

Abstract
While the organometallic chemistry of Pd in its (0), (+II), and (+IV) oxidation states is well-established, organometallic Pd(III) chemistry remains widely unexplored. Few characterized Pd(III) complexes are known, which has inhibited detailed study of the organometallic chemistry of Pd(III). In this review, the potential roles of both mono- and dinuclear Pd(III) complexes in organometallic chemistry will be discussed. While not widely recognized, Pd in the (+III) oxidation state may play a significant role in a variety of known Pd-catalyzed reactions.
Keywords: Pd(III), C–H functionalization, metal-metal bonding, bimetallic redox chemistry
1 Introduction
Palladium is among the most widely used metals for catalysis in organic chemistry, and the fundamental organometallic chemistry of palladium in the (0), (+II), and (+IV) oxidation states has been well-studied [1]. Organometallic Pd(I) complexes, while relatively less common, have been employed as precatalysts in organic synthesis [2-4]. By comparison, the organometallic chemistry of Pd(III) remains in its infancy. Few authentic Pd(III) complexes are known and the potential role of Pd(III) in catalysis is only now beginning to be elucidated. Herein, we will review the organometallic chemistry of Pd(III) and discuss the relevance of Pd(III) intermediates to Pd-catalyzed processes. We will review the organometallic chemistry of well-defined, isolated Pd(III) complexes, as well as organometallic chemistry in which the potential role of Pd(III) intermediates is currently more speculative. Based on the reports discussed herein, Pd(III) may be much more prevalent in Pd-catalyzed processes than has generally been recognized.
2 Mononuclear Pd(III) Chemistry
2.1 Mononuclear Pd(III) Werner-type Complexes
Palladium(II) has a d8 electronic configuration and mononuclear Pd(II) complexes are generally square planar (Figure 1) [5]. Metal-based oxidation of mononuclear Pd(II) complexes by one-electron should result in paramagnetic, low-spin d7, tetragonally distorted octahedral Pd(III) complexes [6]. Further one-electron oxidation should afford octahedral d6 Pd(IV) complexes [5].
Fig. 1.

Electronic structure of mononuclear Pd(II), Pd(III), and Pd(IV)
Unlike complexes based on Pt(III) [7-15], compounds containing Pd(III) are rare. A compound with the empirical formula PdF3 was originally proposed to contain Pd(III) [16,17], but was subsequently shown to by more appropriately formulated as Pd2+[PdF6]2− and contain Pd(II) and Pd(IV) [18,19]. The polyatomic anions PdF4− and PdF63− have been prepared by solid-state synthesis [20-22]. Mononuclear coordination complexes containing Pd(III) have been observed by electrochemical measurements as well as EPR spectroscopy [23-30], although assignment of these complexes as containing Pd(III), and not Pd(II) with a singly oxidized ligand framework, has been the source of continuing discussion [31-34]. In 2010, the (+III) oxidation state of Pd was stabilized in a mixed Ni/Pd oligomeric M–X–M–X chain of the formulation [Ni1-xPdx(chxn)Br]Br2 (chxn = (1R,2R)-cyclohexanediamine) by electrochemical oxidation of a mixture of [Ni(chxn)2]Br2 and [Pd(chxn)2]Br2 [35,36].
Two mononuclear, Werner-type complexes based on Pd(III) have been characterized by X-ray crystallography. The X-ray crystal structure of mononuclear Pd(III) complex 1, in which the Pd(III) center is supported by two 1,4,7-trithiacyclononane ligands, was reported in 1987 (Figure 2) [37-39]. An analogous complex (2), supported by 1,4,7-triazacyclononane ligands, was reported a year later [40]. Both complexes contain distorted octahedral Pd centers, as expected for low-spin, d7 Pd(III) [6]. Detailed examination of the electrochemical and spectroscopic properties of Pd(III) complexes supported by macrocyclic polydentate ligands has indicated that the unpaired electron in 1 and 2 resides predominantly in the 4 dz2 orbital [41-46].
Fig. 2.
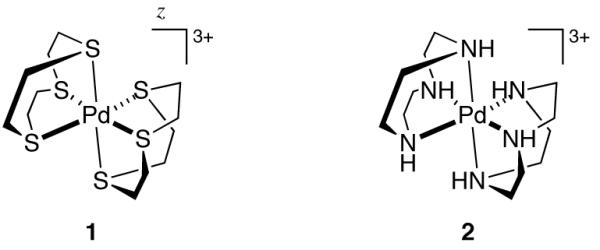
Crystallographically characterized examples of mononuclear Pd(III) Werner-type complexes
2.2 Mononuclear Pd(III) Complexes in Catalysis
Pd(III) in Pd-catalyzed Oxidative C–H Coupling Reactions
Palladium is a versatile transition metal for the catalysis of various carbon–carbon and carbon–heteroatom cross-coupling reactions [1]. Traditional C–C cross-coupling reactions employ pre-functionalized substrates – typically containing C–X (X = Cl, Br, I, OTf) and C–M (M = B, Sn, Zn, Mg) bonds respectively – and are generally accepted to proceed through Pd(0)/Pd(II) catalysis cycles [47]. Direct oxidative coupling of arene C–H bonds has the potential to generate the products of traditional cross-coupling chemistry without the need for pre-functionalized reaction partners (Figure 3a) [48-53]. A general catalysis cycle for such an oxidative coupling of aryl C–H bonds is outlined in Figure 3b. Initial C–H palladation at Pd(II) (3) would generate an aryl Pd(II) complex (4). Subsequently, a second C–H palladation could afford a heteroleptic biaryl Pd(II) complex (5), poised to undergo C–C bond-forming reductive elimination. The catalysis cycle is closed by re-oxidation of the thus formed Pd(0) (6) to Pd(II) (3) by an external oxidant. While biaryl Pd(II) intermediate 5 is typically proposed to undergo direct C–C reductive elimination, in the presence of an external oxidant, 5 could potentially be oxidized to a higher-valent species prior to the C–C bond forming event [54,55].
Fig. 3.
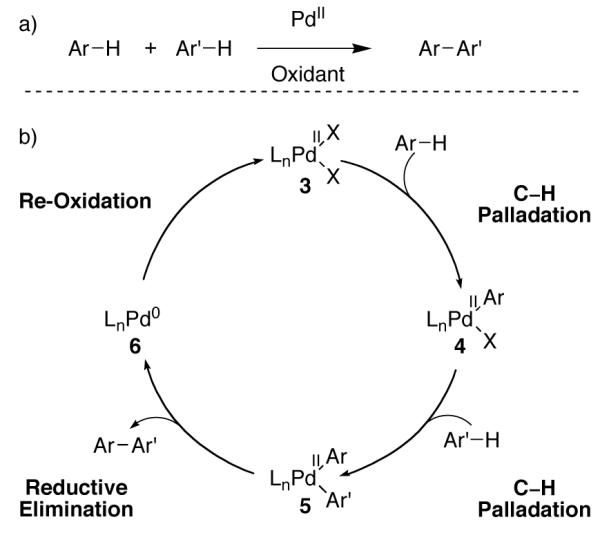
Generic Pd(0)/Pd(II) catalysis cycle for oxidative C–H cross-coupling
Single electron oxidation of biaryl Pd(II) complexes to afford Pd(III) species was observed during the electrochemical oxidation of bis-mesityl Pd(II) complex 7 (Figure 4) [56]. Complex 7, which can be viewed as a model for oxidative C–H coupling intermediate 5 (Figure 3), undergoes a one-electron oxidation at 0.57 V, assigned to the Pd(II)/Pd(III) redox couple.
Fig. 4.
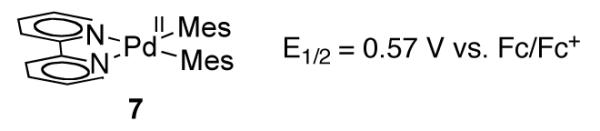
Bis-mesityl Pd(II) complex 7, an analog of intermediate 5, shows a reversible one-electron oxidation wave for the Pd(II)/Pd(III) couple at 0.57 V
Oxidative C–H coupling reactions are frequently carried out in the presence of Ag(I) additives [57-64]. As described below, due to: 1) the demonstrated availability of one-electron oxidation processes for compounds such as 7 [56], 2) the propensity of Ag(I) to facilitate one-electron oxidative cleavage of Pd–C bonds [65] (vide infra), and 3) the frequency with which Ag(I) additives are employed in Pd-catalyzed cross-coupling chemistry [57-64], the reaction chemistry of Ag(I) salts with organometallic Pd(II) complexes has received experimental scrutiny.
In 2001, Milstein studied the reactivity of Pd(II) aryl complex 8 with one-electron oxidants galvinoxyl radical and AgOTf (Figure 5) [66]. Treatment of Pd(II) aryl complex 8 with AgOTf resulted in the formation of biphenyl along with Pd(II) triflate 10. Similar reactivity was observed upon treatment of 8 with galvinoxyl radical. Milstein proposed that this oxidant-induced reductive coupling of aryl ligands proceeds through Pd(III) intermediate 9. The authors speculated that Pd(III) aryl intermediate 9 may be better formulated as a Pd(II) complex with a pendant aryl radical ligand, generated by inner-sphere ligand-to-metal electron transfer. No organic product resulting from coupling of free organic radicals with solvent were observed, suggesting that biphenyl is not produced by radical combination of phenyl radicals generated by Pd–C bond homolysis. This observation led to the suggestion that intermediate 9 may be an aryl-bridged dinuclear complex, which can liberate biphenyl without the intermediacy of free radical chemistry [67].
Fig. 5.

