

Abstract
An idea to use 4′-C-substituted-2′-deoxynucleoside derivatives was proposed based on a working hypothesis to solve the problems of existing acquired immune deficiency syndrome chemotherapy (highly active antiretroviral therapy). Subsequent studies have successfully proved the validity of the idea and resulted in the development of 2′-deoxy-4′-C-ethynyl-2-fluoroadenosine and 2′-deoxy-4′-C-ethynyl-2-chloroadenosine, nucleoside reverse transcriptase inhibitors, which have supremely high activity against all human immunodeficiency viruses including multidrug-resistant HIV and low toxicity.
Keywords: AIDS, anti-HIV agent, nucleoside reverse transcriptase inhibitor, drug-resistant HIV, 4′-C-substituted-2′-deoxynucleoside
Preface
I would like to describe the development of 2′-deoxy-4′-C-ethynyl-2-fluoroadenosine and its 2-chloro congener that have high anti-HIV activity and low toxicity and prevent the emergence of resistant HIV mutants, because the development is one of my contributions to the collaboration studies, with Dr. Takeshi Kitahara, a Professor Emeritus of The University of Tokyo, on Invention and Application of Molecules with Novel Biological Functions for receiving The Japan Academy Prize in 2010.
Introduction
Since the discovery of 3′-C-azido-3′-deoxythymidine (AZT) as the anti-human immunodeficiency virus (HIV) agent by Dr. Hiroaki Mitsuya in 1985,1) many 2′,3′-dideoxy nucleoside (ddN) analogs have been developed as nucleoside reverse transcriptase inhibitors (NRTIs). However, resistant HIV mutants against all these NRTIs have emerged rapidly and easily.
A highly active antiretroviral therapy (HAART) using two or more nucleoside reverse transcriptase inhibitors (NRTIs) and protease inhibitors has dramatically improved the quality of life and prognosis of patients infected by HIV.2,3) However, the existing HAART has several critical problems that remain to be solved. These problems include: (i) the emergence of new drug-resistant HIV mutants, (ii) the need to take large dosages of drugs, and (iii) drug side effects. Therefore, the development of new, highly potent anti-HIV drugs that prevent the emergence of drug-resistant mutants and have few side effects is urgently needed.
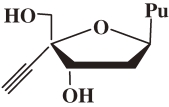
These problems prompted me to speculate on the reason for the emergence of HIV mutant resistant to clinical NRTIs, which belong to the family of ddN. I proposed a working hypothesis for the chemical structure of a new highly anti-HIV active NRTIs that might prevent the emergence of drug-resistant mutants. The design of 4′-C-substituted-2′-deoxynucleoside (4′SdN) was based on the working hypothesis. The working hypothesis for the decrease of toxicity of nucleoside derivatives was also proposed based on my past findings of the relationship between the biological activity and structure of nucleoside derivatives. Study on the synthesis and biological evaluation of 4′SdNs have proved the validity of the working hypotheses and resulted in the development of 2′-deoxy-4′-C-ethynyl-2-fluoroadenosine (4′Ed2FA) and 2′-deoxy-4′-C-ethynyl-2-chloroadenosine (4′Ed2ClA) (Fig. 1 ). These NRTIs are supremely active against all HIVs and have prevented the emergence of drug-resistant mutants for many years since their development and expected to prevent the emergence of drug-resistant mutants, have a low toxicity, and stable to enzymatic catabolism. Hence, 4′Ed2FA and 4′Ed2ClA are important keys for their further development of potential therapeutics to solve the problems being encountered by the present HAART.
Figure 1.

Structures of 2′-deoxy-4′-C-ethynyl-2-fluoroadenosine (4′Ed2FA) and its 2-chloro congener (4′Ed2ClA).
Working hypotheses of the study
1. Design of the nucleoside derivatives that might be effective for drug-resistant HIV-mutants and possibly prevent the emergence of new drug-resistant HIV mutants.
The structures of the clinical NRTIs are shown in Fig. 2. All belong to the ddN family. The ddN structure has been considered essential for the nucleoside derivatives to be the chain terminator of the reverse transcriptase (RT)-catalyzed biosynthesis of proviral DNA. However, HIV mutants resistant to all ddN-NRTIs have emerged rapidly and easily.
Figure 2.

Structures of clinical nucleosides reverse transcriptase inhibitors (NRTIs), physiologic 2′-deoxynucleoside (4′SdN). HIV = human immunodeficiency virus; dN = 2′-deoxynucleoside, 4′SdN = 4′-C-substituted-2′-deoxynucleoside.
The emergence of HIV-mutants resistant to ddNs indicates that HIV-mutants acquired the ability to distinguish ddNs from physiological 2′-deoxy nucleosides (dNs) and do not accept ddNs into the active center of RT and/or cut off the incorporated ddNs from the proviral DNA terminus.
Therefore, the anti-HIV nucleosides that might prevent the emergence of drug-resistant HIV mutants must satisfy the following two conditions.
-
1.
To prevent the discrimination by HIV, the modified nucleosides should have a structure resembling those of physiologic nucleosides as closely as possible so that RT mistakes them for physiologic nucleosides. Since the striking difference of the ddN and dN is whether they have 3′-OH, the modified nucleosides must have 3′-OH.
-
2.
In spite of having 3′-OH, the nucleoside must be the chain terminator of RT-catalyzed biosynthesis of proviral DNA.
Based on the following hypothesis, 4′SdN (Fig. 2) was designed as a nucleoside that could satisfy the above mentioned two conditions:
-
①
It would be difficult for HIV to discriminate 4′SdN from dN because 4′SdN has all the functional groups of dN.
-
②
The introduction of a substituent at 4′-position makes the 3′-OH into a very unreactive neopentyl-type secondary alcohol. Thus, the 3′-OH of 4′SdN will be used for HIV to mistake 4′SdN for dN, but is too unreactive to be used for the elongation of proviral DNA by RT. Therefore, 4′SdNs could be the chain terminators of proviral DNA biosynthesis.
-
③
Steric hindrance between 3′-OH and 4′-substituent changes the conformation of the furanose ring of 4′SdN preferably to the 3′-endo conformation (N-type). This results in 4′SdN being less susceptible to both acidic and enzymatic glycolysis than dN and ddN. (In glycolysis, the oxygen atom of the furanose ring participates to form a coplanar oxocarbonium ion, but the conformational change makes it difficult for the oxygen atom to form a coplanar oxocarbonium ion.)
