

Abstract
An initial response of Staphylococcus aureus to encounter with cell wall-active antibiotics occurs by transmembrane signaling systems that orchestrate changes in gene expression to promote survival. Histidine kinase two-component sensor-response regulators such as VraRS contribute to this response. In this study, we examined VraS membrane sensor phosphotransfer signal transduction and explored the genetic consequences of disrupting signaling by engineering a site-specific vraS chromosomal mutation. We have used in vitro autophosphorylation assay with purified VraS[64-347] lacking its transmembrane anchor region and tested site-specific kinase domain histidine mutants. We identified VraS H156 as the probable site of autophosphorylation and show phosphotransfer in vitro using purified VraR. Genetic studies show that the vraS(H156A) mutation in three strain backgrounds (ISP794, Newman, and COL) fails to generate detectable first-step reduced susceptibility teicoplanin mutants and severely reduces first-step vancomycin mutants. The emergence of low-level glycopeptide resistance in strain ISP794, derived from strain 8325 (ΔrsbU), did not require a functional σB, but rsbU restoration could enhance the emergence frequency supporting a role for this alternative sigma factor in promoting glycopeptide resistance. Transcriptional analysis of vraS(H156A) strains revealed a pronounced reduction but not complete abrogation of the vraRS operon after exposure to cell wall-active antibiotics, suggesting that additional factors independent of VraS-driven phosphotransfer, or σB, exist for this promoter. Collectively, our results reveal important details of the VraRS signaling system and predict that pharmacologic blockade of the VraS sensor kinase will have profound effects on blocking emergence of cell wall-active antibiotic resistance in S. aureus.
Staphylococcus aureus is a major human pathogen that causes a variety of diseases ranging from relatively minor skin infections to invasive and systemic disease with significant morbidity and mortality (17, 44). Of particular concern are S. aureus strains carrying one of several allotypes of a mobile genetic element, the SCCmec cassette, which encodes mecA, and a low-affinity penicillin-binding protein variant, PBP2A (12). The expression of PBP2A renders S. aureus insensitive to a broad range of β-lactams, including methicillin, hence the name MRSA (for methicillin-resistant S. aureus). Various MRSA strains have become endemic in hospitals and in the community prompting worldwide efforts for detection and infection control.
Glycopeptide antibiotics (teicoplanin and vancomycin) are considered first-line drugs for the treatment of infections due to MRSA. The emergence of resistance (most often termed reduced sensitivity) to glycopeptides poses a major challenge to the treatment of MRSA infections since few clinically proven and effective alternative therapies exist (29).
Altered sensitivity to glycopeptides occurs by two mechanisms termed exogenous and endogenous. The exogenous mechanism (VanA type) conferring high-level resistance (vancomycin MIC ≥ 16 μg/ml) occurs by the horizontal acquisition of the vanA multigene complex from Enterococcus faecalis encoded on Tn1546 and results in the alteration of peptidoglycan terminal stem peptide from d-Ala-d-Ala to d-Ala-d-Lac, a structure to which glycopeptides no longer bind efficiently and therefore fail to block transglycosylase and transpeptidase cell wall cross-linking (47, 57). Worldwide, only a few examples of VRSA have been reported (55).
In contrast, endogenous resistance to glycopeptides is much more prevalent. S. aureus displaying intermediate glycopeptide resistance (termed VISA if referring to vancomycin-intermediate S. aureus and GISA for glycopeptide-intermediate S. aureus encompassing both vancomycin and teicoplanin) are thought to arise stepwise from so-called heterogenous (hVISA and hGISA) precursor populations through selection of mutation(s) during the course of exposure to glycopeptides (28, 29, 43, 47). Rare subpopulations of bacteria displaying higher levels of resistance presumably serve as a reservoir driving the eventual emergence of glycopeptide resistance. Subpopulations of this type are difficult to detect, and no routine clinical laboratory tests exist that are standardized and reliable for their detection (29, 61).
The MIC breakpoint defining the transition from sensitive to intermediate resistant for glycopeptide intermediate-resistant S. aureus (GISA and VISA) is not universally agreed upon; however, relatively minor alterations in reduced sensitivity to glycopeptides (minor changes in MIC) are now frequently associated with clinical failure, requiring recourse to alternative pharmacotherapy (29).
The genetic basis of endogenous glycopeptide resistance is poorly understood. Mutation in genes such as tcaA, graRS, and vraRS have been described and are known to be causal or strongly correlated to the emergence of VISA and GISA (15, 30, 45, 48, 53). In some, but not all cases, morphological changes associated with the emergence of glycopeptide resistance include a thickened cell wall, reduced cross-linking, and decreased autolytic activity, suggesting that complex alterations in cell wall biosynthesis and turnover underlie the resistance mechanism (27, 47, 57).
Transcriptomic studies demonstrated that an encounter with cell wall-active antibiotics elicits a cell wall stress response in S. aureus (22, 40, 46, 49, 51, 73). The precise mechanisms that are responsible for the detection of cell wall damage are also poorly understood, and there are known significant interstrain variations (49).
In several studies, the transcriptional induction of the vraRS two-component sensor (TCS) system, which is part of a four-gene operon, is significantly induced after encounter with cell wall-active drugs such as oxacillin, vancomycin, teicoplanin, and d-cycloserine (22, 40, 49, 69, 77). The vraRS operon is also strongly induced after reduced transcription of pbp2, which encodes the bifunctional penicillin-binding protein PBP2 (22). The VraRS TCS is highly conserved in the low-percent G+C Gram-positive family Firmicutes. In S. aureus, the VraRS TCS is thought to regulate numerous genes, some necessary for cell wall biosynthesis and proteolytic quality control (40). At least two other S. aureus TCS systems—WalKR (YycFG) and GraRS—have been implicated in modulating resistance to cell wall-active antibiotics (16, 18, 30, 53, 63).
TCS systems are widespread in bacteria and represent environmental sensing systems that integrate a broad range of input stimuli to effector proteins, often including transcription factors (20, 21). A typical TCS system is composed of a membrane sensor histidine kinase and a cognate response regulator. Environmental signal captured by the receptor kinase results first in histidine autophosphorylation. In a second step, phosphotransfer from the histidine kinase to a conserved aspartate in the receiver domain of the cognate response regulator ultimately culminates in alterations in downstream gene expression, or altered enzymatic activity, appropriate to the applied stimulus.
In the present study, we dissected the VraRS phosphotransfer sensing mechanism. We identified the key elements of VraS-VraR phosphotransfer in vitro and examined genetic consequences in vivo by using a site-specifically engineered vraS autophosphorylation-defective chromosomal point mutation. Our results reveal a crucial role for VraS-VraR signaling in mediating the emergence of endogenous glycopeptide resistance.
MATERIALS AND METHODS
Bacterial strains and plasmids.
