on sections of the small intestine. Simultaneous inactivation of Dll1 and Dll4 or RBP-J resulted in the loss of expression of all three SC markers described above. Insets show high magnification (400x) of outlined crypt regions. Note that for Olfm4 in situ hybridization on Dll1-Dll4 mutant mice a region with a few positive SC (presumably none deleted) was chosen on purpose to indicate successful probe labeling.
Goblet cell metaplasia in adult Notch signaling deficient mice is not driven by Klf4 upregulation
Finally, we wanted to investigate the function of a candidate playing a role in driving goblet cell metaplasia following loss of Notch signaling. Several reports suggest that the zinc finger transcription factor Klf4 acts as a master regulator for goblet cell differentiation during fetal life. Klf4 is upregulated as a consequence of Notch deficiency and is thereby thought to drive goblet cell metaplasia12-14. Since Klf4 null mice die shortly after birth, we intercrossed mice carrying floxed alleles of the Klf4 gene31 with vil-Cre-ERT2 transgenic mice. Two-week old Klf4flox/floxvil-Cre-ERT2 (hereafter Klf4vil-Cre-ERT2) and corresponding littermate controls lacking the vil-Cre-ERT2 transgene (control) were analyzed 2 weeks after the last tamoxifen injection. Successful inactivation of the Klf4 gene was confirmed by Southern blot analysis and/or by qRT-PCR. Surprisingly, histological analysis with Alcian blue staining did not reveal any differences in goblet cell numbers between control and Klf4 mutant animals in either the small intestine or the colon (Figure S5). Proliferation of crypt progenitors, Paneth and enteroendocrine cell compartments within the small intestine were also similar between the two groups of mice (Figure S5).
Although under homeostatic conditions Klf4 seems to be dispensable for goblet cell fate specification and differentiation within the adult intestine, it was still conceivable that reactivation of Klf4 expression in the crypt compartment after loss of Notch signaling might trigger goblet cell differentiation. If this hypothesis is correct, one would expect that loss of Klf4 in Notch deficient intestine should reduce or suppress the goblet cell metaplasia. We investigated this possibility by generating double mutant mice intercrossing RBP-Jflox/flox mice with Klf4 mutant animals (RBP-J Klf4vil-Cre-ERT2). Gene inactivation in double mutant mice and control RBP-Jvil-Cre-ERT2 mice was induced and analyzed 4 days post inactivation. Interestingly, the phenotype of the small intestine and colon of RBP-J Klf4vil-Cre-ERT2 double deficient mice was indistinguishable from RBP-Jvil-Cre-ERT2 animals (Figure 6A-C) suggesting that Klf4 does not play a regulatory role in goblet cell metaplasia or in loss of proliferation within the crypt. This observation was further confirmed by analyzing intestine of Klf4vil-Cre-ERT2 and control mice that were treated with the pharmacological Notch inhibitor dibenzazepine (DBZ). Both control and Klf4 mutant DBZ-treated animals displayed goblet cell metaplasia and loss of the proliferative crypt compartment (Figure 6C). These findings demonstrate that Klf4 is not a mediator of goblet cell differentiation induced by loss of Notch signaling in the adult intestine.
Discussion
It is now well established that both Wnt and Notch signaling are essential for the maintenance of the proliferative crypt compartment. Both pathways are also involved in cellular differentiation processes of the intestinal epithelium3. Loss of Notch signaling due to genetic inactivation of the RBP-J gene, or as consequence of using pharmacological γ-secretase inhibitors (GSI), or blocking antibodies, results in the loss of proliferating crypt progenitors which convert into postmitotic goblet cells5, 7. In recent years aberrant Notch signaling has been linked to numerous diseases, in particular to cancer, which led to increased attention of the Notch cascade as a potential therapeutic target. Although GSI inhibitors are currently considered and tested in clinical trials for several conditions, the intestinal toxicity due to their indiscriminate blockade of Notch receptors in the gut, with consequent loss of SC, represents a serious restriction32. New therapeutic strategies such as the development of inhibitory antibodies against individual Notch receptors and/or ligands are currently being developed and tested in preclinical models7, 33-35. It is therefore important to understand which components of the Notch cascade mediate crypt progenitor maintenance and how their inhibition results in goblet cell metaplasia. In this context we previously established that the Notch1 and Notch2 receptors function redundantly in the small intestine, since only the concomitant genetic ablation of both receptors results in goblet cell metaplasia and loss of crypt progenitors6. These genetic results were recently confirmed showing that antibody-mediated inhibition of Notch1 and Notch2 causes severe intestinal toxicity while selective blocking of Notch1 inhibited tumor growth in pre-clinical models without major gut toxicity7.