-
④
Further, the electron-withdrawing 3′-OH makes 4′SdN more acid stable than does ddN even with purines. Thus, various purine derivatives can be made in this way.
-
⑤
The lipophilic substituent at the 4′-position imparts more lipophilicity to 4′SdNs, thus enabling them to penetrate the cell membrane efficiently. This possibly enhances their bioavailability.
2. Method of decreasing the toxicity of nucleoside drugs based on my past findings of the relationship between the biological activity and structure of nucleoside derivatives.
If DNA polymerases also mistake 4′SdN for dN, 4′SdN should be highly toxic. However, ddNs have been used as anti-HIV drugs, meaning that DNA polymerases hardly accept ddNs as their substrates. Thus, the ability of DNA polymerases to distinguish substrate is superior to that of RT. (The substrate selectivity between DNA polymerases and RT must be different.)
The structures of the representative nucleoside antibiotic are shown in Fig. 3.4) Most of them are nucleoside derivatives modified at one site of the physiological nucleosides. Though they are highly active against microorganisms, they are highly toxic, too. Therefore, they can not be clinically used. In the 1960s and 1970s, many organic chemists synthesized nucleoside derivatives modified at two or more positions of physiologic nucleosides expecting to get nucleoside derivatives having new and/or higher biological activity. However, none of these modified nucleosides exhibited antimicrobial activity. These results indicated that intracellular enzymes do not recognize the nucleoside derivatives modified at two or more positions of physiologic nucleosides as substrates. Thus, there is a chance of decreasing the toxicity of 4′SdNs further by additional modification.
Figure 3.

Structures of the representative nucleoside antibiotics. The arrows point at the modified.
Results and discussion
Examination of the validity of the working hypotheses with 4′-C-methyl nucleosides.
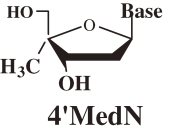
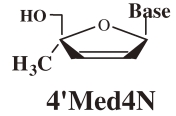
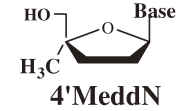
At first, to examine the validity of the working hypothesis, 4′-C-methyl-nucleosides (4′MNs), 4′-C-methyl-2′-deoxynucleosides (4′MdNs), 4′-C-methyl-2′,3′-dideoxynucleosides (4′MddNs), and 4′-C-methyl-2′,3′-didehydrodideoxynucleosides (4′Md4Ns) (Table 1 ) were synthesized and evaluated for their biological activity.5,6) 4′MdN showed remarkable biologoical activity (both anti-HIV activity and toxicity), but 4′MddN and 4′Md4N did not show notable biological activity (Table 1). 4′MNs did not show any anti-HIV activity at all.
Table 1.
Anti-HIV activity of 4′-C-methyl-2′-deoxynucleosides
| Structure | Base | EC50 (µM) | CC50 (µM) | SI (CC50/EC50) |
|---|---|---|---|---|
 |
Ad | 2.6 | 2.6 | 1.0 |
| Th | 7.2 | 104 | ||
| Cy | 0.072 | 0.13 | 1.8 | |
| Purine | 1.9 | >200 | >100 | |
 |
Ad | >500 | >500 | ∼1 |
| Th | 21 | 330 | 16 | |
| Cy | 350 | 350 | ||
 |
Ad | 30 | 400 | 13 |
| Th | >500 | >500 | ∼1 | |
| Cy | 27 | 27 | 1 | |
| AZT | 0.001 | >20 | >2020 | |
| ddA | 47 | >500 | >11 | |
| d4T | 4.1 | >500 | >120 | |
EC50 = 50% effective concentration; CC50 = cytotoxic concentration; SI = selectivity index (CC50/EC50); 4′MdN = 4′-C-methyl-2′-deoxynucleoside; 4′Md4N = 4′-C-methyl-2′,3′-didehydrodideoxynucleoside; 4′MddN = 4′-C-methyl-2′,3′-dideoxynucleoside; AZT = 3′-azido-3′-deoxythymidine; ddA = 2′,3′-dideoxyadenosine; d4T = 2′,3′-didehydro-3′-deoxythymidine.
These results indicate the importance of the 3′-OH for biological activity. Further, we demonstrated that 5′-O-triphosphate of both 4′-C-methyl-2′-deoxycytidine (4′MdCTP) and 4′-C-methyl-d-arabino-furanosyl cytidine (4′MAraCTP) are the chain terminator of calf thymus DNA polymerase α and recombinant rat DNA polymerase β.7) These results indicate that 4′SdN was an NRTI, although further study of 4′MdCTP with RT was not performed.
They were synthesized by glycosidation of 1,2-di-O-acetyl-3,5-di-O-benzyl-4-C-methyl-d-ribo-furanose (4), prepared from d-glucose through 3-O-benzyl-4-C-hydroxymethyl-1,2-O -isopropylidene-α-d-ribo-furanose (1),8) and nucleobases and then deoxygenation of the hydroxyl groups of 4′MNs (Scheme 1 ).
Scheme 1.

General synthetic scheme of 4′-C-substituted-2′-deoxynucleoside (4′SdNs) from d-glucose. 4′SdN = 4′-C-substituted-2′-deoxynucleoside; 4′MN = 4′-C-methylnucleoside; 4′Md4N = 4′-C-methyl-2′,3′-didehydrodeoxynucleoside; 4′MddN = 4′-C-methyl-2′,3′-dideoxynucleoside.
It should be noted that the 4′-C-methyl ribo-furanose derivatives took longer reaction time to complete the glycosidation reaction with silylated nucleoside bases than that of normal ribo-furanose derivatives, indicating the lower reactivity of the 4′-C-methyl ribo-furanoses than normal ribo-furanoses. In addition, the 3J1′,2′ values of 4′-C-methyl ribo-furanosyl nucleoside were larger (4–7 Hz) than those of the normal β-ribo-nucleoside (0–1 Hz), indicating the change of the conformation of the furanose ring of the 4′-C-substituted nucleosides.
Structure–activity relationship (SAR) of 4′SdNs.
Next, to study the SAR of 4′SdNs and develop 4′SdNs having more potent anti-HIV activity and less toxicity than 4′MdNs, 4′SdNs having various kinds of 4′-C-substituents and nucleobases were synthesized and evaluated for their biological activity.9–16) While we were working on our project, the anti-HIV activity of several 4′SdNs was reported by the Syntex group17–21) and others.22,23) Therefore, the anti-HIV activities of 4′SdNs that we studied together with those reported by other groups are listed in Table 2.