The strains and plasmids used in the present study are listed in Table 1. Antibiotics and their suppliers were as follows: teicoplanin (Sanofi Aventis), vancomycin (Sandoz), and oxacillin, d-cycloserine, and carbenicillin (Sigma).
TABLE 1.
Bacterial strains and plasmids used in this study
| Strain or plasmid | Relevant genotype or characteristicsa | Source or reference(s) |
|---|---|---|
| Strains | ||
| E. coli | ||
| ER2566 | IPTG-inducible T7 RNA polymerase restriction-deficient DNA cloning strain | New England Biolabs |
| DH5α | Gibco-BRL | |
| S. aureus | ||
| COL | MRSA, rsbU+ | 24 |
| RN4220 | 8325-4, r− m+, restriction-defective laboratory strain | 38 |
| Newman | ATCC 25904, MSSA, rsbU+ | 1 |
| MB211 | 8325, (rsbUVW-sigB)+, tetL nearby | 23 |
| MB212 | 8325, ΔrsbUVW-sigB::ermB | 39 |
| ISP794 | 8325, pig-131 rsbU mutant | 60, 67 |
| ALC1001 | RN6390 sigB::Tn551 | 13 |
| ALC2057 | RN6390 sarA::kan | 14 |
| AR850 | ISP794, ΔrsbUVW-sigB::ermB | This study |
| AR851 | ISP794, sigB::Tn551 | This study |
| AR852 | ISP794, (rsbUVW-sigB)+, tetL nearby | This study |
| AR756 | ISP794, vraSΔ2-160::ermB | This study |
| AR769 | ISP794, vraS(H156A), Kanr nearby | This study |
| AR758 | ISP794, Kanr within intergenic region SA1699-vraR | This study |
| AR943 | ISP794, tetK within intergenic region SA1699-vraR | This study |
| AR828 | ISP794, vraS(H156A), Kanr nearby, Φ11 transduction from AR769 | This study |
| AR878 | ISP794, vraS(H156A), Kanr nearby + pAM1483 | This study |
| AR872 | ISP794, vraS(H156A), Kanr nearby + pAM902 | This study |
| AR848 | Newman, vraS(H156A), Kanr nearby, Φ11 transduction from AR769 | This study |
| AR868 | COL, vraS(H156A), Kanr nearby, Φ80α transduction from AR769 | This study |
| Plasmids | ||
| pCL84 | tetK; S. aureus geh locus integrating plasmid | 42 |
| pUC18 | Multicopy E. coli cloning vector | 76 |
| pUC19-MuSupF | Ampr mini-Mu transposon | 26 |
| pEC2 | ermB cassette | 11 |
| pMK4 | E. coli-S. aureus shuttle vector; Ampr Camr | 70 |
| pBT2 | E. coli-S. aureus thermosensitive shuttle vector; Ampr Camr | 11 |
| pTYB12 | N-terminal fusion Impact intein and chitin-binding domain plasmid | New England Biolabs |
| pKSII+Bluescript | Routine multicopy E. coli cloning vector | Stratagene |
| pAM1118 | pKS+II VraScyt (NdeI-PstI) | This study |
| pAM1158 | pKS+II VraR (NdeI-PstI) | This study |
| pEG1129 | pTYB12-VraScyt (NdeI-PstI) | This study |
| pEG1 | pTYB12-VraScyt H156A | This study |
| pEG2 | pTYB12-VraScyt H238A | This study |
| pEG1055 | pTYB12-VraR (Nde-Pst) | This study |
| pEG3 | pTYB12-VraR-D55A | This study |
| pAR712 | pBT2, vraS-Kanr-SA1699 intergenic ts shuttle vector | This study |
| pAR907 | pBT2, vraS-tetK-SA1699 intergenic ts shuttle vector | This study |
| pAM1284 | pBT2, vraSΔ[2-160]::ermB ts shuttle vector | This study |
| pAR747 | pBT2, vraS(H156A), Kanr nearby ts shuttle vector | This study |
| pAM1483 | pMK4, 3.3-kb entire vraR operon and upstream promoter region, KpnI-PstI | This study |
| pAM1246 | pKS+II, 3.3-kb entire vraR operon and upstream promoter region, KpnI-PstI | This study |
| pAM902 | pMK4, pGlyS gfpuv4 | 72 |
Camr, chloramphenicol resistance; Ampr, ampicillin resistance; Kanr, kanamycin resistance.
Cloning and purification of recombinant VraR and VraS proteins.
The open reading frame of the vraR gene (SA1700 using the N315 ordered sequence tag numbering [41]) and the nucleotide region encoding the cytoplasmic domain of the vraS gene (corresponding to amino acids 65 to 347), hereafter referred to as VraScyt, were amplified by the PCR using S. aureus genomic DNA and primers indicated in Table 2. Fragments of 884 bp (vraS) and 650 bp (vraR) were cleaved with KpnI and PstI and cloned into the KpnI and PstI sites of pKSII+Bluescript, respectively. After sequence verification, the fragments were excised with NdeI-PstI and cloned into the Escherichia coli expression vector pTYB12 (New England Biolabs). Recombinant VraR and VraScyt proteins were then purified by using an N-terminal chitin affinity tag (Impact System; New England BioLabs). E. coli strain ER2566 containing pTYB12-VraR and pTYB12-VraScyt was grown in Luria-Bertani medium containing carbenicillin at 100 μg/ml until reaching an optical density at 600 nm of 0.4, induced with 0.5 mM IPTG (isopropyl-β-d-thiogalactopyranoside), and incubated for an additional 6 h at 30°C with vigorous shaking. Bacteria were harvested by low-speed centrifugation and resuspended in buffer containing 0.1% Tween 20, followed by affinity chromatography and intein-mediated proteolytic cleavage of the affinity tag with 50 mM dithiothreitol (DTT) according to the manufacturer's recommendations. Thiol-induced intein cleavage resulted in the N-terminal attachment of three additional amino acids (Ala-Gly-His) for VraR and four amino acids (Ala-Gly-His-Met) for VraScyt upstream of Ser65. Cleaved protein was eluted from the chitin beads, and the supernatant was concentrated and diafiltered using Centricon-10 spin columns. Protein concentrations assuming monomer molecular weights predicted from the amino acid composition were determined using Bradford reagent (Bio-Rad) and a bovine serum albumin standard. Recombinant proteins were stored at 4°C and were stable for at least 6 months.
TABLE 2.