Treatment of Pd(II) complex 8 with AgOTf generates Pd(II) complex 10 and biphenyl
The oxidatively induced reductive coupling of methyl ligands to afford ethane from Pd(II) dimethyl complex 11 was reported in 2009 by Mayer and Sanford [68]. Treatment of dimethyl Pd(II) complex 11 with ferrocenium hexafluorophosphate ([Cp2Fe]PF6; Fc+), an outer-sphere, single-electron oxidant, led to the formation of ethane along with cationic Pd(II) complex 13 (Figure 6). Based on the electrochemical study of closely related bis-mesityl Pd(II) complex 7 (Figure 4) [56], the observed ethane formation was proposed to proceed via initial single-electron oxidation of 11 to Pd(III) complex 12.
Fig. 6.

Treatment of Pd(II) complex 11 with [Cp2Fe]PF6 (Fc+) affords ethane and 13
Three mechanisms for the formation of ethane from 12 were considered (Figure 7). In mechanism A, Pd(III) complex 12 undergoes Pd–C bond homolysis to afford Pd(II) complex 13 and methyl radicals, which subsequently combine to generate ethane. Similar Pd–C bond homolysis following single electron oxidation was proposed by Trogler during an independent study of the oxidation chemistry of Pd(II) methyl complexes with Fc+ [65]. In mechanism B, direct reductive elimination from 12 affords ethane and mononuclear Pd(I) complex 14, which further reacts with 11 and Fc+ to generate Pd(II) complex 13. Analogous chemistry has been proposed for coupling reactions from organonickel complexes [69-75]. In mechanism C, two equivalents of complex 12 undergo disproportionation to afford Pd(II) complex 13 and Pd(IV) intermediate 15. Subsequent reductive elimination from 15 generates ethane and Pd(II) complex 13. Similar disproportionation has been proposed from Pt(III) dimethyl complexes [76].
Fig. 7.

Mechanisms considered for the formation of ethane from Pd(III) complex 12
The observation that ethane formation is uninhibited by radical traps such as 1,4-cyclohexadiene and styrene suggests that ethane is not formed by combination of free methyl radicals and is inconsistent with free radical pathway A. To differentiate between pathways B and C, the oxidation of 11 with Fc+ was carried out at low temperature in order to observe potential reaction intermediates. Treatment of 11 with Fc+ at −80 °C led to the observation of Pd(II) complex 13 and Pd(IV) complex 15 (Figure 8). Subsequent warming to −30 °C led to the formation of ethane. Based on these observations, the authors concluded that pathway C is likely responsible for the formation of ethane in the reaction of 11 with Fc+.
Fig. 8.

Pd(II) complex 13 and Pd(IV) complex 15 were observed following oxidation of 11 with Fc+ at −80 °C
The chemistry of complex 11 with AgPF6 was evaluated because Ag(I) is a common additive in Pd-catalyzed oxidative C–H coupling reactions [57-64] and a potential one-electron oxidant, similar to Fc+. Treatment of dimethyl Pd(II) complex 11 with AgPF6 resulted in the immediate formation of an intermediate (16), as observed by 1H NMR spectroscopy, which subsequently generated 13, ethane, and Ag mirror (Figure 9). The authors proposed that Ag(I) acts as an inner-sphere one-electron oxidant. Initial coordination of Ag(I) to Pd to generate 16, followed by electron transfer, would furnish proposed Pd(III) intermediate 12. Disproportionation of Pd(III) intermediate 12 to Pd(II) complex 13 and Pd(IV) intermediate 15 followed by reductive elimination from Pd(IV) complex 15, as was proposed in the oxidation of 11 with Fc+, would then generate the observed reaction products.
Fig. 9.

Treatment of 11 with AgPF6 results in the formation of an intermediate, assigned as 16, which subsequently generates ethane and 13
Pd(III) in Pd-catalyzed Oxidative Carbon–Heteroatom Bond Forming Reactions
In 2009, two reports disclosed the use of single-electron oxidants in intramolecular Pd-catalyzed C–H amidation reactions. Yu and coworkers disclosed a Pd-catalyzed N-triflyl indoline (18) synthesis from N-triflyl phenethylamines (17) using single-electron oxidant Ce(SO4)2 (Figure 10) [77]. The authors proposed that this reaction proceeds through initial oxidation of Pd(II) to Pd(III). Glorius and coworkers reported a Pd-catalyzed N-acyl indoline (20) synthesis from N-acyl anilines (19) in the presence of AgOAc (Figure 11) [78]. While the authors favored a Pd(0)/Pd(II) catalysis cycle, they noted that the intermediacy of higher-valent Pd species could not be discounted given that AgOAc can serve as a single-electron oxidant.
Fig. 10.
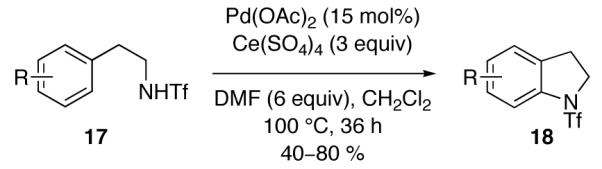
Pd-catalyzed C–H amidation reported by Yu
Fig. 11.
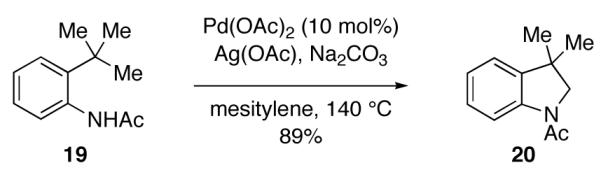
Pd-catalyzed C–H amidation reported by Glorius
Many questions remain to be addressed regarding the mechanisms of these amidation reactions. Primarily, are high-valent Pd intermediates involved, or are classical Pd(0)/Pd(II) catalysis cycles operative? If single-electron oxidation affords Pd(III) intermediates, does C–N bond formation proceed directly from Pd(III) or are Pd(IV) species, generated by either disproportionation of Pd(III) or further oxidation of Pd(III) to Pd(IV), the competent intermediates for C–N bond formation? Are potential high-valent intermediates mononuclear or is more than one palladium present during oxidation? Answers to these questions will provide insight into the potential role of high-valent Pd complexes in C–H amidation reactions.
Pd(III) in Kumada and Negishi Coupling Reactions
Knochel has noted remarkable rate accelerations for both Kumada [79] and Negishi [80] coupling reactions when performed in the presence of isopropyl iodide (Figure 12).
Fig. 12.

Isopropyl iodide catalyzed Kumada coupling reported by Knochel
Oxidative addition of Pd(0) to isopropyl iodide has been shown to proceed via initial single electron transfer to generate transient Pd(I) intermediates and isopropyl radicals [81,82]. On the basis of a positive isopropyl iodide-induced radical-clock experiment (Figure 13), Knochel suggested that the observed rate acceleration in Kumada coupling reactions is due to the participation of a radical pathway and proposed a radical mechanism involving Pd(I) and Pd(III) radical chain carriers (Figure 14). The proposal of Pd(III) intermediates in Kumada coupling reactions is, to the best of our knowledge, the only proposal of Pd(III) intermediates under reducing conditions.
Fig. 13.