In this study we identified Dll1 and Dll4 as being the key physiological ligands mediating Notch signaling in the intestinal epithelium of the mouse. Intestine-specific inactivation of individual ligands such as Dll4 and Jag1 did not show any obvious phenotype and loss of Dll1 mimicked loss of Notch1 by producing a moderate increase in goblet cell numbers without noticeable perturbation of the proliferative compartment. These results suggested a functional redundancy amongst the ligands similar to the one observed within the receptors. Just as loss of Notch1 was not fully compensated by Notch2, loss of Dll1 is not fully compensated by Dll4. In contrast, loss of Dll4 is completely counterbalanced by Dll1 indicating that Dll1 contributes more to Notch1 signaling in the intestinal epithelium than Dll4. It is currently unclear whether these differences between Dll1 and Dll4 are mediated by different expression patterns within the crypt compartment or by differences in their ability to bind different Notch receptors, as has been shown within the hematopoietic system36.
Jag1 has been shown to be downstream of Wnt signaling in multiple organs including the intestine37. Moreover, deletion of a single Jag1 allele in APCMin/+ mice resulted in reduced tumor growth and human adenomas derived from Familial Adenomatous Polyposis patients show high levels of Jag1 that correlated with activated Notch1 and Notch2 in tumor cells containing nuclear β-catenin37. Although Jag1 is indeed a target gene of β-catenin mediated Wnt signaling, and Wnt signaling is essential for the maintenance of the crypt compartment3, Jag1 is not a functionally important Notch ligand in the mouse intestine. We find that Dll4, and not Jag1, is acting redundantly with Dll1. Human colorectal tumors, colorectal cancer cell lines and adenomas of APCMin/+ mice all exhibit increased levels of β-catenin mediated Wnt signaling compared to their normal intestinal epithelium. It is therefore possible that elevated Jag1 expression under pathological conditions could contribute to disease progression whereas the physiological levels of Jag1 in the crypt compartment are not functionally significant.
Dll4 is best known for its essential role during development of embryonic vasculature and arteriogenesis38, 39. The finding that Dll4 is preferentially expressed in tumor vasculature as opposed to normal blood vessels40 rendered Dll4 an attractive therapeutic target for tumor angiogenesis. Studies targeting blood vessel formation employing blocking antibodies to Dll4 revealed substantial tumor growth reduction in cancer cell line-based xenograft models34, 35. Unlike blocking Notch signaling using a γ-secretase inhibitor, neutralizing anti-Dll4 antibodies had no measurable effect on goblet cell differentiation and/or proliferation of crypt progenitor cells34. This led to the interpretation that Dll4-mediated Notch signaling would be largely restricted to the vascular compartment34. Although our genetic studies confirm that ablation of Dll4 in the intestinal epithelium does not perturb proliferation or goblet cell differentiation in the crypt, they show that Dll4-mediated Notch signaling is indeed occurring in the intestine and that the absence of a measurable phenotype in Dll4 mutant mice is due to redundant Dll1-mediated Notch signaling.
To date, Notch signaling has been mostly described to function at the level of intestinal progenitors but not necessarily at the SC level. Label retaining assays combined with irradiation-induced regeneration of the intestinal epithelium positioned SC at the +4 position, just above the Paneth cells29. Recent in vivo lineage tracing experiments revealed that slow cycling Bmi1+ cells localize to the +4 position and give rise to all four epithelial lineages of the gut28 suggesting that these cells represent SC. CBC cells were identified as another pool of SC based on Lgr5 expression and lineage tracing experiments. In contrast to the +4 cells Lgr5+ cells are actively cycling, long lived and also give rise to all four epithelial lineages1. Moreover, a single Lgr5+ cell has the potential to form a self-renewing mini-gut in culture41. Whether these two pools of SC represent separate entities, or whether one is the descendent of the other needs further investigations. Independent of these issues, our Dll1-Dll4 and RBP-J mutant mice combined with the lineage tracing experiments for Notch1 show that Notch signaling is essential for the maintenance of the Lgr5+ SC and that intestinal SC have active Notch1 signaling. Since Notch1 and Notch2 have redundant functions within the small intestine, it is conceivable that SC may also have active Notch2 signaling. Future studies are needed to verify this hypothesis. Which cell provides Dll1 and Dll4 ligands to trigger Notch signaling in stem and progenitor cells is currently not well defined. In the absence of effective anti-Dll1 and Dll4 antibodies, in situ hybridization and Dll1lacZ/+ and Dll4lacz/+ knock-in mice identify individual cells within the crypt and secretory cells as ligand presenting cells20-22, 42.