Table 2.
Anti-HIV activity of 4′-C-substituted-2′-deoxynucleosides
| Compound | EC50 (mM)a) | CC50 (mM) | S.I. |
|---|---|---|---|
| 4′-C-cyanothymidine | 0.002 | 1 | 500 |
| 4′-C-azidothymidine | 0.01 | 8 | 300 |
| 4′-C-ethynylthymidine | 0.83 | >400 | >482 |
| 4′-C-ethynylarabinofuranosylthymine | 119 | >400 | >3.4 |
| 4′-C-azidomethylthymidine | 2.1 | 333 | 159 |
| 4′-C-methylthymidine | 7.2 | 104 | 14 |
| 4′-C-ethylthymidine | >400 | 400 | ND |
| 4′-C-methoxythymidine | 8.49 | 200 | 24 |
| 4′-C-vinylthymidine | >400 | >400 | ND |
| 4′-C-hydroxymethylthymidine | 7.0 | >400 | >57 |
| 4′-C-propylthymidine | >100 | >100 | ND |
| 4′-C-cyano-2′-deoxycytidine | 0.0012 | 0.17 | 142 |
| 4′-C-azido-2′-deoxycytidine | 0.004 | 0.21 | 52 |
| 4′-C-ethynyl-2′-deoxycytidine | 0.0048 | 2.2 | 458 |
| l-4′-C-ethynyl-2′-deoxycytidine | >400 | >400 | ND |
| 4′-C-ethynyl-2′-deoxy-5-fluorocytidine | 0.030 | >100 | >3333 |
| 4′-C-ethynylarabinofuranosylcytidine | 0.043 | 2.0 | 46.5 |
| 4′-C-methyl-2′-deoxycytidine | 0.015 | 1.0 | 66.7 |
| 4′-C-fluoromethyl-2′-deoxycytidine | 0.0068 | 0.12 | 18 |
| 4′-C-methyl-2′-deoxyadenosine | 2.6 | 2.6 | 1 |
| 4′-C-azido-2′-deoxyadenosine | 0.13 | 50 | 385 |
| 4′-C-ethynyl-2′-deoxyadenosine | 0.098 | 16 | 1630 |
| 2′,3′-dideoxy-3′-thia-l-cyrtidine (3TC) | 0.10 | >100 | >1000 |
| 3′-azido-3′-deoxythymidine (AZT) | 0.0032 | 29.4 | 9190 |
a) Anti-HIV activity was determined by MTT assay. MT-4 cells and HIV-1LAI were employed.
ND: not determined.
The SARs of 4′-C-substituted nucleosides against HIV are summarized as follows:
-
1.
The estimated relative order of anti-HIV activity is as follows: CN ≥ C≡CH > N3 > CH=CH2 > Me = Et > C≡C–CH3. Interestingly, the order is the reverse of the -ΔG° values between equatorial and axial substituents on a cyclohexane ring: CN < F < C≡CH < CH=CH2 < Me ≤ Et < tBu. Thus, these results indicate that the sterically less demanding substituent at the 4′-position gives more potent anti-HIV activity.
-
2.
Purine analogs are generally less toxic than pyrimidine. Although 2′-deoxy-4′-C-ethynyl-5-fluorocytidine, which is a nucleoside derivative modified at two positions of physiologic 2′-deoxycytidine, gave a very acceptable Selectivity Index (SI = CC50/EC50) with MT-4 cells, it was toxic with other cells (Kohgo, Yamasa Corporation, private communication).
-
3.
Arabino analogs are less active and less toxic compared with their corresponding 2′-deoxy counterparts.
-
4.
4′SddNs do not show anti-HIV activity.
-
5.
The l-isomers of 4′SdN have no anti-HIV activity,14) although it is known that the l-enantiomer of 2′,3′-dideoxy-3′-thia-l-cytidine (3TC) is as active as the d-enantiomer and less toxic than the d-isomer.24) This may be due to that the l-isomers are too much modified to be recognized by RT as its substrates.
Synthesis of purine derivatives of 4′-C-cyano-2′-deoxynucleoside (4′CNdNs) and 4′-C-ethynyl-2′-deoxynucleoside (4′EdNs) and their anti-HIV activity.
The mentioned results led us to study the biological activity of purine derivatives of 4′CNdN and 4′EdN.25) Although 4′SdNs in our project had been synthesized by the glycosidation of 4-C-substituted-d-ribo-furanose derivatives and nucleobases, this synthetic route incurred some problems, as follows:
-
1.
Preparation of 4-C-substituted-d-ribo-furanoses and their conversion to the desired 4′SdNs require multistep reactions, and their total yields are low.
-
2.
4-C-Substituited-d-ribo-furanose derivatives have low reactivity in glycosidation reactions, especially when the substituent is an electron-withdrawing group such as a cyano group. (In contrast, the low reactivity of the anomeric position of 4-C-substituted furanose derivatives indicates that 4′SdN will be more stable to acidic hydrolysis than dN, ddN, and d4N will.)
These problems prompted us to develop another method of preparing purine derivatives of 4′SdN, which starts from dNs (Scheme 2 ).25)
Scheme 2.

Synthetic scheme of purine derivatives of 4′-C-cyano-2′-deoxynucleoside and 4′-C-ethynyl-2′-deoxynucleoside from 2′-deoxynucleosides. TFA = trifluoroacetic acid; DMF = dimethylformamide; rt = room temperature.
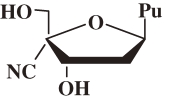
The biological activities of the purine derivatives of 4′CNdN and 4′EdN are summarized as follows (Table 3 ):
Table 3.
Anti-HIV activity of purine derivatives of 4′-C-cyano-2′-deoxy (4′CNd) and 4′-C-ethynyl-2′-deoxynucleosides
| Structure | Base | EC50 (µM)a) | CC50 (µM) | S.I. |
|---|---|---|---|---|
 |
A | 0.051 | 12 | 235 |
| I | 0.051 | 23 | 451 | |
| 2AAb) | 0.00079 | 0.034 | 43 | |
| G | 0.000188 | 0.034 | 181 | |
 |
A | 0.098 | 16 | 1630 |
| I | 0.15 | 216 | 1440 | |
| 2AA | 0.0003 | 0.82 | 2733 | |
| G | 0.0014 | 1.5 | 975 | |
| AZT | 0.0032 | 29.4 | 9190 | |
a) Anti-HIV activity was determined by MTT assay. MT-4 cells and HIV-1LAI were employed.
b) 2-aminoadenine.