Oligonucleotide PCR primers used in this study
| Gene, mutant, protein, or method | Primer or probe | Primer sequence (5′-3′)a |
|---|---|---|
| VraR wild-type protein | VraR-Kpn-Nde | GGGGTACCAAGGAGGAACACATATGACGATTAAAGTATTG |
| VraR-Pst | CAAACTGCAGCTATTGAATTAAATTATGTTGGAATGC | |
| VraR-D55A protein | VraR D55A-A | GATTTAATTTTAATGGCTTTACTTATGGAAG |
| VraR D55A-B | CTTCCATAAGTAAAGCCATTAAAATTAAATC | |
| VraScyt wild-type protein | VraScyt-KpnNde PCR | GGGGTACCAAGGAGGAACACATATGGGTTCGGTACTCGCATACAAAATC |
| VraScyt-Pst PCR | CAAACTGCAGTTAATCGTCATACGAATCCTC | |
| VraS-H156A protein | VraS H156A-A | GCTCGAGAACTTGCCGATTCTGTTAGTC |
| VraS H156A-B | GACTAACAGAATCGGCAAGTTCTCGAGC | |
| VraS-H238A protein | VraS H238A-A | ATGAAAGTTGTGGCTGAAATACAAGATTTTAAAG |
| VraS H238A-B | CTTTAAAATCTTGTATTTCAGCCACAACTTTCAT | |
| VraSΔ[2-160]::ermB | SA1703 Kpn-Bam | GGGGTACCGGATCCATGAACTATGTTGAACGTTATATTGAACAG |
| Primer 2 Bam | CGGGATCCGTTCATCGATAAATCACCTCTACG | |
| Primer 3 Bam | CGGGATCCCAGCAACTTTTTGCGGCAAGTATGA | |
| SA1699 staEco-Pst | CGGAATTCCTGCAGATGTCGAAAAATCACTCTTCTTCAAAATACC | |
| VraS-H156A chromosomal | SA1703KpnBamHI | GGGGTACCGGATCCATGAACTATGTTGAACGTTATATTGAACAG |
| mutant | SA1699EcoPstI | CGGAATTCCTGCAGATGTCGAAAAAGGATCACTCTTCTTCAAAATACC |
| Kan marked vraR-SA1699 | 3BKpnBamHI | GGGGTACCGGATCCCAGCAACTTTTTGCGGCAAGTATGATGC |
| intergenic | 12ABgl2 | GAAGATCTCGTAAGTAACTTTTCTTAATTCGATACG |
| 12BBgl2 | GAAGATCTCCAATCACAATATAACATCAAATAGACAC | |
| SA1699EcoPstI | CGGAATTCCTGCAGATGTCGAAAAAGGATCACTCTTCTTCAAAATACC | |
| Upstream vraR operon | EYKpn | GGGGGTACCACTTTGATCCAAAAGACAAAACA |
| promoter | Tet marker upF | CGGGATCCGCTTCACAGAAATTCTAGAAC |
| Tet marker downR | ACGCGTCGACTTTTATTACCTACAACTTCTTTA | |
| Kan marker upF | CGGGATCCGATAAACCCAGCGAACCATTTG | |
| Kan marker downR | CGGGATCCATCGATACAAATTCCTCGTAGG | |
| qRT-PCR | VraR-450 forward | TGCTTACAGAACGAGAAATGGAAA |
| VraR-535 reverse | CCGTTTTAATAGTAATATGCGATGCA | |
| VraR-473T (TAMRA-FAM) | TGATTGCGAAAGGTTACTCAAATCAAGAAAT |
Underlined regions represent restriction enzyme sequences.
QuikChange PCR mutagenesis (Stratagene) was used to create additional protein expression vectors by a similar strategy as described above for mutants VraR-D55A, VraScyt-H156A, and VraScyt-H238A (Table 2). All protein expression vector constructs were sequence verified prior to use. This purification strategy was used for the preparation of each mutant recombinant protein.
Autophosphorylation and phosphotransfer assay.
To assess histidine autophosphorylation, 5 μM concentrations of either VraScyt, VraScyt-H156A, or VraScyt-H238A were incubated with 20 μCi of [γ-32P]ATP (Hartmann Analytics, FP-301, 111 TBq/mmol) at 22°C in a 70-μl labeling reaction containing 25 mM Tris-HCl (pH 7.0), 2.5 mM MgCl2, 100 mM KCl, 0.1 mM DTT, 5% glycerol, and 20 μΜ cold ATP. Aliquots (10 μl) were removed after 5, 15, and 45 min and transferred to an equal volume of 2× sodium dodecyl sulfate (SDS) sample loading buffer and applied to 12% polyacrylamide-SDS protein gels (without heating). Protean II minigels (Bio-Rad) were electrophoresed at 11 W for 2 to 3 h. The gels were dried and autoradiographed (Amersham Hyperfilms). For phosphotransfer reactions between VraS and VraR, an VraScyt or VraScyt-H238A labeling protocol identical to that described above was used. After 45 min of VraS autophosphorylation, 40 μl was mixed with either purified VraR or with VraR-D55A to a final concentration of 17.5 μΜ. Aliquots (10 μl) were removed and mixed with an equal volume of 2× SDS sample buffer and applied to SDS protein gels as described above. Control phosphorylation reactions run in parallel and stained with Coomassie brilliant blue showed that all recombinant proteins were stable under the conditions of the in vitro assay.
Total RNA extraction.
Overnight bacterial cultures were diluted 1:100 and grown at 37°C for 1 h in Mueller-Hinton broth (MHB) with shaking. When indicated, oxacillin (1 μg/ml; ISP794 MIC = 2 μg/ml; thus, conducted at 0.5 MIC) was added, and bacteria were grown for an additional hour. Bacteria were harvested, and RNA extraction and verification of the absence of contaminating DNA was performed as described previously (59). Purified RNA samples were analyzed by using the RNA NanoLab chip on a 2100 Bioanalyzer (Agilent, Palo Alto, CA).
Real-time RT-PCR.
mRNA levels were determined by quantitative RT-PCR (qRT-PCR) by using a one-step reverse transcriptase qPCR master mix kit (Eurogentec, Seraing, Belgium), as described previously (59). Appropriate vraR and vraS primers and probes were designed by using Primer Express software (version 1.5; Applied Biosystems) and obtained from Eurogentec. The mRNA levels of target genes extracted from the different strains were normalized to 16S rRNA levels, which were assayed in each round of qRT-PCR as internal controls as described previously (74). The statistical significance of strain-specific differences in normalized cycle threshold (CT) values of each transcript was evaluated by using a Student paired t test, and data were considered significant when P was <0.05.
Northern blotting.