Observation of isopropyl iodide-induced radical clock (R = 2,6-di-i-Pr-C6H3)
Fig. 14.
Mechanism proposed to account for the rate acceleration of cross-coupling observed in the presence of radical promoters
Pd(III) Intermediates in O2 Insertion Reactions
Goldberg and coworkers proposed Pd(III) intermediates during an investigation of the reaction of dimethyl Pd(II) complex 24 with O2 (Figure 15) [83].
Fig. 15.
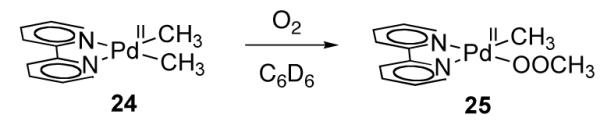
Pd(II) peroxide 25 is obtained by insertion of O2 into the Pd–C bond of 24
Consistent with a radical chain mechanism, the rate of O2 insertion was found to be sensitive to light, and the addition of radical initiator AIBN was required in order to observed reproducible reaction rates. Based on analysis of the kinetics of O2 insertion into the Pd–C bond of 24, a mechanism involving mononuclear Pd(III) intermediates was proposed (Figure 16). Palladium(III) intermediate 27, formed by combination of dimethyl Pd(II) complex 24 with peroxy radical 26 [84], generates the observed Pd(II) peroxide 25 by homolytic Pd–C cleavage to reduce Pd(III) complex 27 and generate radical chain carrier Me·.
Fig. 16.

Proposed mechanism for the insertion of O2 into the Pd–C bond of 24
The mechanism of O2 insertion into the Pd–C bond of 24 differs from the autoxidation of organic substrates due to the ability of Pd to attain high oxidation state intermediates. In hydrocarbon autoxidation, peroxy radical abstracts hydrogen without the intermediacy of hypervalent intermediates. In the oxidation of 24, the coordination number of Pd is proposed to increase from four to five upon reaction of Pd(II) complex 24 with peroxy radical (26).
2.3 Organometallic Chemistry of Isolated Pd(III) Complexes
The first example of organometallic chemistry from isolated, well-defined mononuclear Pd(III) complexes was reported by Mirica in 2010 [85]. Organometallic Pd(III) complexes 30 and 31 were prepared by controlled bulk-electrolysis of Pd(II) precursors 28 and 29, respectively (Figure 17). Complex 33 was prepared by chemical oxidation of 32 with Fc+. X-ray crystallographic analysis of 30, 31, and 33 revealed tetragonally distorted octahedral complexes, consistent with the expected Jahn-Teller distortion for mononuclear, low-spin Pd(III) complexes [6]. A combination of EPR spectroscopy and computational results suggests that the unpaired electron resides in the 4 dz2 orbital, consistent with the MO description of octahedral Pd(III) in Figure 1.
Fig. 17.
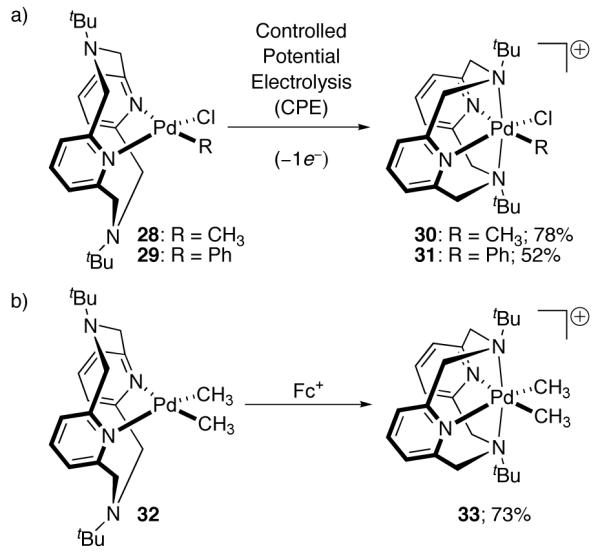
Mononuclear Pd(III) complexes 30 and 31 were prepared by Controlled Potential Electrolysis (CPE) of 28 and 29, respectively. Complex 33 was prepared by oxidation of 32 with Fc+
Photolysis of 30 afforded a mixture of ethane, methane, and methyl chloride, along with Pd(II) complex 34 (Figure 18).
Fig. 18.
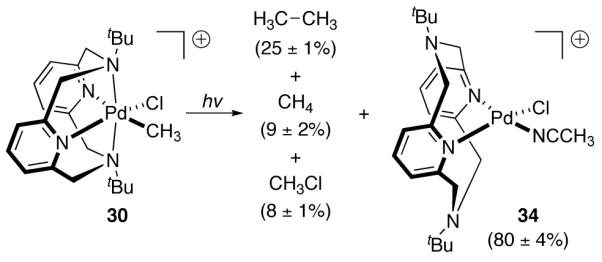
Photolysis of Pd(III) complex 30 generated ethane, methane, methyl chloride, and Pd(II) complex 34
The addition of radical scavengers, such as TEMPO, suppressed the formation of ethane, methane, and methyl chloride, instead leading only to the observation of TEMPO-Me (35) and Pd(II) complex 34 (Figure 19). The observed reaction with radical scavengers is consistent with photo-induced homolytic Pd–C bond cleavage as the operative pathway for the formation of the observed organic products, although the observed organic products may also arise from radical combination of Me· with 30 to afford a transient Pd(IV) intermediate, which subsequently generates the observed products.
Fig. 19.
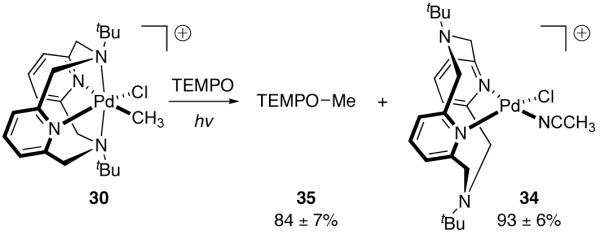
Photolysis in the presence of radical scavenger TEMPO suppressed formation of ethane, methane, and methyl chloride
3 Dinuclear Pd(III) Chemistry
3.1 Dinuclear Pd(III) Complexes
Palladium(II) has a d8 electronic configuration; the HOMO of mononuclear Pd(II) complexes is typically the dz2 orbital (Figure 1) [5]. When two palladium nuclei are held in proximity such that electronic communication between the two metals is possible, mixing of the d-orbitals gives rise to the qualitative molecular orbital picture in Figure 20 [86]. Bonding antibonding interactions result from mixing of the dz2, dxy, dxz and dyz orbitals; the dx2 −y2 is predominantly metal–ligand bonding and does not significantly participate in metal–metal bonding interactions.
Fig. 20.

Qualitative molecular orbital diagram for electronically coupled dinuclear Pd complexes based on d-orbital mixing. Oxidation of dinuclear Pd(II) complexes can result in the formation of metal–metal bonds
Dinuclear Pd(II) Complexes
Both the metal-metal σ and σ* orbitals should be filled for dinuclear Pd(II) complexes (Figure 20). Based on these considerations alone, no attractive metal–metal interaction is expected; there is no Pd–Pd bond [87]. Second-order, symmetry-allowed mixing of the Pd dz2 orbital with the 5pz and the 5s orbital, however, perturbs the molecular orbital diagram based only on d orbital interactions [88-91]. In 2010, an evaluation of the bonding interactions between the Pd centers in acetate-bridged, dinuclear Pd(II) complex 36 was reported (Figure 21) [88]. Based on DFT calculations, the authors predicted a weak attractive interaction between the Pd centers in 36 and computed a Pd–Pd bond order of 0.11.
Fig. 21.
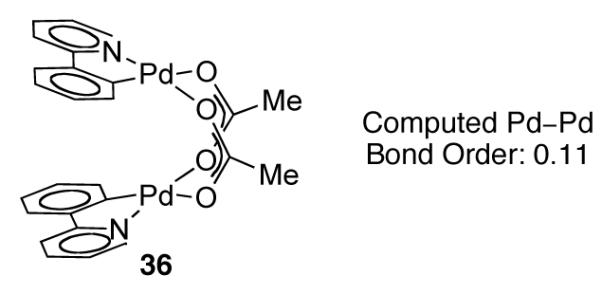
Acetate-bridged dinuclear Pd(II) complex 36 is computed to have a Pd–Pd bond order of 0.11, arising from mixing of the 5pz and 5s orbitals with the 4d orbitals
Pd(II)/Pd(III) Mixed Valence Complexes
Oxidation of dinuclear Pd(II) complexes by one electron is predicted to afford dinuclear Pd(II)/Pd(III) mixed valence complexes with a Pd–Pd bond order of 0.5 (Figure 20) [92]. Dinuclear Pd(II)/Pd(III) mixed valence complexes have been detected by EPR spectroscopy, although, similar to mononuclear systems, the unpaired electron is not always metal-centered [87,93]. Two examples of dinuclear Pd(II)/Pd(III) complexes bearing a metal-centered unpaired electron have been reported [94,95]. In 1988, Bear reported the EPR spectrum of tetra-bridged dinuclear complex 38, prepared by electrochemical oxidation of 37 (Figure 22a) [95]. In 2007, Cotton reported the only crystallographically characterized mixed valence Pd(II)/Pd(III) complex (40; Figure 22b) [94]. Both 38 and 40 are paramagnetic and have EPR spectra consistent with a metal-based oxidation. The metal–metal distance in 40 is 0.052 Å shorter than the corresponding distance in Pd(II) complex 39, consistent with a Pd–Pd bond order of 0.5.
Fig. 22.