-
1.
Some of the purine derivatives of 4′CNdN have high anti-HIV activity, but none of them gives an acceptable SI.
-
2.
All the purine derivatives of 4′EdN have both high anti-HIV activity and an acceptable SI.
Anti-HIV activity of 4′SdNs against drug-resistant HIV mutants.13,14,25)
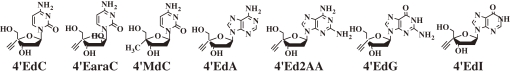
Many 4′SdNs showed very high anti-HIV activity against wild-type HIV. However, the most important point of our study is whether they are active against drug-resistant HIV-mutants. The anti-HIV activity of selected 4′SdNs against HIV mutants resistant to various NRTIs is listed in Table 4.
Table 4.
Anti-HIV activity of selected 4′-C-substituted-2′-deoxynucleoside against wild type HIV and drug-resistant HIVs
 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Compound | EC50 (µM)a) | |||||||||
| HXB2b) | KH65R | L74V | 41/215 | M184V | M184I | 41/69/ 125/SG | MDRc) | Y181C | CC50 (µM) | |
| 4′EdC | 0.0012 | 0.0008 | 0.0013 | 0.006 | 0.0024 | 0.0026 | 0.015 | 0.0012 | 0.0021 | >200 |
| 4′EaraC | 0.0071 | 0.015 | 0.026 | 0.026 | 0.71 | 0.48 | 0.17 | 0.0079 | 0.016 | >200 |
| 4′MedC | 0.0058 | 0.0071 | 0.0062 | ND | 0.2 | 0.74 | ND | 0.0033 | ND | >200 |
| 4′EdA | 0.008 | 0.0033 | 0.004 | 0.012 | 0.047 | 0.022 | 0.065 | 0.0062 | 0.011 | >200 |
| 4′Ed2AA | 0.0014 | 0.00035 | 0.0007 | 0.0017 | 0.0059 | 0.0027 | 0.0041 | 0.001 | 0.0008 | >200 |
| 4′EdG | 0.007 | 0.001 | 0.0012 | 0.019 | 0.008 | 0.0041 | 0.0068 | 0.0048 | 0.01 | 52 |
| 4′EdI | 0.81 | 0.25 | 0.61 | 1.3 | 1.6 | 1.5 | 2.2 | 0.51 | ND | >200 |
| AZT | 0.022 | 0.02 | 0.02 | 0.3 | 0.01 | 0.017 | 1.6 | 15.3 | 0.014 | >100 |
| 3TC | 0.71 | ND | ND | ND | >100 | >100 | 9.9 | 1.1 | ND | >100 |
| ddC | 0.2 | 3.0 | 1.5 | ND | 2.2 | ND | 1.3 | 5.5 | ND | >100 |
| ddI | 3.9 | 12.7 | 19.5 | 3.6 | 10.1 | ND | 12.2 | 25 | ND | >100 |
a) Anti-HIV activity was determined with MAGI assay, ND: not determined. b) wild type HIV. c) multidrug-resistant HIV.
It is noteworthy that the three cytidine derivtives maintained their activity against the drug-resistant HIV mutants, although the activity of 4′-C-ethynyl d-arabino-furanosyl cytosine (4′EaraC) and 4′MdC decreased significantly against M184V, M184I, and 41/69/125/SG. The three purine derivatives, 2′-deoxy-4′-C-ethynyladenosine (4′EdA), 2′-deoxy-4′-C-ethynyl-2-aminoadenosine (4′Ed2AA), and 2′-deoxy-4′-C-ethynylguanosine (4′EdG) except for 2′-deoxy-4′-C-ethynylinosine (4′EdI) were highly potent against all drug-resistant HIV-mutants (4′EdI was much less active than the former three derivatives, especially against M184V). Additionally, the three were also active against a non-nucleoside reverse transcriptase inhibitor-resistant Y181C. Further, the three purine derivatives were highly potent against the HIVs isolated from seven heavily drug-experienced patients with acquired immune deficiency syndrome (AIDS) as efficiently as against wild-type HIV.15,16,26) Thus, 4′EdA, 4′Ed2AA, and 4′EdG were highly potent against all the existing HIVs.
These results let us suppose that the three purine 4′EdNs could even prevent the emergence of drug-resistant HIVs. It should be noted that 4′EdG showed toxicity to Hela cells at 52 µM.
Mouse toxicity of purine 4′EdNs.
Because the three purine derivatives of 4′EdNs showed high activity against all HIVs and acceptable SIs, the mouse toxicity of these 4′EdNs was next examined (Table 5 ).25,26)
Table 5.
Toxicity of purine derivatives of 4′-C-ethynyl-2′-deoxynucleosides to micea)
| Intraveneous administration | Oral administration | |||
|---|---|---|---|---|
| Dose (mg/Kg) | Mortality (%) | Dose (mg/Kg) | Mortality (%) | |
| 4′EdA and 4′EdI | 100 | 0 | 100 | 0 |
| 10 | 0 | 10 | 0 | |
| 3 | 0 | 3 | 0 | |
| 1 | 0 | 1 | 0 | |
| 4′Ed2AA | 100 | 100 (1 day)b) | 100 | 100 (1 day) |
| 10 | 100 (2 days) | 10 | 100 (2 days) | |
| 3 | 0 | 3 | 100 (2 days) | |
| 1 | 0 | 1 | 0 | |
| 4′EdG | 100 | 100 (1 day) | 100 | 100 (1 day) |
| 10 | 100 (2 days) | 10 | 100 (4 days) | |
| 3 | 100 (4 days) | 3 | 100 (4 days) | |
| 1 | 0 | 1 | 0 | |
a) Six-week-old ICR male mice were employed.
b) Numbers in parentheses represent survival days of mice after administration.
All eight mice survived after a single dosage of 3–100 mg kg−1 of 4′EdA and 4′EdI by both intravenous and oral administrations, but all mice died after a single dosage of 3 mg kg−1 of 4′Ed2AA and 4′EdG irrespective of the administration method (Table 5). Thus, it seemed that 4′EA and 4′EI were not toxic, but 4′E2AA and 4′EdG were highly toxic.
However, in mice, it was found that 4′EdA and 4′Ed2AA were easily converted to 4′EdI and 4′EdG, respectively, by adenosine deaminase.25,26) These results showed that the actual toxicity of 4′EdA and 4′Ed2AA to animals is hard to estimate.