For transcript analysis, 6 μg of total RNA was separated in a 1% agarose formaldehyde gel and blotted onto a nylon membrane (Hybond-N Amersham) using 20× SSC (1× SSC is 0.15 M NaCl plus 0.015 M sodium citrate) buffer (Quantum Biotechnologies) with a semidry blot apparatus (Bio-Rad). The membrane was prehybridized with QuikHyb buffer (Stratagene) for 2 h at 65°C. An [α-32P]UTP (Hartmann Analytics, FP-110, 15 TBq/mmol)-labeled vraR riboprobe was generated from pAM1158. After plasmid linearization with Acc65I and gel purification, an [α-32P]UTP-labeled complementary antisense transcript was produced by in vitro transcription using T7 polymerase essentially as described previously (35). Unincorporated nucleotide was removed by passage over a microspin ProbeQuant G-50 column (GE Healthcare). The riboprobe mixture was treated with DNase I (Promega RQ1) to eliminate template DNA, extracted with phenol-chloroform-isoamyl alcohol (25:24:1), and precipitated with ethanol in the presence of 16 μg of glycogen carrier. The pellet was washed with ice-cold 70% ethanol, dried, and resuspended in a minimal volume of Tris-EDTA. An aliquot was tested for probe purity on a 6% polyacrylamide 8 M urea sequencing gel. The prehybridized membrane was incubated overnight with the vraR riboprobe at 65°C in the same QuikHyb buffer. Washes were done as follows: a first wash at 50°C with 2× SSC-0.1% SDS for 15 min, a second wash with 1× SSC-0.1% SDS for 15 min at 65°C, and then three washes with 0.1× SSC-0.1% SDS for 15 min at 70°C. The membrane was transferred to 3MM paper without drying, sealed, and autoradiographed (Amersham Hyperfilms).
Construction of AR756 containing a vraS disruption.
To facilitate the construction of a site-specific codon change VraS H156A by allelic exchange, a chromosomal vraS disruption mutant was first constructed by deletion of the vraS coding sequence corresponding to amino acids 2 to 160 (inclusive) and insertion of an erythromycin resistance gene using the temperature-sensitive vector pBT2 (11). Briefly, a 1,076-bp upstream fragment and a 1,185-bp downstream fragment were separately amplified by using the primer pairs presented in Table 2; the products were then mixed and reamplified with the outside primers to create the fusion PCR-generated internal deletion of vraS and cloned into pUC18-digested by using KpnI and PstI. The erythromycin resistance ermB cassette was obtained by BamHI digestion of a mini-Mu transposon derivative of pUC19-MuSupF (26) harboring the ermB gene subcloned with linkers from pEC2 (11). The BamHI ermB fragment was cloned into the unique BglII site. A plasmid with the transcriptional orientation of ermB oriented in the same sense as the native vraR operon transcript was chosen, digested with KpnI, and partially digested with PstI, and then the 3.7-kb fragment was cloned into pBT2 to yield pAM1284. Plasmid pAM1284 was electroporated into the restriction-defective strain RN4220 and then transferred by electroporation into ISP794, selecting for erythromycin resistance at 30°C. ISP794 containing pAM1284 was grown overnight at 30°C, followed by growth with applied marker selection for 6 days with dilution passages at 42°C, a nonpermissive temperature for pBT2 replication. Bacteria were plated on agar containing 5 μg of erythromycin/ml and then replica streaked on 15-μg/ml chloramphenicol plates to screen for chloramphenicol-sensitive colonies. Double-crossover events corresponding to vraS gene disruption was confirmed by PCR. The resulting strain was named AR756.
Generation of AR769 containing the vraS(H156A) mutant.
The site-specific chromosomal two-nucleotide change for the creation of vraS(H156A) was constructed using the thermosensitive plasmid pBT2 as follows. A KpnI-BglII overlap PCR fragment of ∼2,700 bp was amplified from ISP794 using the primers SA1703KpnBamHI, H156A-B, H156A-A, and 12ABgl2 (Table 2). The overlap PCR fragment spanning the region from SA1703 gene to the vraR-SA1699 intergenic region contains the nucleotide changes in positions 466 (C to G) and 467 (A to C) of the vraS open reading frame, resulting in the H156A codon change. A second downstream PCR fragment was amplified by using the primer pair 12BBgl2 and SA1699EcoPst (Table 2) incorporating BglII and PstI restriction sites covering ∼1,300 bp from the vraR-SA1699 intergenic region and the adjacent SA1699 gene. The fragments were cloned together in a three-piece ligation with KpnI-PstI-digested pBT2. The unique BglII restriction site thus engineered in the vraR-SA1699 intergenic region was used to insert a kanamycin resistance marker obtained by PCR amplification from strain ALC2057 and incorporating terminal BamHI restriction sites (60). The site-specific vraS(H156A) codon change gene targeting plasmid was named pAR747 and was fully sequence verified.
Plasmid pAR747 was passaged through the nonrestricting strain RN4220, followed by recovery and electroporation into AR756, selecting for kanamycin resistance (Kanr) at 30°C. A single colony was isolated and subjected to kanamycin selection for 6 days with regular dilution and subculture passages at 42°C. Bacteria were plated on agar containing 40 μg of kanamycin/ml and then replica streaked on 15-μg/ml chloramphenicol and 5-μg/ml erythromycin plates to screen for the Kanr and erythromycin-sensitive (Erys) double-crossover events corresponding to the expected vraS allelic exchange. A single colony was grown and confirmed by PCR and sequencing. The resulting strain harboring vraS(H156A) and its nearby linked Kanr marker to facilitate bacteriophage-mediated cotransduction was named AR769. Bacteriophage Φ80α was used to backcross the kanamycin-linked vraS(H156A) allele to ISP794 to yield AR828 (>95% cotransduction). For both AR769 and AR828, the entire chromosomal 3.3-kb vraR four-gene operon, including 600 bp of upstream promoter sequence from the SA1703 ATG start codon, was sequence verified. Both strains had only the desired vraS(H156A) codon change. Bacteriophage Φ80α and Φ11 were used to backcross the kanamycin-linked vraS(H156A) allele to strains COL and Newman, yielding strains AR868 and AR848, respectively. The presence of the vraS(H156A) allele in each derivative strain was verified by sequencing.
Construction of AR758 and AR943 containing a kanamycin- or tetracycline-marked wild-type vraR operon.
Identical targeted insertions of only the kanamycin resistance gene, or a tetracycline resistance gene, inserted nearby the vraR operon in the intergenic region between vraR and SA1699, were obtained using pBT2 as described above. Briefly, the regions 1,200 bp downstream and 1,300 bp upstream from chromosomal location 1946712 (using sequence coordinates based on N315 annotation [41]) were amplified by using the primer pairs described in Table 2. The kanamycin resistance marker was obtained by PCR amplification as described above. A BglII tetracycline resistance marker cassette was obtained as described previously (60). The desired double-crossover events were identified by screening for kanamycin/tetracycline-resistant but chloramphenicol-sensitive colonies. Correct insertion of the markers into the intergenic region was confirmed by PCR and sequencing.
Constructions of rsbU+ and sigB mutant derivatives of ISP794.
The wild-type rsbU+ allele was restored in ISP794 by Φ80α bacteriophage transduction of the tetL-linked (rsbUVW-sigB)+ operon from donor strain MB211 and selection for tetracycline to yield AR852. ISP794 derivatives lacking a functional sigB gene (AR851) or disrupted for the entire rsbUVW-sigB operon (AR850) were constructed by Φ80α bacteriophage transduction using the donor strain ALC1001 or MB212, respectively. The restoration of rsbU+ in strain ISP794 was monitored both by the reappearance of yellow-orange pigmentation and by the presence of a functional σB monitored by using a qRT-PCR assay of the σB-dependent transcription of asp23 (23, 39, 59).