The gastrointestinal toxicity of GSIs, resulting from loss of SC/progenitors, which limits the clinical utility of these drugs, is avoided in mice in which Math1 is conditionally inactivated in the intestine, but at the expense of loss of secretory cells11, 43. This demonstrates the absolute requirement of Math1 for conversion of SC to secretory cells in crypts deficient in Notch signaling. Multiple studies have suggested that goblet cell metaplasia in GSI-treated mice is mediated by derepression of Klf412-14, positioning Klf4 downstream of Math1. However, our genetic loss-of-function analysis failed to support such a role for Klf4 in the intestine and demonstrate that Klf4 is dispensable for this process.
In conclusion, our results identified Dll1 and Dll4 as the physiologically relevant Notch1 and Notch2 ligands within the small intestine of the mouse. These two ligands cooperate and exhibit a partial functional redundancy with Dll1 contributing more to Notch signaling then Dll4 in the intestinal epithelium. Collectively, our experiments reveal that intestinal SC activate Notch1, that Notch signaling is required for SC maintenance and that Klf4 is not an essential mediator of goblet cell differentiation.
Supplementary Material
Acknowledgments
We would like to acknowledge C.S. Nowell for discussion and critical reading of the manuscript, the animal and histology facilities at EPFL for their assistance.
Funding: This work was in part supported by the Swiss National Science Foundation, the Swiss Cancer League, EuroSyStem, OptiStem (F.R.) and EMBO (V.R.), and the National Institute of Diabetes, Digestive and Kidney Diseases (K.H.K.), Cancer Research UK (JL), and the National Institutes of Health grant DK066408 (ZL, SC, RK)
Abbreviations
- SC
stem cells
- bHLH
basic helix-loop-helix
- p1
post-natal day 1
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest: The authors have no conflicting financial interests.
References
- 1.Barker N, van Es JH, Kuipers J, et al. Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature. 2007;449:1003–7. doi: 10.1038/nature06196. [DOI] [PubMed] [Google Scholar]
- 2.van der Flier LG, Haegebarth A, Stange DE, et al. OLFM4 is a robust marker for stem cells in human intestine and marks a subset of colorectal cancer cells. Gastroenterology. 2009;137:15–7. doi: 10.1053/j.gastro.2009.05.035. [DOI] [PubMed] [Google Scholar]
- 3.van der Flier LG, Clevers H. Stem cells, self-renewal, and differentiation in the intestinal epithelium. Annu Rev Physiol. 2009;71:241–60. doi: 10.1146/annurev.physiol.010908.163145. [DOI] [PubMed] [Google Scholar]
- 4.Kopan R, Ilagan MXG. The canonical Notch signaling pathway: unfolding the activation mechanism. Cell. 2009;137:216–33. doi: 10.1016/j.cell.2009.03.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.van Es JH, van Gijn ME, Riccio O, et al. Notch/gamma-secretase inhibition turns proliferative cells in intestinal crypts and adenomas into goblet cells. Nature. 2005;435:959–63. doi: 10.1038/nature03659. [DOI] [PubMed] [Google Scholar]
- 6.Riccio O, van Gijn ME, Bezdek AC, et al. Loss of intestinal crypt progenitor cells owing to inactivation of both Notch1 and Notch2 is accompanied by derepression of CDK inhibitors p27(Kip1) and p57(Kip2) EMBO Rep. 2008 doi: 10.1038/embor.2008.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wu Y, Cain-Hom C, Choy L, et al. Therapeutic antibody targeting of individual Notch receptors. Nature. 2010;464:1052–7. doi: 10.1038/nature08878. [DOI] [PubMed] [Google Scholar]
- 8.Jensen J, Pedersen EE, Galante P, et al. Control of endodermal endocrine development by Hes-1. Nat Genet. 2000;24:36–44. doi: 10.1038/71657. [DOI] [PubMed] [Google Scholar]
- 9.Suzuki K, Fukui H, Kayahara T, et al. Hes1-deficient mice show precocious differentiation of Paneth cells in the small intestine. Biochem Biophys Res Commun. 2005;328:348–52. doi: 10.1016/j.bbrc.2004.12.174. [DOI] [PubMed] [Google Scholar]