a) Electrochemical oxidation of 37 afforded mixed valence Pd(II)/Pd(III) complex 38. b) One-electron oxidation of 39 with AgPF6 afforded 40. The Pd–Pd distance in 40 is consistent with a Pd–Pd bond order of 0.5
Elegant electrochemical studies related to Bear’s report of dinuclear Pd(II)/Pd(III) mixed valence complex 38 emphasize an important difference between the oxidation chemistry of mono- and dinuclear complexes. The electrochemistry of complex 38, bearing four bridging ligands, and 41, in which two ligands are bridging and two are chelating, was studied (Figure 23) [95]. Complex 41 displays oxidation behavior consistent with two electronically isolated Pd centers; a single two-electron oxidation wave was observed at 1.02 V. By contrast, complex 38 displays electrochemical behavior consistent with electronically coupled metal centers; a one-electron oxidation wave is observed at 0.65 V. Based on these electrochemical measurements, it was concluded that the electronic interaction between two palladium centers raises the HOMO energy by 370 mV and renders the dinuclear complex easier to oxidize than the complex with non-interacting metal centers. It is not clear why the Pd centers in 41 are electronically non-coupled, as many dinuclear Pd complexes bridged by two ligands display electrochemical behavior consistent with electronically coupled systems [92,96].
Fig. 23.

The HOMO of 38, in which the Pd centers are electronically coupled, is 370 mV higher in energy than the HOMO of 41, in which the palladium centers are not electronically coupled
Dinuclear Pd(III) Complexes
Oxidation of dinuclear Pd(II) complexes by two electrons can result in dinuclear Pd(III) complexes with a metal–metal σ bond (Figure 20). Cotton reported the first dinuclear Pd(III) complex (43) in 1998 (Figure 24) [97]. Complex 43, obtained by oxidation of dinuclear Pd(II) complex 42 with PhICl2, is diamagnetic, consistent with the formation of a spin-paired Pd–Pd bond. Further, the metal–metal bond distance in 43 is 2.39 Å, a contraction of 0.16 Å as compared to Pd(II) complex 42 (Pd–Pd = 2.55 Å).
Fig. 24.
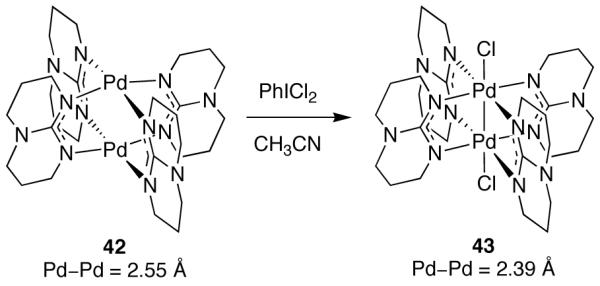
In 1998, Cotton reported the first dinuclear Pd(III) complex (43), obtained by oxidation of 42 with PhICl2
The first organometallic Pd(III) complexes (44–46) were reported in 2006 (Figure 25) [98]. Complexes 44–47 have been used as precatalysts for both the diborylation of terminal olefins and diborylation/cross-coupling tandem reactions (Figure 26) [99]. The role of the Pd(III) complexes in these reactions has not been established; the diborane reagents employed have been shown to immediately reduce the dinuclear Pd(III) complexes to Pd(II) species, and thus the Pd(III) complexes may be a precatalyst for lower-valent active catalysts.
Fig. 25.

Early examples of organometallic dinuclear Pd(III) complexes (R = Ph)
Fig. 26.
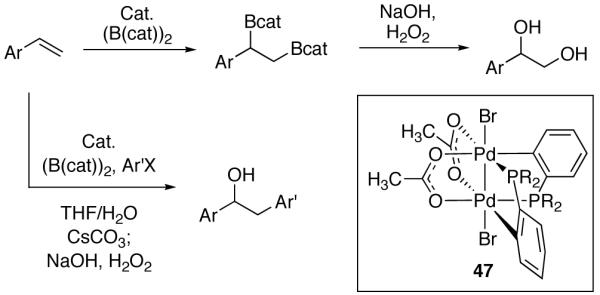
Dinuclear Pd(III) precatalysts in diborylation and borylation/cross-coupling tandem reactions (R = Ph; (B(cat))2 = bis(pinacolato)diboron)
3.2 Dinuclear Pd(III) Complexes in Catalysis
Pd-Catalyzed C–H Acetoxylation
Palladium-catalyzed aromatic C–H acetoxylation was first reported in 1966 [100,101]. In 1971, Henry proposed Pd(IV) intermediates in the Pd-catalyzed acetoxylation of benzene with K2Cr2O7 in AcOH [102]. Subsequent reports by Stock [103] and Crabtree [104] also discussed the possible intermediacy of Pd(IV) complexes in the acetoxylation of benzene (Figure 27a). In 2004, Sanford reported the regioselective ortho-acetoxylation of 2-arylpyridines and proposed a reaction mechanism involving aromatic C–H metallation at Pd(II), oxidation of the resulting aryl Pd(II) intermediate to a Pd(IV) complex, and product-forming C–O reductive elimination (Figure 27b) [105-108].
Fig. 27.
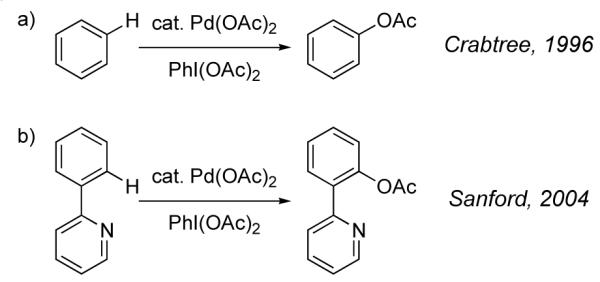
Pd-catalyzed aromatic C–H acetoxylation reactions with PhI(OAc)2
In 2009, we suggested that Pd-catalyzed aromatic C–H acetoxylation may proceed via dinuclear Pd(III) complexes instead of via mononuclear Pd(IV) intermediates [109]. Based on dinuclear Pd(II) complex 36, the product of cyclometallation of 2-phenylpyridine (48) with Pd(OAc)2 [110], a synthesis cycle based on dinuclear Pd(III) complexes was established (Figure 28). Oxidation of 36 with PhI(OAc)2, a common oxidant in Pd-catalyzed aromatic acetoxylation, afforded dinuclear Pd(III) complex 50. Complex 50 was observed to undergo C–O reductive elimination under pseudocatalytic conditions to generate 49 in 91% yield. The critical dinuclear Pd(III) intermediate (50) was crystallographically characterized; the Pd–Pd distance in 50 was measured to be 2.555 Å (compared with 2.872 Å for 36 [111]), consistent with the formation of a Pd–Pd single bond. Dinuclear Pd(III) complex 50 was found to be a kinetically competent catalyst in the acetoxylation of 2-phenylpyridine with PhI(OAc)2.
Fig. 28.

A synthesis cycle for the acetoxylation of 2-phenylpyridine (48) based on dinuclear Pd(III) complex 50 has been established
Pd-Catalyzed C–H Chlorination
Fahey reported the Pd-catalyzed directed aromatic C–H chlorination of azobenzene using Cl2 in 1970 (Figure 29) [112,113], and Sanford reported the Pd-catalyzed directed aromatic C–H chlorination of 2-arylpyridines with NCS in 2004 (chlorination reaction shown in Figure 30) [105, 114-116].
Fig. 29.