Genetic assays for the emergence of glycopeptide resistance.
Tests for the emergence of glycopeptide resistance were performed essentially as described previously (60). Briefly, overnight cultures of each strain to be tested were grown in MHB at 37°C with vigorous agitation. Each bacterial culture was subsequently normalized using sterile 0.9% (wt/vol) NaCl to McFarland standard 2 by using a Densimat apparatus (bioMérieux, France). Aliquots (500 μl, 1.5 × 108 CFU) were spread on MHB agar containing various concentrations of freshly prepared teicoplanin or vancomycin, followed by incubation for 48 h at 37°C. To determine the relative efficiency of colony formation, serial dilutions of each culture were also plated on MHB plates without drug. For each strain background—ISP794, Newman, or COL—an emergence assay was performed using drug concentrations determined from pilot experiments to be at or above the broth macrodilution MIC. (Specifically, for teicoplanin these consisted of an ISP794 MIC of 1 to 2 μg/ml, with an emergence assay performed at 2 μg/ml; a Newman MIC of 1 to 2 μg/ml [although the emergence assay was performed at 4 μg/ml because pilot experiments showed that plates were nearly confluent if selection at 2 μg/ml was applied]; and a COL MIC of 8 μg/ml, with an emergence assay performed at 8 μg/ml. For vancomycin, they were an ISP794 MIC of 1 to 2 μg/ml, with an emergence assay performed at 2 μg/ml; a Newman MIC of 2 to 4 μg/ml, with an emergence assay performed at 2 μg/ml; and a COL MIC of 2 to 4 μg/ml, with an emergence assay performed at 2 μg/ml.) Broth macrodilution MIC assays were performed as described previously (60). The data were tabulated as the number of viable colonies at each drug concentration tested and normalized per McFarland unit. The sum of CFU obtained on selective media in multiple experiments was reported, together with the mean emergence frequency defined as the ratio of normalized McFarland standard 1 CFU obtained on selective media divided by the number of CFU obtained on nonselective media. A subset of colonies was retested by replica plating on selective agar plates to estimate the percentage of false positives arising in each experiment. The raw data are reported without correction, and thus calculated emergence frequencies represent an upper limit.
Genetic complementation of vraS(H156A).
Strain AR828 was transformed with the empty E. coli-S. aureus shuttle plasmid pMK4 or pMK4 containing the entire cloned 3.3-kb vraR operon, together with native upstream promoter sequences (pAM1483). The genomic vraR operon fragment was obtained by PCR amplification using the primers EYKpn and VraR-Pst (Table 2) and Pfx polymerase (Invitrogen). The entire vraR operon was sequence verified using the appropriate primers. The functional restoration of induction of the vraR operon after challenge with oxacillin was performed as described above and monitored by qRT-PCR assay using a vraR TaqMan probe.
RESULTS
Prediction of the VraS H-box region and protein purification.
Phylogenetic and functional studies of histidine kinase sensors have revealed several highly conserved motifs within the cytoplasmic kinase domain (21). The motifs comprise two distinct domains: a HisKA domain containing the H-box and a conserved histidine residue that serves as the site of autophosphorylation, as well as a C-terminal domain termed HATPase within which are found boxes named N1, G, F, G2, and G3 that comprise the ATP-binding pocket (54).
Multiple sequence alignment using T-Coffee and CLUSTAL W2 algorithms (http://www.expasy.org/tools/proteome) were performed using the S. aureus VraS sequence and a panel of bacterial HK sensors. We identified one region, ARELH156DSVSQ, which was similar to the H-box region of eight HK sensors subclassified as type III (36). In addition, sequence context inspection of all other histidines revealed a second potential histidine autophosphorylation site, VVH238EI, which was highly similar to the VSH249EI found in the S. aureus TCS HssRS system (68). A schematic diagram summarizing the predicted domain architecture of VraS is shown in Fig. 1.
FIG. 1.

Schematic diagram of VraS and VraR highlighting their domain architecture and the location of amino acids mutated in the present study. The predicted transmembrane region (TM), histidine kinase (HisKA), and histidine kinase ATP-binding domain (HATPase) are shown. VraScyt indicates the point of truncation used for the purification of the cytoplasmic and soluble portion of the protein used in the present study. The VraR response regulator is depicted and shows the receiver domain and position of phosphorylated aspartate, along with the C-terminal DNA-binding domain. The sequence alignment shows the H-box sequence context of VraS-H156, together with other conserved motifs within the ATP-binding domain. Sequences used were from Swiss-Prot accession: Staphylococcus aureus VraS, Q99SZ7; Bacillus subtilis LiaS, O32198; Escherichia coli UhpB, P09835; and Haemophilus influenzae NarQ, P44604.
We cloned the region comprising the predicted cytoplasmic domain of VraS, amino acids 65 to 347, hereafter referred to as VraScyt. The N-terminal 64 amino acids of VraS are very rich in hydrophobic residues, and this region is thought to comprise the transmembrane anchor sequence (Fig. 1). The precise topological configuration of the transmembrane region is unknown, as are the amino acids responsible for sensing external signals. To purify VraScyt, we designed PCR primers (Table 2) that incorporated both NdeI and PstI restriction sites appropriately positioned to permit cloning in pTYB12 (NEB). A recombinant hybrid VraScyt protein was produced in E. coli as an N-terminal intein fusion. Protein was purified by chitin affinity chromatography, cleaved in situ using DTT, eluted, and concentrated (see Materials and Methods). Thiol-induced cleavage resulted in the release of VraScyt containing four additional N-terminal amino acids: Ala, Gly, and His due to the intein self-cleavage recognition sequence, together with a Met introduced by the NdeI restriction site. A similar strategy was used to purify two variants of VraScyt in which histidines H156 and H238 had been separately replaced by alanine and named H156A and H238A (Fig. 1).
As a substrate for the detection of phosphotransfer by VraScyt, we used the same intein fusion protein strategy to purify wild-type VraR and a point mutant, VraR-D55A. Purified VraR and VraRD55A also possessed an additional N-terminal three amino acids, Ala-Gly-His, after thiol cleavage.
Aliquots of purified proteins were examined by Coomassie blue staining after electrophoresis in 12% polyacrylamide-SDS gels and judged to be >99% pure (Fig. 2). Purified wild-type and mutant VraScyt proteins all comigrated with an apparent mass of 34 kDa, which is consistent with the predicted 34-kDa molecular mass for this fragment. Purified wild-type VraR and VraR-D55A both comigrated in SDS-PAGE with an apparent molecular mass of 22 kDa, which is slightly smaller than the predicted 24 kDa for this fragment. Purified proteins were stable for at least several months at 4°C.
FIG. 2.