- 10.Chen X, Johns DC, Geiman DE, et al. Kruppel-like factor 4 (gut-enriched Kruppel-like factor) inhibits cell proliferation by blocking G1/S progression of the cell cycle. J Biol Chem. 2001;276:30423–8. doi: 10.1074/jbc.M101194200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.van ESJH, de Geest N, van de Born M, et al. Intestinal stem cells lacking the math1 tumor suppressor are refractory to notch inhibitors. nature communications. 2010;1:1–5. doi: 10.1038/ncomms1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ghaleb AM, Aggarwal G, Bialkowska AB, et al. Notch inhibits expression of the Kruppel-like factor 4 tumor suppressor in the intestinal epithelium. Mol Cancer Res. 2008;6:1920–7. doi: 10.1158/1541-7786.MCR-08-0224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zheng H, Pritchard DM, Yang X, et al. KLF4 gene expression is inhibited by the notch signaling pathway that controls goblet cell differentiation in mouse gastrointestinal tract. Am J Physiol Gastrointest Liver Physiol. 2009;296:G490–8. doi: 10.1152/ajpgi.90393.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Real PJ, Tosello V, Palomero T, et al. Gamma-secretase inhibitors reverse glucocorticoid resistance in T cell acute lymphoblastic leukemia. Nat Med. 2009;15:50–8. doi: 10.1038/nm.1900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Katz JP, Perreault N, Goldstein BG, et al. The zinc-finger transcription factor Klf4 is required for terminal differentiation of goblet cells in the colon. Development. 2002;129:2619–28. doi: 10.1242/dev.129.11.2619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ghaleb AM, McConnell BB, Kaestner KH, et al. Altered intestinal epithelial homeostasis in mice with intestine-specific deletion of the Kruppel-like factor 4 gene. Dev Biol. 2010 doi: 10.1016/j.ydbio.2010.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.van Es JH, de Geest N, van de Born M, et al. Intestinal stem cells lacking the Math1 tumour suppressor are refractory to Notch inhibitors. Nat Commun. 2010;1:1–5. doi: 10.1038/ncomms1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu Z, Obenauf AC, Speicher MR, et al. Rapid identification of homologous recombinants and determination of gene copy number with reference/query pyrosequencing (RQPS) Genome Res. 2009;19:2081–9. doi: 10.1101/gr.093856.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vooijs M, Ong CT, Hadland B, et al. Mapping the consequence of Notch1 proteolysis in vivo with NIP-CRE. Development. 2007;134:535–44. doi: 10.1242/dev.02733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sander GR, Powell BC. Expression of notch receptors and ligands in the adult gut. J Histochem Cytochem. 2004;52:509–16. doi: 10.1177/002215540405200409. [DOI] [PubMed] [Google Scholar]
- 21.Crosnier C, Vargesson N, Gschmeissner S, et al. Delta-Notch signalling controls commitment to a secretory fate in the zebrafish intestine. Development. 2005;132:1093–104. doi: 10.1242/dev.01644. [DOI] [PubMed] [Google Scholar]
- 22.Benedito R, Duarte A. Expression of Dll4 during mouse embryogenesis suggests multiple developmental roles. Gene Expr Patterns. 2005;5:750–5. doi: 10.1016/j.modgep.2005.04.004. [DOI] [PubMed] [Google Scholar]
- 23.Hozumi K, Negishi N, Suzuki D, et al. Delta-like 1 is necessary for the generation of marginal zone B cells but not T cells in vivo. Nat Immunol. 2004;5:638–44. doi: 10.1038/ni1075. [DOI] [PubMed] [Google Scholar]
- 24.Koch U, Fiorini E, Benedito R, et al. Delta-like 4 is the essential, nonredundant ligand for Notch1 during thymic T cell lineage commitment. J Exp Med. 2008;205:2515–23. doi: 10.1084/jem.20080829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mancini SJ, Mantei N, Dumortier A, et al. Jagged1-dependent Notch signaling is dispensable for hematopoietic stem cell self-renewal and differentiation. Blood. 2005;105:2340–2. doi: 10.1182/blood-2004-08-3207. [DOI] [PubMed] [Google Scholar]
- 26.el Marjou F, Janssen KP, Chang BH, et al. Tissue-specific and inducible Cre-mediated recombination in the gut epithelium. Genesis. 2004;39:186–93. doi: 10.1002/gene.20042. [DOI] [PubMed] [Google Scholar]