Chlorination of azobenzene reported by Fahey
Fig. 30.

The resting state of Pd-catalyzed chlorination of benzo[h]quinoline (51) is succinate-bridged dinuclear Pd complex 53. Pd–Pd bond length in 53: 2.8628(4) Å
In 2010, we reported an investigation of the mechanism of the Pd(OAc)2-catalyzed chlorination of benzo[h]quinoline (51) with NCS (N-chlorosuccinimide) (Figure 30) [96,117]. Elucidation of the salient features of the mechanism operative in catalysis was enabled by identification of the catalyst resting state, which was found to be succinate-bridged dinuclear Pd(II) complex 53. The two palladium centers in 53 are held in proximity (Pd–Pd = 2.863 Å) by the bridging succinate ligands as established by x-ray crystallography.
Using resting state 53 as the catalyst for the chlorination reaction shown in Figure 30, the rate law of chlorination was determined to be: rate = k [53] [NCS] [AcO−], which implies that oxidation is the turnover limiting step in catalysis. Further, the observed first-order dependence on dinuclear resting state 53 implies that two Pd centers participate in oxidation. The unexpected co-catalysis by acetate ions – generated by acetate for succinate exchange during formation of 53 – is consistent with a rate-determining transition state for oxidation in which acetate and NCS each interact with one of the Pd centers of resting state 53, generating a dinuclear Pd(III) intermediate (Figure 31).
Fig. 31.

Proposed acetate-assisted bimetallic oxidation of 53 would afford dinuclear Pd(III) complex 54 immediately following oxidation
The experimentally derived rate law for chlorination is consistent with dinuclear Pd(III) complex 54 being the immediate product of oxidation during catalysis. Complex 54 has one apical chloride ligand and one apical acetate ligand and thus, upon thermolysis, could undergo either C–Cl reductive elimination, to generate 52, or C–O reductive elimination, to generate 55 (Figure 32). We evaluated and confirmed the kinetic and chemical competence of 54 as an intermediate for chlorination. Chemoselective C–Cl reductive elimination from 54 was observed upon warming 54 above −78 °C (200 : 1 ratio of 52 to 55). The observed ratio of 52 to 55 in the thermal decomposition of preformed 54 is consistent with the product distribution from the Pd(OAc)2-catalyzed chlorination of benzo[h]quinoline (51). Using 10 mol% Pd(OAc)2, a 200 : 1 ratio of 52 to 55 was observed (Figure 32).
Fig. 32.

The ratio of 52 to 55 obtained by thermal decomposition of dinuclear Pd(III) complex 54 is similar to the ratio of 52 to 55 obtained by Pd(OAc)2-catalyzed chlorination of benzo[h]quinoline (51) with NCS
Pd-Catalyzed C–H Arylation
Deprez and Sanford reported an investigation of the mechanism of Pd(OAc)2-catalyzed arylation of 2-arylpyridine derivatives with diaryliodonium salt 57 in 2009 (Figure 33) [118]. The catalyst resting state was proposed to be mononuclear Pd complex 59. By examining the initial rate of arylation as a function of [Pd(OAc)2], the rate law of arylation was determined to be second order in Pd. In combination with the observation that oxidation is the turnover-limiting step in catalysis, the experimentally determined rate law is consistent with two Pd centers participating in oxidation during catalysis.
Fig. 33.

The catalyst resting state for the Pd(OAc)2-catalyzed arylation of 3-methyl-2-phenylpyridine (56) is proposed to be mononuclear Pd(II) complex 59
Sanford proposed the product of oxidation during catalysis to be one of the two constitutional isomers of a high-valent dinuclear Pd complex shown in Figure 34 and suggested that the second palladium center in either 60 or 61 functions as an auxiliary ligand to the metal center that mediates the C–C bond formation.
Fig. 34.

Formulations of the high-valent, dinuclear Pd complex proposed by Sanford in the arylation of 2-arylpyridine derivatives
3.3 Role of Dinuclear Core During Redox Chemistry
Carbon–heteroatom reductive elimination from dinuclear transition metal complexes, as was proposed by us [96,109] as the product-forming step in Pd-catalyzed C–H acetoxylation and chlorination reactions, is rare. The two formulations of the high-valent, dinuclear Pd intermediate in arylation proposed by Sanford (60 and 61) highlight that reductive elimination from dinuclear Pd structures could, in principle, proceed with either redox chemistry at both metals (bimetallic reductive elimination; reductive elimination from 60) or with redox chemistry at a single metal (monometallic redox chemistry; reductive elimination from 61). While structures 60 and 61 do not differ in composition, they do differ in their respective potentials for metal-metal redox cooperation to be involved in C–C bond forming reductive elimination.
In 2010, we reported a study regarding the role of the dinuclear core during C–Cl reductive elimination from 62, an analog of the dinuclear Pd complexes that have been proposed in catalysis (Figure 35) [96,119]. Experimental results established that reductive elimination from 62 proceeds without fragmentation of the dinuclear core; C–Cl bond formation proceeds from a dinuclear complex.
Fig. 35.
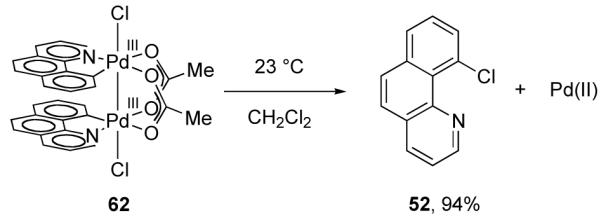
C–Cl reductive elimination from dinuclear Pd(III) complex 62
To probe the role of the dinuclear core during reductive elimination (i.e. mono- versus bimetallic reductive elimination), the electron binding energies of each Pd center were computed as a function of reaction progress. The electron binding energy is a measure of the energy required to remove an electron from a particular orbital to infinite separation. The electron binding energy of an electron decreases during reduction and increases during oxidation. For metal centers with similar ligand environments, electron binding energy is well correlated with formal oxidation state [120-124].
The computed electron binding energies of Pda and Pdb during the low energy reductive elimination pathway from A, the computed structure of 62, monotonically decrease during reduction from Pd(III) (A) to Pd(II) (D). The observed trends are consistent with simultaneous redox chemistry at both metal centers during C–Cl reductive elimination (Figure 36).
Fig. 36.

Electron binding energies as a function of reaction progress for C–Cl reductive elimination from dinuclear Pd(III) structure A
For comparison, the electron binding energies of the palladium centers during a hypothetical pathway involving a Pd(II)/Pd(IV) mixed valence intermediate were also computed (Figure 37). For hypothetical reductive elimination via a Pd(II)/Pd(IV) mixed valence complex (E), the electron binding energies of Pda and Pdb diverge during Pd(III)/Pd(III) to Pd(II)/Pd(IV) disproportionation (A → E). Subsequent reductive elimination is accompanied by a convergence of the electron binding energies as both Pd centers are becoming Pd(II) (E → D). That the electron binding energy profiles for the computed reductive elimination pathway (Figure 36) and hypothetical monometallic reductive elimination pathways (Figure 37) are different is consistent with the assertion of metal-metal redox synergy during reductive elimination from 62.
Fig. 37.