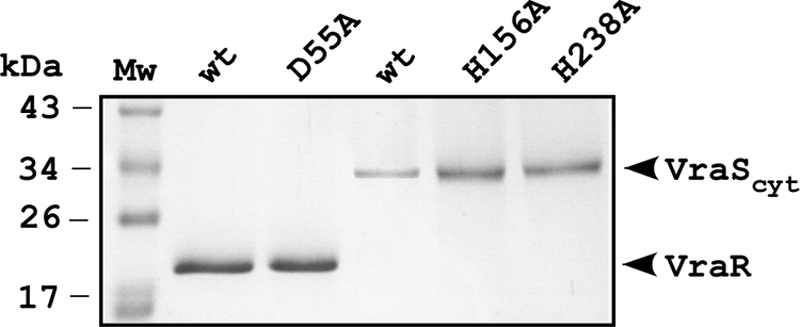
Purified VraR and VraS and their indicated mutant derivatives used in the present study. Proteins were resolved on 12% polyacrylamide-SDS gels and stained with Coomassie brilliant blue. Molecular mass standards are indicated in kilodaltons for the marker ladder.
Determination of the site of VraS autophosphorylation.
To examine the ability of each VraScyt protein to undergo autophosphorylation in vitro, samples were incubated in the presence of [γ-32P]ATP, and aliquots were removed at various times, resolved on 12% polyacrylamide SDS gels, dried, and autoradiographed. The results are shown in Fig. 3 A.
FIG. 3.

In vitro autophosphorylation and phosphotransfer assay using purified VraS and VraR. (A) VraS autophosphorylation time course assay in the presence of [γ-32P]ATP. Reactions were assembled for the indicated times and applied to 12% polyacrylamide-SDS gels, dried, and autoradiographed. Wild-type VraS lacking the N-terminal transmembrane domain VraScyt and two purified histidine mutants, H156A and H238A, are shown. (B) VraS-VraR phosphotransfer assay. Autophosphorylation reactions were performed as described for panel A, and then VraR was added. Aliquots were removed at the indicated times and resolved on SDS-protein gels as described above. Note that a minor phosphorylated degradation product of VraS migrates at a position above phosphorylated VraR. S, VraS; R, VraR.
We observed a progressive increase in the intensity of radiolabeled wild-type VraScyt indicating autophosphorylation that reached an apparent plateau at 45 min. We observed a similar autophosphorylation of VraScyt H238A, which also showed a plateau after 45 min of incubation. The relative intensity of VraScyt and H238A autophosphorylation seen in the representative autoradiogram is most likely due to experimental conditions, or perhaps to altered protein specific activity. We did not further explore the exact cause of this discrepancy. In contrast, to the results obtained with VraScyt and H238A, we observed no detectable autophosphorylation of the H156A variant under identical conditions and replicate experiments.
We conclude from this analysis that VraS H156 is essential for autophosphorylation of the purified VraScyt fragment in vitro and that this site most probably represents the site of autophosphorylation. The arrangement and position of conserved amino acid residues in the N, G1, G2, and G3 regions of the VraS cytoplasmic domain fragment are coincident when comparing VraS alignment with other sensor kinase proteins such as Escherichia coli NarX and UhpB or Haemophilus influenzae NarQ (Fig. 1). The absence of an F region, together with the 108-amino-acid spacing between H156 and N strongly suggests the classification of VraS as HK subtype type III and kinase type unorthodox (36).
VraS histidine kinase phosphotransfer assay.
Alignment of response regulators from multiple bacteria has consistently revealed a highly conserved acidic pocket positioned at the top of the characteristic α5β5 protein fold of the receiver domain. An aspartate side chain within the pocket serves as an acceptor for phosphotransfer reaction from the cognate histidine kinase receiver domain. Inspection of the VraR domain revealed that D55A was most likely the site of phosphorylation. Using purified proteins and in vitro phosphotransfer assay, we demonstrated that phosphorylated VraScyt can transfer its phosphate to VraR but not to VraR-D55A (Fig. 3B). While the present study was in progress, VraR-D55A was independently identified by mass spectrometry as the site of VraR phosphorylation (3), thus confirming these results. Collectively, these findings define key molecular details of the essential amino acid residues, VraS-H156 and VraR-D55, comprising the phosphorelay network of the VraRS TCS system.
Targeted engineering of vraS(H156A) point mutation in the S. aureus chromosome.
To examine the functional consequences of uncoupling VraS-VraR phospho-signaling in vivo, we next constructed S. aureus strains (AR769 and AR828) in which the vraS(H156A) mutation tagged with a nearby kanamycin resistance marker was stably introduced into the bacterial chromosome. Another strain, AR758, containing only the kanamycin marker within the VraR-SA1699 intergenic region, but otherwise wild type for the entire vraSR operon, was designed in parallel. The two-step strategy used for the genetic engineering by allelic exchange of this targeted codon mutation is depicted in Fig. 4.
FIG. 4.

Schematic showing the various genetic steps used for the construction of a chromosomal mutant encoding vraS(H156A) and marked with a nearby kanamycin resistance cassette in the VraR-SA1699 (N315 ordered sequence tag numbering) intergenic region. A second strain harboring only the kanamycin resistance marker, but otherwise wild type for the entire VraSR four-gene operon, was designed in parallel. A thermosensitive shuttle plasmid, pAR747, was introduced into AR756. The correct double-crossover event yielded the desired vraS-H156 mutation by allelic exchange. The vraS(H156A) allele was then backcrossed into strain ISP794 by bacteriophage-mediated transduction using selection for the tightly linked nearby kanamycin marker to give AR828. The entire operon and upstream promoter sequences were completely sequence verified. Arrows indicate the transcription direction. Kpn and Pst denote the positions of the restriction sites used for the construction of pAR747 targeting vector.
A design feature that permitted efficient screening for the kanamycin-linked vraS(H156A) mutation in strain AR769 was the prior construction of an intermediate strain, AR756, that contained an internal deletion within vraS (Fig. 4B). Kanamycin selection with the thermosensitive pAR747 targeting plasmid resulted in a subset of colonies having lost the ermB marker by allelic exchange, thus guaranteeing the chromosomal insertion of the desired vraS(H156A) mutation (Fig. 4B).
Growth curves for ISP794, AR758, and the mutant strains AR769 and AR828 were identical and indicated that under our routine laboratory conditions, neither the introduction of a kanamycin marker in the VraR-SA1699 intergenic region nor the H156A codon change in VraS resulted in detectable altered fitness (data not shown). Strains AR769 and AR828 were both used interchangeably for subsequent in vivo experiments.
Northern blot analysis.
To assess the effect of VraS-VraR phosphotransfer uncoupling by the H156A mutation in vivo, transcriptional induction of the vraR operon was monitored by Northern blot and qRT-PCR analysis for strains ISP794 and AR828 after challenge with subinhibitory amounts of cell wall-active antibiotics.