Anti-HIV activity of 4′EdA derivatives stable to adenosine deaminase.
The fact that both 4′EdA and 4′Ed2AA are deaminated by adenosine deaminase prompted us to prepare 4′EdA derivatives stable to the enzyme. It has been known that the adenine derivatives having a halogen atom at the 2-position of the base are stable to adenosine deaminase.27,28) Therefore, at first, 4′Ed2FA (Fig. 1) was synthesized and evaluated for anti-HIV activity28–31) and stability to both adenosine deaminase and acidic conditions.
Because 4′Ed2FA is a nucleoside derivative modified at two positions (4′-position and 2-position) of physiologic 2′-deoxyadenosine, the toxicity of 4′Ed2FA is expected to be lower than that of 4′EdA. As shown in Table 6 , 4′Ed2FA is highly potent against all HIVs including multidrug-resistant and M184V HIV mutants and has an acceptable SI (110,000).28–31)
Table 6.
Anti-HIV activity of 2′-deoxy-4′-C-ethynyl-2-fluoroadenosine (4′Ed2FA)
| HIV-1 | Compound | EC50 (nM) | CC50 (nM) | SI |
|---|---|---|---|---|
| Wild-type | 4′Ed2AA | 0.34 | 900 | 2,600 |
| 4′Ed2FA | 0.068 | 7,500 | 110,000 | |
| AZT | 3.2 | 29,400 | 9,190 | |
| M184V | 4′Ed2FA | 3.1 | 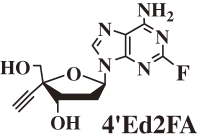 |
m.p. (decomp.) 224.4–224.6 ℃ [α]D +4.64 (c = 0.5, DMSO) |
| AZT | 16.0 | |||
| MDR | 4′Ed2FA | 0.15 | ||
| AZT | 15,300.00 | |||
EC50 = 50% effective concentration; CC50 = 50% cytotoxic concentration; SI = selectivity index = CC50/EC50
Expectedly, 4′Ed2FA was completely stable to adenosine deaminase under the conditions where 4′EdA was completely deaminated in 60 min and, further, fairly stable under acidic conditions. Thus, in 120 min only a small part (3%) of 4′Ed2FA was decomposed under the acidic conditions of gastric juices (pH 1.06) at 24 ℃, while 2′,3′-dideoxyadenosine (ddA) was completely decomposed in 5 min.
Appearance of two papers claiming that 3′-OH of 4′EdNs is the cause of the toxicity of 4′EdNs.
While we were working on our project, two papers on the role of 3′-OH of 4′SdNs appeared in which the authors claimed that 3′-OH was the cause of the toxicity of 4′SdNs. Tanaka and coworkers reported32) that 4′-C-ethynyl-2′,3′-didehydro-3′-deoxythymidine (4′Ed4T) was more potent against wild-type HIV and multi-drug resistant HIV mutant but a little less potent against M184V-mutant and much less toxic than d4T. Further, they claimed that 3′-OH of 4′SdNs was the cause of the toxicity.
In contrast, Marquez and coworkers reported33) that 3′-OH of 4′SdNs played an important role in the phosphorylation of 5′-OH by cellular kinases, but was the cause of the toxicity of 4′SdNs. This determination was based on their results that 4′-C-ethynyl-2′,3′-dideoxycytidine (4′EddC) was inactive against HIV in cellular systems, but its 5-O-triphosphate (4′EddCTP) was more active than 3′-azido-3′-deoxythymidine-5′-O-phosphate (AZTTP) against the RT of wild-type HIV. They also reported that 4′EddCTP was much less active against RT of the M184V mutant than against the RT of wild-type HIV. In addition, the l-isomer of 4′EddCTP was not active against the RT of the M184V mutant.
Anti-HIV activity of 2′,3′-dideoxy (dd-) and 2′,3′-didehydrodideoxy (d4) analogs of 4′Ed2FA.
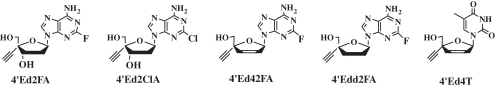
The two papers previously cited caused us to synthesize the dd- and d4-analogs of 4′Ed2FA and evaluate their anti-HIV activity.31) The anti-HIV activities of 2′,3′-dideoxy-4′-C-ethynyl-2-fluoroadenosine (4′Edd2FA) and 2′,3′-didehydrodideoxy-4′-C-ethynyl-2-fluoroadenosine (4′Ed42FA) together with that of 2′-deoxy-4′-C-ethynyl-2-chloroadenosine (4′Ed2ClA) are listed in Table 7.
Table 7.
Anti-HIV activity of 4′-C-substituted-2′-deoxy-2-haloadenosines
 | ||||
|---|---|---|---|---|
| Compound | Anti-HIV activity (MAGI assay, µg) | |||
| HIV-1wild | HIV-1MDR | HIV-1M184V | SI | |
| 4′Ed2FA | 0.00020 | 0.00014 | 110,000 | |
| 4′Ed2ClA | 0.0019 | 0.0084 | 0.01 | 330,000 |
| 4′Ed42FA | 0.80 | 0.15 | 1.8 | |
| 4′Edd2FA | 0.94 | 8.7 | 97 | |
| AZT | 0.17 | 74.3 | 0.13 | |
| 3TC | 1.0 | 2.8 | >100 | |
| 4′Ed4T | 1.5 | 1.1 | 17 | >50,000 |
| d4T | 7.6 | 64 | 5.6 | |
MAGI = multinuclear activation of galactosidase indicator; HIV = human immunodeficiency virus; AZT = 3′-azido-3′-deoxythymidine; 3TC = 2′,3′-dideoxy-3′-thia-l-cytidime, d4T = 2′,3′-didehydrodideoxythymidine.
Although both 4′Ed42FA and 4′Edd2FA showed some activity against wild-type HIV, they significantly lost any activity against drug-resistant HIVs. 4′Ed2ClA is highly active against all HIVs, however, its activity is lower than that of 4′Ed2FA. These results indicated that the 3′-OH played important roles not only for the phosphorylation of 5′-OH, but also for the activity against drug-resistant HIVs. Further, these results together with those reported by Tanaka and coworkers32) indicated that the 4′-C-ethynyl group played an important role for the anti-HIV activity. Recently, it was reported that the active center of the RT of HIVs has a narrow lipophylic cavity to accept preferably the ethynyl group, thus 4′-C-ethynyl nucleoside derivatives have potent anti-HIV activity.34)
Toxicity of 4′Ed2FA to mice and inhibition of DNA polymerases.