Electron binding energy as a function of reaction progress for monometallic reductive elimination via a Pd(II)/Pd(IV) mixed valence structure
In addition, the energetic barrier to reductive elimination from 62 has been calculated as a function of metal–metal bond length. As the metal-metal distance is increased, orbital overlap, which mediates redox communication during reductive elimination, is reduced. The barrier to reductive elimination is positively correlated with the metal-metal distance; as the metals are increasingly separated, reductive elimination becomes increasingly energetically demanding. These calculations suggest that metal-metal redox synergy, in which the redox chemistry of reductive elimination is shared by two metals, lowers the energetic barrier to reductive elimination versus related processes involving a single metal.
Discussion of High-Valent Pd Intermediates Relevant in Pd-Catalyzed C–H Oxidations
From the seminal studies regarding the oxidation of benzene by Henry [102], Stock [103], and Crabtree [104], to the vast array of Pd-catalyzed aromatic C–H functionalizations reported in the last five years [125-128], Pd-catalyzed aromatic C–H oxidation continues to be an area of intense methodological and mechanistic investigation. Early mechanism proposals for the acetoxylation of aromatic C–H bonds invoked Pd(IV) intermediates. While mononuclear Pd(IV) complexes have been prepared and the intimate mechanisms of reductive elimination from these complexes have been elucidated [107,108,129-132], the relevance of Pd(IV) complexes to catalysis has not yet been established. Frequently, aromatic C–H palladation is the turnover-limiting step in Pd-catalyzed C–H oxidation reactions [109, 133]. When palladation is turnover limiting, reaction kinetics analysis provides detailed information about the mechanism of metallation, not the mechanism of oxidation and cannot provide any information regarding the identity of potential high-valent intermediates.
Elucidation of the catalysis cycle that is operative during a given transformation requires direct investigation of the reaction during catalysis. Elucidation of the mechanism of oxidation relevant to catalysis by kinetics analysis requires a reaction in which oxidation is turnover limiting. In both the chlorination [96,117] and arylation [118] of 2-arylpyridine derivatives, oxidation is the turnover-limiting step of catalysis; in both reactions, kinetics analysis has revealed that two Pd centers are required for oxidation during catalysis. The structures of the proposed dinuclear Pd(III) complexes relevant to chlorination have been established by independent synthesis. A catalysis cycle, which is consistent with the results of all studies in which the identity of high-valent intermediates could be probed, is presented in Figure 38. Following C–H metallation, nucleophile-assisted bimetallic oxidation affords a dinuclear Pd(III) complex. Subsequent reductive elimination from this high-valent dinuclear complex affords the observed organic fragments and Pd(II). Although the results were obtained during study of the chlorination of benzo[h]quinoline, they may be relevant to a variety of other C–H oxidation reactions. While detailed experimentation regarding the specific mechanism of individual reactions remains to be examined, we suggest that a bimetallic redox mechanism based on dinuclear Pd(III) complexes is a viable conceptual framework for Pd-catalyzed aromatic C–H oxidation reactions.
Fig. 38.