Using a radiolabeled vraR probe, we observed a strong transcriptional induction of the vraR operon in the presence of 0.5 MIC oxacillin as an inducer (Fig. 5 A). We detected multiple distinct bands by this Northern blot analysis, including the longest transcript (2.7 to 3.0 kb) sufficient to encode the entire operon (22, 40, 77). In contrast, transcriptional induction was severely reduced in the mutant strain AR828. Similar results were obtained with subinhibitory concentrations of d-cycloserine and teicoplanin as cell wall stress inducers (data not shown).
FIG. 5.
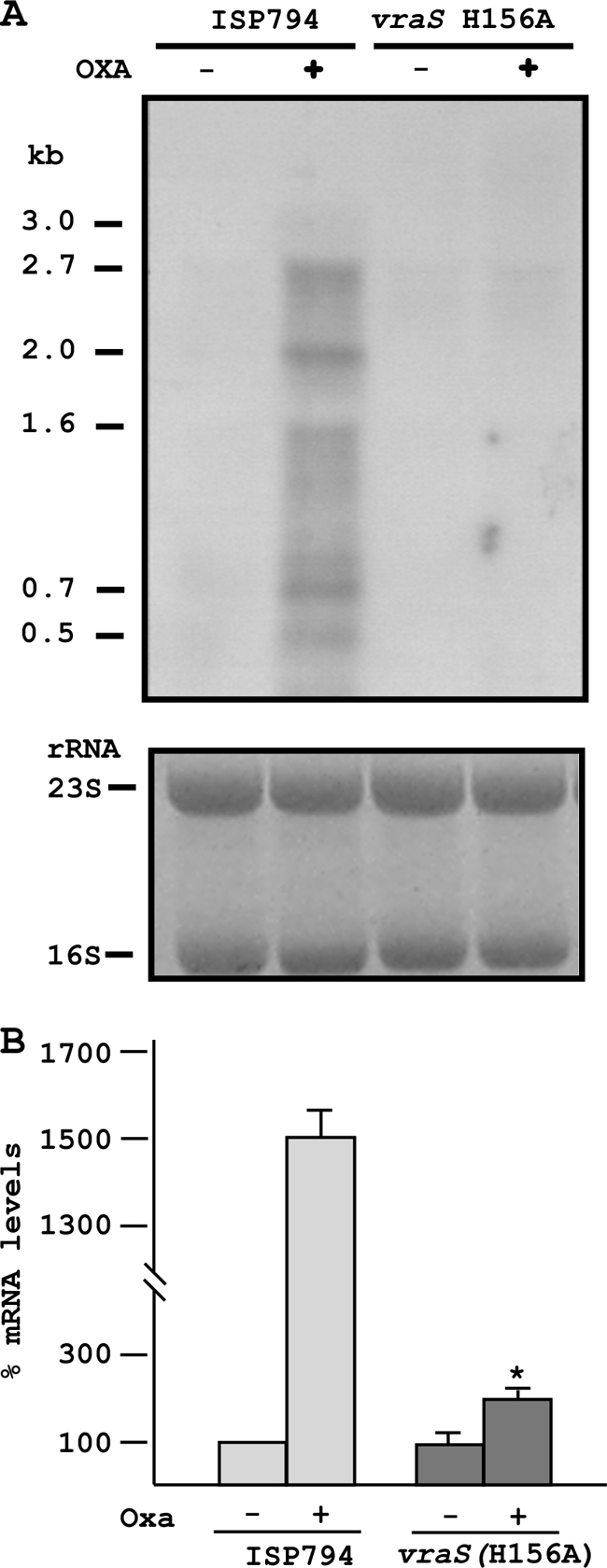
Transcriptional analysis showing the effects of uncoupling of VraS phosphotransfer. (A) Northern blot analysis showing strong oxacillin-induced induction of vraRS operon transcription in wild-type cells but severe reduction in the vraS(H156A) mutant. Note that the vraRS operon is autoregulated. The 2.7-kb band encodes the entire four-gene operon. Estimated lengths of additional transcripts are indicated that may represent strong pause sites or partial transcript degradation. Ethidium bromide-stained 16S and 23S rRNAs are shown as loading controls. (B) Quantitation of VraR mRNA levels by qRT-PCR analysis showing that oxacillin-stimulated transcriptional induction of the vraRS operon is severely reduced but not completely abolished by vraS(H156A). An asterisk denotes Student two-tailed t test analysis of transcript levels in the presence or absence of oxacillin (P < 0.05) from three independent determinations.
Quantification of vraR mRNA levels by qRT-PCR analysis confirmed the oxacillin-stimulated transcriptional induction of the vraR operon. A small, but significant (P < 0.05) residual level of transcriptional induction (∼2-fold) was observed in the vraS(H156A) mutant strain compared to its uninduced control level (Fig. 5B). This result suggests the possibility that additional transcriptional regulatory circuits exist for this operon that do not depend upon VraS-mediated signal transduction.
We next repeated the qRT-PCR assay using RNA extracted from strains harboring the multicopy plasmid pMK4, or pMK4 containing the entire 3.3-kb vraR operon including native upstream promoter sequences (pAM1483). We observed strong transcriptional induction of the vraR operon in the mutant strain harboring pAM1483, but not pMK4 vector alone, in the presence of subinhibitory oxacillin as described above (data not shown). We conclude that the vraS(H156A) mutation is recessive and that only the vraS(H156A) mutation accounts for the observed loss of transcriptional induction in the experiments described above.
Effect of vraS(H156A) on the frequency of emergence of reduced sensitivity first step glycopeptide mutants.
To test the functional consequences in vivo of uncoupling VraS-VraR phospho-signaling, we next examined whether the mutation detectably altered the MIC, or the frequency with which resistant colonies appeared on agar plates supplemented with various amounts of glycopeptide antibiotics. We reasoned that uncoupling the VraS-VraR phosphotransfer sensory system would significantly affect the detection of cell wall stress and thus alter or abolish the emergence of glycopeptide-resistant mutants.
We addressed this hypothesis using three S. aureus strain backgrounds: ISP794 (an 8325 derivative defective in rsbU and consequently lacking a normal alternative stress sigma factor σB pathway), Newman (rsbU+), and COL (an rsbU+ MRSA strain). The teicoplanin and vancomycin MICs were determined for ISP794, Newman, and COL, and the emergence assay was designed to detect the number of viable colonies arising on plates containing drug at the MIC determined for each parental strain (see Materials and Methods). We observed a significant MIC reduction in every case (Table 3). Measurement of the oxacillin MIC for the MRSA strain COL (MIC = 400 μg/ml) and AR868 (MIC 100 μg/ml) indicated that the vraS(H156A) mutation also significantly reduced methicillin resistance in this strain background.
TABLE 3.