- 27.Ireland H, Houghton C, Howard L, et al. Cellular inheritance of a Cre-activated reporter gene to determine Paneth cell longevity in the murine small intestine. Dev Dyn. 2005;233:1332–6. doi: 10.1002/dvdy.20446. [DOI] [PubMed] [Google Scholar]
- 28.Sangiorgi E, Capecchi MR. Bmi1 is expressed in vivo in intestinal stem cells. Nat. Genet. 2008;40:915–20. doi: 10.1038/ng.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Potten CS, Kovacs L, Hamilton E. Continuous labelling studies on mouse skin and intestine. Cell Tissue Kinet. 1974;7:271–83. doi: 10.1111/j.1365-2184.1974.tb00907.x. [DOI] [PubMed] [Google Scholar]
- 30.van der Flier LG, van Gijn ME, Hatzis P, et al. Transcription factor achaete scute-like 2 controls intestinal stem cell fate. Cell. 2009;136:903–12. doi: 10.1016/j.cell.2009.01.031. [DOI] [PubMed] [Google Scholar]
- 31.Swamynathan SK, Katz JP, Kaestner KH, et al. Conditional deletion of the mouse Klf4 gene results in corneal epithelial fragility, stromal edema, and loss of conjunctival goblet cells. Mol Cell Biol. 2007;27:182–94. doi: 10.1128/MCB.00846-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shih Ie M, Wang TL. Notch signaling, gamma-secretase inhibitors, and cancer therapy. Cancer Res. 2007;67:1879–82. doi: 10.1158/0008-5472.CAN-06-3958. [DOI] [PubMed] [Google Scholar]
- 33.Hoey T, Yen W-C, Axelrod F, et al. DLL4 blockade inhibits tumor growth and reduces tumor-initiating cell frequency. Cell Stem Cell. 2009;5:168–77. doi: 10.1016/j.stem.2009.05.019. [DOI] [PubMed] [Google Scholar]
- 34.Ridgway J, Zhang G, Wu Y, et al. Inhibition of Dll4 signalling inhibits tumour growth by deregulating angiogenesis. Nature. 2006;444:1083–7. doi: 10.1038/nature05313. [DOI] [PubMed] [Google Scholar]
- 35.Noguera-Troise I, Daly C, Papadopoulos NJ, et al. Blockade of Dll4 inhibits tumour growth by promoting non-productive angiogenesis. Nature. 2006;444:1032–7. doi: 10.1038/nature05355. [DOI] [PubMed] [Google Scholar]
- 36.Besseyrias V, Fiorini E, Strobl LJ, et al. Hierarchy of Notch-Delta interactions promoting T cell lineage commitment and maturation. J Exp Med. 2007;204:331–43. doi: 10.1084/jem.20061442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rodilla V, Villanueva A, Obrador-Hevia A, et al. Jagged1 is the pathological link between Wnt and Notch pathways in colorectal cancer. Proc Natl Acad Sci U S A. 2009;106:6315–20. doi: 10.1073/pnas.0813221106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Duarte A, Hirashima M, Benedito R, et al. Dosage-sensitive requirement for mouse Dll4 in artery development. Genes Dev. 2004;18:2474–8. doi: 10.1101/gad.1239004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Krebs LT, Shutter JR, Tanigaki K, et al. Haploinsufficient lethality and formation of arteriovenous malformations in Notch pathway mutants. Genes Dev. 2004;18:2469–73. doi: 10.1101/gad.1239204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Patel NS, Dobbie MS, Rochester M, et al. Up-regulation of endothelial delta-like 4 expression correlates with vessel maturation in bladder cancer. Clin Cancer Res. 2006;12:4836–44. doi: 10.1158/1078-0432.CCR-06-0285. [DOI] [PubMed] [Google Scholar]
- 41.Sato T, Vries RG, Snippert HJ, et al. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature. 2009;459:262–5. doi: 10.1038/nature07935. [DOI] [PubMed] [Google Scholar]
- 42.Gregorieff A, Stange DE, Kujala P, et al. The ets-domain transcription factor Spdef promotes maturation of goblet and paneth cells in the intestinal epithelium. Gastroenterology. 2009;137:1333–45. e1–3. doi: 10.1053/j.gastro.2009.06.044. [DOI] [PubMed] [Google Scholar]
- 43.Kazanjian A, Noah T, Brown D, et al. Atonal homolog 1 is required for growth and differentiation effects of notch/gamma-secretase inhibitors on normal and cancerous intestinal epithelial cells. Gastroenterology. 2010;139:918–28. 928, e1–6. doi: 10.1053/j.gastro.2010.05.081. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.