Proposed bimetallic Pd(II)2/Pd(III)2 catalysis cycle
4 Outlook
Herein, we have reviewed the organometallic chemistry of Pd(III), discussing examples of both well-defined Pd(III) complexes that participate in organometallic reactions, as well as examples in which the potential involvement of Pd(III) is more speculative at this time. We have discussed oxidative C–H coupling reactions in which biaryl Pd(II) intermediates may be diverted from the traditional Pd(0)/Pd(II) redox cycle in the presence of one-electron oxidants, allowing access to catalytically relevant mononuclear Pd(III) intermediates. We have also examined the recent proposal that many Pd-catalyzed oxidative C–H functionalization reactions may proceed via dinuclear Pd(III) complexes which undergo product-forming reductive elimination with redox participation of both metal centers. In both oxidative C–H coupling reactions as well as Pd-catalyzed C–H oxidations, Pd(III) intermediates have been discovered in reactions previously believed to proceed via more traditional two-electron, monometallic Pd redox cycles. Based on these proposals, Pd(III), in both mono- and dinuclear complexes, may play a much more prominent role in catalysis than has previously been appreciated. We anticipate that the unique reactivity of both mono- and dinuclear Pd complexes will provide a foundation for the discovery of new reactions mediated by Pd(III) in the future.
Acknowledgement
We gratefully acknowledge financial support for this work from the NIH-NIGMS (GM088237).
References
- 1.Negishi E, editor. Handbook of Organopalladium chemistry for Organic Synthesis. John Wiley & Sons, Inc.; New York: 2002. [Google Scholar]
- 2.Murahashi T, Kurosawa H. Coord Chem Rev. 2002;231:207–228. [Google Scholar]
- 3.Markert C, Neuburger M, Kulicke K, Meuwly M, Pfaltz A. Angew Chem Int Ed. 2007;46:5892–5895. doi: 10.1002/anie.200701248. [DOI] [PubMed] [Google Scholar]
- 4.Hama T, Hartwig JF. Org Lett. 2008;10:1545–1548. doi: 10.1021/ol8002578. [DOI] [PubMed] [Google Scholar]
- 5.Albright TA. Tetrahedron. 1982;38:1339–1388. [Google Scholar]
- 6.Miessler GL, Tarr DA. Inorganic Chemistry. Pearson Education, Inc.; Upper Saddle River, New Jersey: 2004. [Google Scholar]
- 7.Canty AJ, Gardiner MG, Jones RC, Rodemann T, Sharma M. J Am Chem Soc. 2009;131:7236–7237. doi: 10.1021/ja902799u. [DOI] [PubMed] [Google Scholar]
- 8.Bonnington KJ, Jennings MC, Puddephatt RJ. Organometallics. 2008;27:6521–6530. [Google Scholar]
- 9.Roundhill DM, Gray HB, Che CM. Acc Chem Res. 1989;22:55–61. [Google Scholar]
- 10.Matsumoto K, Sakai K. Advances in Inorganic Chemistry. 2000;49:375–427. [Google Scholar]
- 11.Lippert B. Coord Chem Rev. 1999;182:263–295. [Google Scholar]
- 12.Zipp AP. Coord Chem Rev. 1988;84:47–83. [Google Scholar]
- 13.Blake AJ, Gould RO, Holder AJ, Hyde TI, Lavery AJ, Odulate MO, Schröder M. J Chem Soc, Chem Commun. 1987:118–120. [Google Scholar]
- 14.Usón R, Forniés J, Tomás M, Menjón B, Sünkel K, Bau R. J Chem Soc, Chem Commun. 1984:751–752. [Google Scholar]
- 15.Usón R, Forniés J, Tomás M, Menjón B, Bau R, Sünkel K, Kuwabara E. Organometallics. 1986;5:1576–1581. [Google Scholar]
- 16.Nyholm RS, Sharpe AG. J Chem Soc. 1952:3579–3587. [Google Scholar]
- 17.Sharpe AG. J Chem Soc. 1950:3444–3450. [Google Scholar]
- 18.Bartlett N, Rao PR. Proc Chem Soc London. 1964:393–394. [Google Scholar]
- 19.Tressaud A, Wintenberger M, Bartlett N, Hagenmuller P. C R Acad Sc Paris. 1976;282:1069–1072. [Google Scholar]
- 20.Tressaud A, Khairoun S, Dance JM, Grannec J, Portier J, Hagenmuller P. J Fluorine Chem. 1982;21:28. [Google Scholar]
- 21.Tressaud A, Khairoun S, Dance JM, Hagenmuller P. Z Anorg Allg Chem. 1984;517:43–58. [Google Scholar]
- 22.Tressaud A, Khairoun S, Grannec J, Dance JM, Hagenmuller P. J Fluorine Chem. 1985;29:39. [Google Scholar]
- 23.Jasper SA, Huffman JC, Todd LJ. Inorg Chem. 1998;37:6060–6064. doi: 10.1021/ic980681n. [DOI] [PubMed] [Google Scholar]
- 24.Eachus RS, Graves RE. J Chem Phys. 1976;65:5445–5452. [Google Scholar]
- 25.Krigas T, Rogers MT. J Chem Phys. 1971;54:4769–4775. [Google Scholar]
- 26.Raizman A, Barak J, Suss JT. Phys Rev B. 1985;31:5716–5721. doi: 10.1103/physrevb.31.5716. [DOI] [PubMed] [Google Scholar]
- 27.Lane GA, Geiger WE, Connelly NG. J Am Chem Soc. 1987;109:402–407. [Google Scholar]
- 28.Möller E, Kirmse R. Inorg Chim Acta. 1997;257:273–276. [Google Scholar]
- 29.Luca V, Kukkadapu R, Kevan L. J Chem Soc, Faraday Trans. 1991;87:3083–3089. [Google Scholar]
- 30.Warren LF, Hawthorn MF. J Am Chem Soc. 1968;90:4823–4828. [Google Scholar]
- 31.Kirmse R, Stach J, Dietzsch W, Steimecke G, Hoyer E. Inorg Chem. 1980;19:2679–2685. [Google Scholar]
- 32.Pandey KK. Coord Chem Rev. 1992;121:1–42. [Google Scholar]
- 33.Ray K, Weyhermüller T, Neese F, Wieghardt K. Inorg Chem. 2005;44:5345–5360. doi: 10.1021/ic0507565. [DOI] [PubMed] [Google Scholar]
- 34.Motoyama T, Shimazaki Y, Yajima T, Nakabayashi Y, Naruta Y, Yamauchi O. J Am Chem Soc. 2004;126:7378–7385. doi: 10.1021/ja031587v. [DOI] [PubMed] [Google Scholar]
- 35.Yamashita M, Takaishi S. Chem Commun. 2010;46:4438–4448. doi: 10.1039/c002097d. [DOI] [PubMed] [Google Scholar]
- 36.Takaishi S, Wu H, Xie J, Kajiwara T, Breedlove BK, Miyasaka H, Yamashita M. Inorg Chem. 2010;49:3694–3696. doi: 10.1021/ic1001368. [DOI] [PubMed] [Google Scholar]
- 37.Blake AJ, Holder AJ, Hyde TI, Schröder M. J Chem Soc, Chem Commun. 1987:987–988. [Google Scholar]
- 38.Blake AJ, Holder AJ, Hyde TI, Roberts YV, Lavery AJ, Schröder M. J Organomet Chem. 1987;323:261–270. [Google Scholar]
- 39.Matsumoto M, Itoh M, Funahashi S, Takagi HD. Can J Chem. 1999;77:1638–1647. [Google Scholar]
- 40.Blake AJ, Gordon LM, Holder AJ, Hyde TI, Reid G, Schröder M. J Chem Soc, Chem Commun. 1988:1452–1454. [Google Scholar]
- 41.McAuley A, Whitcombe TW. Inorg Chem. 1988;27:3090–3099. [Google Scholar]
- 42.Hunter G, McAuley A, Whitcombe TW. Inorg Chem. 1988;27:2634–2639. [Google Scholar]
- 43.Reid G, Blake AJ, Hyde TI, Schröder M. J Chem Soc, Chem Commun. 1988:1397–1399. [Google Scholar]
- 44.Blake AJ, Reid G, Schröder M. J Chem Soc, Dalton Trans. 1990:3363–3373. [Google Scholar]
- 45.Blake AJ, Crofts RD, de Groot B, Schröder M. J Chem Soc, Dalton Trans. 1993:485–486. [Google Scholar]
- 46.Reid G, Schröder M. Chem Soc Rev. 1990;19:239–269. [Google Scholar]
- 47.Hartwig JF. Organotransition Metal Chemistry. University Science Books; Sausalito, California: 2010. [Google Scholar]
- 48.Ackermann L, Vicente R, Kapdi AR. Angew Chem Int Ed. 2009;48:9792–9826. doi: 10.1002/anie.200902996. [DOI] [PubMed] [Google Scholar]
- 49.Alberico D, Scott ME, Lautens M. Chem Rev. 2007;107:174–238. doi: 10.1021/cr0509760. [DOI] [PubMed] [Google Scholar]
- 50.Campeau LC, Fagnou K. Chem Commun. 2006:1253–1264. doi: 10.1039/b515481m. [DOI] [PubMed] [Google Scholar]
- 51.McGlacken GP, Bateman LM. Chem Soc Rev. 2009;38:2447–2464. doi: 10.1039/b805701j. [DOI] [PubMed] [Google Scholar]
- 52.Pascual S, de Mendoza P, Echavarren AM. Org Biomol Chem. 2007;5:2727–2734. doi: 10.1039/b707940k. [DOI] [PubMed] [Google Scholar]
- 53.Stuart DR, Fagnou K. Science. 2007;316:1172–1175. doi: 10.1126/science.1141956. [DOI] [PubMed] [Google Scholar]
- 54.Kalyani D, Sanford MS. J Am Chem Soc. 2008;130:2150–2151. doi: 10.1021/ja0782798. [DOI] [PubMed] [Google Scholar]
- 55.Kalyani D, Satterfield AD, Sanford MS. J Am Chem Soc. 2010;132:8419–8427. doi: 10.1021/ja101851v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Klein A, Niemeyer M. Z Anorg Allg Chem. 2000;626:1191–1195. [Google Scholar]
- 57.Chen X, Goodhue CE, Yu JQ. J Am Chem Soc. 2006;128:12634–12635. doi: 10.1021/ja0646747. [DOI] [PubMed] [Google Scholar]
- 58.Cho SH, Hwang SJ, Chang S. J Am Chem Soc. 2008;130:9254–9256. doi: 10.1021/ja8026295. [DOI] [PubMed] [Google Scholar]
- 59.Stuart DR, Villemure E, Fagnou K. J Am Chem Soc. 2007;129:12072–12073. doi: 10.1021/ja0745862. [DOI] [PubMed] [Google Scholar]
- 60.Takahashi M, Masui K, Sekiguchi H, Kobayashi N, Mori A, Funahashi M, Tamaoki N. J Am Chem Soc. 2006;128:10930–10933. doi: 10.1021/ja060749v. [DOI] [PubMed] [Google Scholar]