Because 4′Ed2FA is stable to adenosine deaminase, its mouse toxicity was examined.29–31,33,35) 4′Ed2FA did not show any acute toxicity to mice by either oral or intravenous administration up to 100 mg kg−1 (Fig. 4 ; Table 8 ).
Figure 4.

Body weight change of mice after a single dosage of 2′-deoxy-4′-C-ethynyl-2-fluoroadenosin, administrated orally or intravenously to ICR mice.
Table 8.
Toxicity of 2′-deoxy-4′-C-ethynyl-2-fluoroadenosine (4′Ed2FA) after a single dosage to ICR mice
| Dose (mg Kg−1) | Survivors/total | |
|---|---|---|
| p.o | i.v. | |
| Placebo | 8/8 | 8/8 |
| 1 | 8/8 | 8/8 |
| 3 | 8/8 | 8/8 |
| 10 | 8/8 | 8/8 |
| 30 | 8/8 | 8/8 |
| 100 | 8/8 | 8/8 |
It is known that the toxicity of NRTIs to animals is caused by their inhibition of mitochondrial DNA polymerase γ. The 50% effective concentration (EC50) of 2′-deoxy-4′-C-ethynyl-2-fluoroadenosine-5-O-triphosphate (4′Ed2FATP) to inhibit the incorporation of 2′-deoxyadenosine-5-O-triphosphate (dATP) mediated by human mitochondrion DNA polymerase was 10 µM, which was significantly higher than the 0.2 µM of 2′,3′-dideoxyadenosine-5-O-triphosphate (ddATP).30,31,35) The EC50 values of 4′Ed2FATP against DNA polymerase α and β were higher than 200 µM. These results indicate that the DNA polymerases scarcely recognize 4′Ed2FATP, a derivative modified at two positions of physiologic dATP, as their substrate but that RT does.
Intracellular metabolism of 4′Ed2FA.29)
The amounts of all fractions of intracellular 4′Ed2FA metabolites, (4′Ed2FA-monophosphate, 4′Ed2FA-diphosphate, and 4′Ed2FATP) increased proportionately with an increase in the concentration of intracellular 4′Ed2FA, while compared to AZT diphosphate and AZTTP, only AZT monophosphate markedly increased with an increase in intracellular AZT concentration. The intracellular half-life (T1/2) of 4′Ed2FATP was ∼18 h in complete expansion media (CEM) cells, MT4 cells, and multinuclear activation of galactosidase indicator (MAGI)-CCR5 cells (T1/2 of AZTTP was 3 h). About 50% of the cells were protected against the infection of HIV for 24 h after removal of extracellular 4′Ed2FA in both MT4 cells and MAGI cells cultured in the presence of 0.1 µM of 4′Ed2FA.30,31,36) These results indicate that 4′Ed2FA, 2′-deoxy-4′-C-ethynyl-2-fluoroadenosine-5-O-diphosphate (4′Ed2FADP), and 4′Ed2FATP are very stable against intracellular enzymatic catabolism.
Summary
A study of the synthesis and biological evaluation of 4′SdNs was conducted according to the proposed working hypotheses based on the fundamentals of both organic chemistry and biochemistry. All scientific evidences proved the validity of the working hypotheses and resulted in the development of 4′Ed2FA, which is highly potent against all HIVs, is stable to intracellular enzymatic catabolism and acidic degradation, has a very long intracellular T1/2, does not greatly inhibit DNA polymerase γ, and does not have acute mouse toxicity.
These results strongly suggest that 4′Ed2FA deserves further study for the development of a highly potent therapeutic agent for HIV infection (AIDS), and which may solve the problems of the existing HAART.
It should be noted that 4′Ed2ClA is also highly potent against all HIVs, though it is a little less potent than 4′Ed2FA, and has better SI than 4′Ed2FA, Therefore, 4′Ed2ClA deserves also for further study.
The above study together with the recent studies by others37,38) on the development of anti-HCV active and/or anti-tumor active modified nucleosides which are chain-terminators of RNA polymerase, prompted the author to propose the structure of modified nucleosides expected to have high antiviral activity and low toxicity.39)
Acknowlegement
The work described in this paper was achieved through the tremendous efforts of the many coworkers whose names appear in the cited references.
Profile
Hiroshi Ohrui was born in 1942. He received his B.S. (1965) and Ph.D. degrees (1971) from The University of Tokyo under the direction of Professor Masanao Matsui. He joined the Institute for Physical and Chemical Research (RIKEN) in 1966 as a researcher. In 1981, he moved to Tohoku University as Associate Professor in the Department of Food Chemistry, Faculty of Agriculture. In 1997, he was promoted to Full Professor, Graduate School of Life Science. In 2006, he moved to Yokohama College of Pharmacy as Professor. He worked for Dr. J. J. Fox at Sloan-Kettering Institute for Cancer Research (1972–1973) as Research Associate, and for Dr. J. G. Moffatt at Syntex Research (1973–1974) as a postdoctoral fellow. He has received several awards including The Agricultural Society of Japan Award for Young Scientists (1974), Inoue Prize for Science (2001), Japan Prize of Agricultural Sciences (2004), The Japan Society for Analytical Chemistry Award (2004), and the Japan Academy Prize (2010). His research interests cover organic synthesis, chiral discrimination, and medicinal chemistry.
References
- 1).Mitsuya H., Weinhold K., Furman P., St Clair M., Lehrman S., Gallo R., Bolognesi D., Barry D., Broder S. (1985) 3′-Azido-3′-deoxythymidine (BW A509U): An antiviral agent that inhibits the infectivity and cytopathic effect of human T-lymphotropic virus type III/lymphadenopathy-associated virus in vitro. Proc. Natl. Acad. Sci. USA 82, 7096–7100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2).Murphy E.L., Collier A.C., Kalish L.A., Mintz L., Wallach F.R., Nemo G.J. (2001) Highly active antiretroviral therapy decreases mortality and morbidity in patients with advanced HIV disease. Ann. Intern. Med. 135, 17–26 [DOI] [PubMed] [Google Scholar]
- 3).Hung C.-C., Hsiao C.-F., Chen M.-Y., Hsieh S.-M., Chang S.-Y., Sheng W.-H., Sun H.-Y., Chang S.-C. (2006) Improved survival of persons with human immunodeficiency virus type 1 infection in the era of highly active antiretroviral therapy in Taiwan. Jpn. J. Infect. Dis. 59, 222–228 [PubMed] [Google Scholar]
- 4).Shuhadonlnik, R.J. (1970) Nucleoside Antibiotics. Wiley-International, New York. [Google Scholar]
- 5).Waga T., Nishizaki T., Miyakawa I., Ohrui H., Meguro H. (1993) Synthesis of 4′-C-methylnucleosides. Biosci. Biotechnol. Biochem. 57, 1433–1438 [DOI] [PubMed] [Google Scholar]
- 6).Waga T., Ohrui H., Meguro H. (1996) Synthesis and biological evaluation of 4′-C-methyl nucleoside. Nucleosides Nucleotides 15, 287–304 [Google Scholar]
- 7).Yamaguchi T., Tomikawa A., Hirai T., Kawaguchi T., Ohrui H., Saneyoshi M. (1997) Antileukemic activities and mechanism of action of 2′-deoxy-4′-methylcytidine and related nucleosides. Nucleosides Nucleotides 16, 1347–1350 [Google Scholar]
- 8).Youssefyeh R.D., Verheyden J.P.H., Moffatt J.G. (1979) Synthesis of 4′-C-hydroxymethyl ribofuranosyl nucleosides. J. Org. Chem. 44, 1301–1321 [Google Scholar]
- 9).Kitano K., Miura S., Ohrui H., Meguro H. (1997) Synthesis of 4′-C-fluoromethylnucleosides as potential antineoplastic agents. Tetrahedron 53, 13315–13322 [Google Scholar]
- 10).Kitano K., Machida H., Miura S., Ohrui H. (1999) Synthesis of novel 4′-C-methyl-pyrimidine nucleosides and their biological activities. Bioorg. Med. Chem. Lett. 9, 827–830 [DOI] [PubMed] [Google Scholar]
- 11).Kohgo S., Horie H., Ohrui H. (1999) Synthesis of 4′-C-ethynyl-β-d-arabino- and 4′-C-ethynyl-2′-deoxy-β-d-ribo-pentofuranosyl pyrimidines, and their biological evaluation. Biosci. Biotechnol. Biochem. 63, 1146–1149 [DOI] [PubMed] [Google Scholar]
- 12).Kohgo S., Kodama E., Shigeta S., Saneyoshi M., Machida H., Ohrui H. (1999) Synthesis of 4′-substituted nucleosides and their biological evaluation. Nucleic Acids Symp. Ser. 42, 127–128 [DOI] [PubMed] [Google Scholar]
- 13).Ohrui H., Kohgo S., Kitano K., Sakata S., Kodama E., Yoshimura K., Matsuoka M., Shigeta S., Mitsuya H. (2000) Synthesis of 4′-C-ethynyl-β-d-arabino- and 4′-C-ethynyl-2′-deoxy-β-d-ribo-pentofuranosyl pyrimidines and -purines and evaluation of their anti-HIV activity. J. Med. Chem. 44, 4516–4625 [DOI] [PubMed] [Google Scholar]
- 14).Kohgo S., Mitsuya H., Ohrui H. (2001) Synthesis of the l-enantiomer of 4′-C-ethynyl-2′-deoxycytidine. Biosci. Biotechnol. Biochem. 65, 1879–1882 [DOI] [PubMed] [Google Scholar]
- 15).Kodama E., Kohgo S., Kitano K., Machida H., Gatanaga H., Shigeta S., Matsuoka M., Ohrui H., Mitsuya H. (2001) 4′-Ethynyl nucleoside analogs: potent inhibitor of multidrug-resistant human immunodeficiency virus variants in vitro. Antimicrob. Agents Chemother. 45, 1539–1546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16).Ohrui H., Mitsuya H. (2001) 4′-C-Substituted-2′-deoxynucleosides: A family of antiretroviral agents which are potent against drug-resistant HIV variants. Curr. Drug Targets Infect. Disord. 1, 1–10 [DOI] [PubMed] [Google Scholar]
- 17).Maag H., Rydzewski R.M., McRoberts M.J., Crawford-Ruth D., Verheyden J.P.H., Prisbe E.J. (1992) Synthesis and anti-HIV activity of 4′-azido- and 4′-methoxynucleosides. J. Med. Chem. 35, 1440–1451 [DOI] [PubMed] [Google Scholar]
- 18).O-Yang C., Wu H.Y., Fraser-Smith E.B., Walker K.A.M. (1992) Synthesis of 4′-cyanothymidine and analogues as potent inhibitor of HIV. Tetrahedron Lett. 33, 37–40 [Google Scholar]
- 19).O-Yang C., Kurz W., Eugui E.M., McRoberts M.J., Verheyden J.P.H., Kurz L.J., Walker K.A.M. (1992) 4′-Substituted nucleosides as inhibitors of HIV: an unusual oxetane derivative. Tetrahedron Lett. 33, 41–43 [Google Scholar]
- 20).Prisbe, F.J., Verheyden, J.P.H. and Rydzewski, R.M. (1993) 4′-Substituted 2′-deoxy nucleosides as inhibitors of HIV. Nucleosides & Nucleotides as Antitumor & Antiviral Agents (eds. Chu, C.K. and Baker, D.C.). Plenum Press, New York, pp. 101–108. [Google Scholar]
- 21).Chen M.S., Suttmann R.T., Papp E., Cannon P.D., McRoberts M., Baxh C., Copeland W.C., Wang T.S. (1993) Selective action of 4′-azidothymidine triphosphate on reverse transcriptase of human immunodeficiency virus type 1 and human DNA polymerase alpha and beta. Biochemistry 32, 6002–6010 [DOI] [PubMed] [Google Scholar]
- 22).Wang G., Seifert W.E. (1996) Synthesis and evaluation of oligodeoxynucleotides containing 4′-C-substituted thymidines. Tetrahedron Lett. 37, 6515–6518 [Google Scholar]
- 23).Nomura M., Shuto S., Tanaka M., Sasaki T., Shigeta S., Matsuda A. (1999) Nucleosides and nucleotides. 185. Synthesis and biological activities of 4′-α-C-branched-chain sugar pyrimidine nucleosides. J. Med. Chem. 42, 2901–2908 [DOI] [PubMed] [Google Scholar]
- 24).Beach J.W., Jeong L.S.A., Alves J., Pohl D., Kim H.O., Chang C.-N., Doong S.-L., Schinazi R.F., Cheng Y.-C., Chu C.K. (1992) Synthesis of enantiomerically pure (2′R,5′S)-(−)-1-(2-hydroxymethyloxathiolan-5-yl)cytosine as a potent antiviral agent against hepatitis B virus (HBV) and human immunodeficiency virus (HIV). J. Org. Chem. 57, 2217–2219 [Google Scholar]
- 25).Kohgo S., Yamada K., Kitano K., Iwai Y., Sakata S., Ashida N., Hayakawa H., Nameki D., Kodama E., Matsuoka M., Mitsuya H., Ohrui H. (2004) Design, efficient synthesis, and anti-HIV activity of 4′-C-cyano- and 4′-C-ethynyl-2′-deoxy purine nucleosides. Nucleosides Nucleotides Nucleic Acids 23, 671–690 [DOI] [PubMed] [Google Scholar]
- 26).Hayakawa H., Kohgo S., Kitano K., Ashida N., Kodama E., Mitsuya H., Ohrui H. (2004) Potential of 4′-C-substituted nucleosides for the treatment of HIV. Antivir. Chem. Chemother. 15, 169–187 [DOI] [PubMed] [Google Scholar]
- 27).Montgomery J.A., Hewson K. (1969) Nucleosides of 2-fluoroadenine. J. Med. Chem. 12, 498–504 [DOI] [PubMed] [Google Scholar]
- 28).Brockman R.W., Schabel F.M., Jr., Montgomery J.A. (1977) Biologic activity of 9-β-d-arabinofuranosyl-2-fluoroadenine, a metabolically stable analog of 9-β-d-arabinofuranosyladenine. Biochem. Pharmacol. 26, 2193–2196 [DOI] [PubMed] [Google Scholar]
- 29).Patent: International Publication Number, 2005/090349, PCT/JP2005/005374.