- 61.Potavathri S, Dumas AS, Dwight TA, Naumiec GR, Hammann JM, DeBoef B. Tetrahedron Lett. 2008;49:4050–4053. doi: 10.1016/j.tetlet.2008.04.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Campeau LC, Parisien M, Jean A, Fagnou K. J Am Chem Soc. 2006;128:581–590. doi: 10.1021/ja055819x. [DOI] [PubMed] [Google Scholar]
- 63.Lebrasseur N, Larrosa I. J Am Chem Soc. 2008;130:2926–2927. doi: 10.1021/ja710731a. [DOI] [PubMed] [Google Scholar]
- 64.René O, Fagnou K. Org Lett. 2010;12:2116–2119. doi: 10.1021/ol1006136. [DOI] [PubMed] [Google Scholar]
- 65.Seligson AL, Trogler WC. J Am Chem Soc. 1992;114:7085–7089. [Google Scholar]
- 66.Kraatz HB, van der Boom ME, Ben-David Y, Milstein D. Isr J Chem. 2001;41:163–171. [Google Scholar]
- 67.Ozawa F, Fujimori M, Yamamoto T, Yamamoto A. Organometallics. 1986;5:2144–2149. [Google Scholar]
- 68.Lanci MP, Remy MS, Kaminsky W, Mayer JM, Sanford MS. J Am Chem Soc. 2009;131:15618–15620. doi: 10.1021/ja905816q. [DOI] [PubMed] [Google Scholar]
- 69.Coronas JM, Muller G, Rocamora M. J Organomet Chem. 1986;301:227–236. [Google Scholar]
- 70.Ceder RM, Granell J, Muller G, Font-Bardía M, Solans X. Organometallics. 1996;15:4618–4624. [Google Scholar]
- 71.Higgs AT, Zinn PJ, Simmons SJ, Sanford MS. Organometallics. 2009;28:6142–6144. [Google Scholar]
- 72.Tsou TT, Kochi JK. J Am Chem Soc. 1979;101:7547–7560. [Google Scholar]
- 73.Matsunaga PT, Hillhouse GL, Rheingold AL. J Am Chem Soc. 1993;115:2075–2077. [Google Scholar]
- 74.Koo K, Hillhouse GL. Organometallics. 1995;14:4421–4423. [Google Scholar]
- 75.Higgs AT, Zinn PJ, Sanford MS. Organometallics asap. 2010.
- 76.Johansson L, Ryan OB, Rømming C, Tilset M. Organometallics. 1998;17:3957–3966. [Google Scholar]
- 77.Mei TS, Wang X, Yu JQ. J Am Chem Soc. 2009;131:10806–10807. doi: 10.1021/ja904709b. [DOI] [PubMed] [Google Scholar]
- 78.Neumann JJ, Rakshit S, Dröge T, Glorius F. Angew Chem Int Ed. 2009;48:6892–6895. doi: 10.1002/anie.200903035. [DOI] [PubMed] [Google Scholar]
- 79.Manolikakes G, Knochel P. Angew Chem Int Ed. 2009;48:205–209. doi: 10.1002/anie.200803730. [DOI] [PubMed] [Google Scholar]
- 80.Kienle M, Knochel P. Org Lett. 2010;12:2702–2705. doi: 10.1021/ol1007026. [DOI] [PubMed] [Google Scholar]
- 81.Kramer AV, Labinger JA, Bradley JS, Osborn JA. J Am Chem Soc. 1974;96:7145–7147. [Google Scholar]
- 82.Kramer AV, Osborn JA. J Am Chem Soc. 1974;96:7832–7833. [Google Scholar]
- 83.Boisvert L, Denney MC, Hanson SK, Goldberg KI. J Am Chem Soc. 2009;131:15802–15814. doi: 10.1021/ja9061932. [DOI] [PubMed] [Google Scholar]
- 84.Kamaraj K, Bandyopadhyay D. Organometallics. 1999;18:438–446. [Google Scholar]
- 85.Khusnutdinova JR, Rath NP, Mirica LM. J Am Chem Soc. 2010;132:7303–7305. doi: 10.1021/ja103001g. [DOI] [PubMed] [Google Scholar]
- 86.Cotton FA, Murillo CA, Walton RA, editors. Multiple Bonds Between Metal Atoms. Springer Science and Business Media, Inc.; New York: 2005. [Google Scholar]
- 87.Cotton FA, Matusz M, Poli R, Feng X. J Am Chem Soc. 1988;110:1144–1154. [Google Scholar]
- 88.Bercaw JE, Durrell AC, Gray HB, Green JC, Hazari N, Labinger JA, Winkler JR. Inorg Chem. 2010;49:1801–1810. doi: 10.1021/ic902189g. [DOI] [PubMed] [Google Scholar]
- 89.Xia BH, Che CM, Zhou ZY. Chem-Eur J. 2003;9:3055–3064. [Google Scholar]
- 90.Pan QJ, Zhang HX, Zhou X, Fu HG, Yu HT. J Phys Chem A. 2007;111:287–294. doi: 10.1021/jp065183b. [DOI] [PubMed] [Google Scholar]
- 91.Yip HK, Lai TF, Che CM. J Chem Soc, Dalton Trans. 1991:1639–1641. [Google Scholar]
- 92.Berry JF, Cotton FA, Ibragimov SA, Murillo CA, Wang X. Inorg Chem. 2005;44:6129–6137. doi: 10.1021/ic050876c. [DOI] [PubMed] [Google Scholar]
- 93.Cotton FA, Matusz M, Poli R. Inorg Chem. 1987;26:1472–1474. [Google Scholar]
- 94.Berry JF, Bill E, Bothe E, Cotton FA, Dalal NS, Ibragimov SA, Kaur N, Liu CY, Murillo CA, Nellutla S, North JM, Villagrán D. J Am Chem Soc. 2007;129:1393–1401. doi: 10.1021/ja067328y. [DOI] [PubMed] [Google Scholar]
- 95.Yao CL, He LP, Korp JD, Bear JL. Inorg Chem. 1988;27:4389–4395. [Google Scholar]
- 96.Powers DC, Ritter T. Nat Chem. 2009;1:302–309. doi: 10.1038/nchem.246. [DOI] [PubMed] [Google Scholar]
- 97.Cotton FA, Gu J, Murillo CA, Timmons DJ. J Am Chem Soc. 1998;120:13280–13281. [Google Scholar]
- 98.Cotton FA, Koshevoy IO, Lahuerta P, Murillo CA, Sanaú M, Ubeda MA, Zhao Q. J Am Chem Soc. 2006;128:13674–13675. doi: 10.1021/ja0656595. [DOI] [PubMed] [Google Scholar]
- 99.Penno D, Lillo V, Koshevoy IO, Sanaú M, Ubeda MA, Lahuerta P, Fernández E. Chem Eur J. 2008;14:10648–10655. doi: 10.1002/chem.200800931. [DOI] [PubMed] [Google Scholar]
- 100.Davidson JM, Triggs C. Chem Ind (London) 1966:457. [Google Scholar]
- 101.Tisue T, Downs WJ. J Chem Soc D. 1969:410a. [Google Scholar]
- 102.Henry PM. J Org Chem. 1971;36:1886–1890. [Google Scholar]
- 103.Stock LM, Tse K, Vorvick LJ, Walstrum SA. J Org Chem. 1981;46:1757–1759. [Google Scholar]
- 104.Yoneyama T, Crabtree RH. J Mol Catal A: Chem. 1996;108:35–40. [Google Scholar]
- 105.Dick AR, Hull KL, Sanford MS. J Am Chem Soc. 2004;126:2300–2301. doi: 10.1021/ja031543m. [DOI] [PubMed] [Google Scholar]
- 106.Desai LV, Hull KL, Sanford MS. J Am Chem Soc. 2004;126:9542–9543. doi: 10.1021/ja046831c. [DOI] [PubMed] [Google Scholar]
- 107.Dick AR, Kampf JW, Sanford MS. J Am Chem Soc. 2005;127:12790–12791. doi: 10.1021/ja0541940. [DOI] [PubMed] [Google Scholar]
- 108.Racowski JM, Dick AR, Sanford MS. J Am Chem Soc. 2009;131:10974–10983. doi: 10.1021/ja9014474. [DOI] [PubMed] [Google Scholar]
- 109.Powers DC, Geibel MAL, Klein JEMN, Ritter T. J Am Chem Soc. 2009;131:17050–17051. doi: 10.1021/ja906935c. [DOI] [PubMed] [Google Scholar]
- 110.Gutierrez MA, Newkome GR, Selbin J. J Organomet Chem. 1980;202:341–350. [Google Scholar]
- 111.Dinçer M, Özdemir N, Günay ME, Çetinkaya B. Acta Crystallogr, Sect E: Struct Rep Online. 2008;64:M381. doi: 10.1107/S1600536808001736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Fahey DR. J Chem Soc D. 1970:417. [Google Scholar]
- 113.Fahey DR. J Organomet Chem. 1971;27:283–292. [Google Scholar]
- 114.Whitfield SR, Sanford MS. J Am Chem Soc. 2007;129:15142–15143. doi: 10.1021/ja077866q. [DOI] [PubMed] [Google Scholar]
- 115.Kalyani D, Dick AR, Anani WQ, Sanford MS. Org Lett. 2006;8:2523–2526. doi: 10.1021/ol060747f. [DOI] [PubMed] [Google Scholar]
- 116.Kalyani D, Dick AR, Anani WQ, Sanford MS. Tetrahedron. 2006;62:11483–11498. [Google Scholar]
- 117.Powers DC, Xiao DY, Geibel MAL, Ritter T. J Am Chem Soc. 2010;132 doi: 10.1021/ja1054274. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Deprez NR, Sanford MS. J Am Chem Soc. 2009;131:11234–11241. doi: 10.1021/ja904116k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Powers DC, Benitez D, Tkatchouk E, Goddard WA, Ritter T. J Am Chem Soc. 2010;132 doi: 10.1021/ja1036644. asap. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Riggs WM. Anal Chem. 1972;44:830–832. doi: 10.1021/ac60312a041. [DOI] [PubMed] [Google Scholar]
- 121.Leigh GJ. Inorg Chim Acta. 1975;14:L35–L36. [Google Scholar]
- 122.Chatt J, Elson CM, Hooper NE, Leigh GJ. J Chem Soc, Dalton Trans. 1975:2392–2401. [Google Scholar]
- 123.Cisar A, Corbett JD, Daake RL. Inorg Chem. 1979;18:836–843. [Google Scholar]
- 124.Corbett JD. Inorg Chem. 1983;22:2669–2672. [Google Scholar]
- 125.Lyons TW, Sanford MS. Chem Rev. 2010;110:1147–1169. doi: 10.1021/cr900184e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Xu L-M, Li B-J, Yang Z, Shi Z-J. Chem Soc Rev. 2010:712–733. doi: 10.1039/b809912j. [DOI] [PubMed] [Google Scholar]
- 127.Muñiz K. Angew Chem Int Ed. 2009;48:9412–9423. doi: 10.1002/anie.200903671. [DOI] [PubMed] [Google Scholar]
- 128.Chen X, Engle KM, Wang DH, Yu JQ. Angew Chem Int Ed. 2009;48:5094–5115. doi: 10.1002/anie.200806273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Canty AJ. Acc Chem Res. 1992;25:83–90. [Google Scholar]
- 130.Byers PK, Canty AJ, Crespo M, Puddephatt RJ, Scott JD. Organometallics. 1988;7:1363–1367. [Google Scholar]
- 131.Fu Y, Li Z, Liang S, Guo QX, Liu L. Organometallics. 2008;27:3736–3742. [Google Scholar]
- 132.Furuya T, Benitez D, Tkatchouk E, Strom AE, Tang P, Goddard WA, Ritter T. J Am Chem Soc. 2010;132:3793–3807. doi: 10.1021/ja909371t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Stowers KJ, Sanford MS. Org Lett. 2009;11:4584–4587. doi: 10.1021/ol901820w. [DOI] [PMC free article] [PubMed] [Google Scholar]