- 30).Nakata H., Amano M., Koh Y., Kodama E., Yang G., Bailey C.M., Kohgo S., Hayakawa H., Matsuoka M., Anderson K.S., Cheng Y.C., Mitsuya H. (2007) Activity against human immunodeficiency virus type 1, intracellular metabolism, and effects on human DNA polymerases of 4′-ethynyl-2-fluoro-2′-deoxyadenosine. Antimicrob. Agents Chemother. 51, 2701–2708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31).Kawamoto A., Kodama E., Sarafianos S.G., Sakagami Y., Kohgo S., Kitano K., Ashida N., Iwai Y., Hayakawa H., Nakata H., Mitsuya H., Arnol E., Matsuoka M. (2008) 2′-Deoxy-4′-C-ethynyl-2-halo-adenosines active against drug-resistant human immunodeficiency virus type 1 variants. Int. J. Biochem. Cell Biol. 40, 2410–2420 [DOI] [PubMed] [Google Scholar]
- 32).Haraguchi K., Takeda S., Tanaka H., Nitanda T., Baba M., Dutschman G.E., Cheng Y.-C. (2003) Synthesis of a highly active new anti-HIV agent 2′,3′-didehydro-3′-deoxy-4′-ethynylthymidine. Bioorg. Med. Chem. Lett. 13, 3775–3777 [DOI] [PubMed] [Google Scholar]
- 33).Siddiqui M.A., Hughes S.H., Boyer P.L., Mitsuya H., Van Q.N., George C., Saranfinanos S.G., Marquez V.E.A. (2004) 4′-C-Ethynyl-2′,3′-dideoxynucleoside analogue, highlights the role of the 3′-OH in anti-HIV active 4′-C-ethynyl-2′-deoxy nucleosides. J. Med. Chem. 47, 5041–5048 [DOI] [PubMed] [Google Scholar]
- 34).Boojamra C.G., Parrish J.P., Sperandio D., Gao Y., Petrakovsky O.V., Lee S.K., Markevitch D.Y., Vela J.E., Laflamme G., Chen J.M., Ray A.S., Barron A.C., Sparacino M.L., Desai M.C., Kim C.U., Cihlar T., Mackman R.L. (2009) Design, synthesis, and anti-HIV activity of 4′-modified carbocyclic nucleoside phosphonate reverse transcriptase inhibitors. Bioorg. Med. Chem. 17, 1739–1746 [DOI] [PubMed] [Google Scholar]
- 35).Ohrui, H., Kohgo, S., Kitano, K., Hayakawa, H., Kodama, E., Matsuoka, M. and Mitsuya, H. (2005) 4′-C-Ethynyl-2′-deoxy-2-fluoroadenosine, a nucleoside derivative potent against HIV-1 with no acute mouse toxicity: Highlights of the role of 3′-OH for biological activity. Abstract of the 18th International Conference on Antiviral Research. Barcelona, April 10–14, p. 16. [Google Scholar]
- 36).Ohrui H. (2006) 2′-Deoxy-4′-C-ethynyl-2-fluoroadenosine, a nucleoside reverse transcriptase inhibitor, is highly potent against all human immunodeficiency viruses type 1 and has low toxicity. Chem. Rec. 6, 133–143 [DOI] [PubMed] [Google Scholar]
- 37).Eldrup A.B., Prhavc M., Brooks J., Bhat B., Prakash T.P., Song Q., Bera S., Bhat N., Dande P., Cook P.D., Bennet C.F., Carroll S.S., Ball R.G., Bosserman M., Burlein C., Colwell L.F., Fay J.F., Flores O.A., Getty K., LaFemina R.L., Leone J., MacCoss M., McMaster D.R., Tomassini J.E., Langen D.V., Wolanski B., Olsen D.B. (2004) Structure-activity relationship of heterobase-modified 2′-C-methyl ribonucleosides as inhibitors of hepatitis C virus RNA replication. J. Med. Chem. 47, 5284–5297 [DOI] [PubMed] [Google Scholar]
- 38).Hattori H., Tanaka M., Fukushima M., Sasaki T., Matsuda A. (1996) (3-C-Ethynyl-β-d-ribo-pentofuranosyl)-cytosine, 1-(3-C-Ethynyl-β-d-ribo-pento-furanosyl)uracil, and their nucleobase analogs as new potential multifunctional antitumor nucleosides with a broad spectrum of activity. J. Med. Chem. 39, 5005–5011 [DOI] [PubMed] [Google Scholar]
- 39).Ohrui H. (2009) A proposed structure of modified nucleosides expected to have high antiviral activity and low toxicity. Nucleosides Nucleotides Nucleic Acids 28, 772–775 [DOI] [PubMed] [Google Scholar]