Effect of vraS(H156A) mutation on glycopeptide MIC
| Strain | MIC (μg/ml)a |
|
|---|---|---|
| Teicoplanin | Vancomycin | |
| ISP794 | 1-2 | 1-2 |
| AR828 | 0.5 | 0.5 |
| Newman | 1-2 | 2-4 |
| AR848 | 0.5 | 0.5-1 |
| COL | 4-8 | 2-4 |
| AR868 | 0.5 | 0.5-2 |
MIC assays were performed by using broth macrodilution. Note that discrepancies (ranges) have been indicated for glycopeptide MICs that are method dependent (75).
It is worthwhile to emphasize that the MIC values we report throughout our studies here were determined using broth macrodilution; this method may give higher values than other susceptibility testing methods. Indeed, a recent study conducted in our laboratory indicates that microdilution methods tend to underestimate glycopeptide resistance (75). Glycopeptide MIC values for both COL and Newman have also been shown to be sensitive to experimental conditions (7, 66). Some caution is warranted, therefore, for cross-comparison of MIC values in the published literature since it is becoming clear that inoculum size, growth medium, time of incubation, and method are important parameters that collectively impact reported MIC values.
Next, for each parental strain tested, we observed the consistent appearance of viable colonies on agar plates containing various amounts of teicoplanin or vancomycin in the emergence assay. The frequency of emergence was computed for each strain and defined as the ratio of the number of colonies appearing using a standardized bacterial inoculum applied to each plate (1.5 × 108 CFU) under selective (TMIC or VMIC) or nonselective (T0 or V0) conditions. We measured a frequency of viable colonies (defined hereafter as emergence) by this method predominantly in a range from 1.1 × 10−6 to 5.0 × 10−8, depending upon the strain and applied selection (Table 4). Similar results were observed when vancomycin was used, although we noted that the emergence frequency was lower for this drug than for teicoplanin under our standard laboratory conditions.
TABLE 4.
Effect of vraS(H156A) mutation on emergence of glycopeptide resistance
| Strain | Description | Relevant genotype | Emergencea of: |
|||
|---|---|---|---|---|---|---|
| Teicoplanin |
Vancomycin |
|||||
| CFU Σtot (n) | FOE (TMIC/T0) | CFU Σtot (n) | FOE (VMIC/V0) | |||
| ISP794 | MSSA, ΔrsbU | 5591 (9) | 1.1 × 10−6 | 37 (3) | 5.0 × 10−8 | |
| AR828 | ISP794, vraS(H156A) | MSSA, ΔrsbU | 0 (9) | NC | 0 (3) | NC |
| AR878 | ISP794, vraS(H156A), pAM1483 | MSSA, ΔrsbU | 238 (3) | 6.6 × 10−7 | 30 (3) | 6.7 × 10−8 |
| AR872 | ISP794, vraS(H156A), pAM902 | MSSA, ΔrsbU | 0 (3) | NC | 0 (3) | NC |
| Newman | MSSA, rsbU+ | 504 (6) | 4.0 × 10−7 | 200 (3) | 2.0 × 10−7 | |
| AR848 | Newman, vraS(H156A) | MSSA, rsbU+ | 0 (6) | NC | 1 (3) | 7.6 × 10−10 |
| COL | MRSA, rsbU+ | 626 (4) | 8.0 × 10−7 | 456 (3) | 3.5 × 10−6 | |
| AR868 | COL, vraS(H156A) | MRSA, rsbU+ | 0 (4) | NC | 4 (3) | 3.1 × 10−8 |
The frequency of emergence (FOE) is expressed as the ratio of CFU under selective (TMIC or VMIC) and nonselective (T0 or V0) conditions computed as the mean from the indicated number (n) of independent experiments. Each emergence assay was performed using MH agar and 1.5×108 applied bacteria per plate. CFU were counted at 48 h and 37°C. False-positive rates of CFU detected in the emergence assay were found to be 5 to 20% depending upon the experiment. NC, not computed.
Colonies that arise in our emergence assay are either false positives and do not grow under selective conditions upon replating or represent stable mutants that do grow at or above the original selective conditions upon replating. We estimated the false-positive rate in multiple experiments by sampling subsets of colonies and determined it to be in the range of 5 to 20%. The tabular data we report, however, reflect the summated raw data from multiple independent experiments without correction for false-positive rate, which was not computed for every experiment. Thus, the resulting calculation of resistance frequencies yields an upper limit. In some cases, we also examined the spread of MIC values of colonies that arose during selection and which were classed as stable. For example, ISP794 selected for growth on MH agar plates in the presence of 2 μg of teicoplanin/ml gave rise to a range of MICs from 2 to 8 μg/ml (data not shown). Thus, colonies that grow at the applied selection concentration and that are not false positive show MIC values at or above the drug concentration used for the initial applied selection.
In striking contrast to our results obtained with strains harboring a wild-type vraS allele, we never observed colonies arising in multiple independent trials using teicoplanin selection for any of the three strains tested containing the vraS(H156A) mutation. When using vancomycin selection, we did not observe the appearance of resistant colonies with strain ISP794; however, we did observe the appearance of several viable colonies derived from strains Newman or COL (Table 4). The appearance of these infrequent colonies, whose vancomycin resistance was confirmed by replating, indicated that during selection with vancomycin the vraS(H156A) mutation drastically reduced (by >2 log10) but did not entirely abolish the emergence of resistant colonies in these strain backgrounds. Control experiments with strains carrying only the kanamycin resistance marker in the vraR-SA1699 intergenic region showed equivalent emergence frequencies of drug-resistant colonies compared to parental strains, thus ruling out any deleterious role for this marker that could account for the observed abolition or significant reduction in viable CFU for strains AR828, AR848, and AR868 exposed to glycopeptides (data not shown).
To confirm that the abolition of the emergence of glycopeptide-resistant mutants was indeed the result of the vraS(H156A) mutation, we performed a complementation analysis using strains AR878 and AR872 containing multicopy plasmid pAM1483 encoding the entire four-gene vraR operon or containing pAM902 control vector harboring green fluorescent protein (Table 4). The results revealed that restoration of wild-type vraS coding sequence concomitantly restored emergence of glycopeptide resistant mutants using either teicoplanin or vancomycin selection (Table 4). Similar results were obtained in a second type of complementation analysis without multicopy plasmid where we restored wild-type vraS in the ISP794 genetic background by allelic exchange using bacteriophage-mediated transduction from AR943 (data not shown).
Collectively, we conclude from these results that the VraS-VraR phosphotransfer uncoupling mutation vraS(H156A) abolished the detectable emergence of teicoplanin-resistant colonies and severely reduced the frequency of emergence of vancomycin resistant colonies. Since the vraS(H156A) mutation also alters COL resistance to oxacillin, we conclude that uncoupling VraS-VraR phosphotransfer impacts S. aureus response to at least two classes of antibiotics and can act both at the level of blocking emergence or altering the sensitivity profile of preexisting drug resistance.
Role of the alternative sigma factor σB in the emergence of glycopeptide resistance.
The ISP794 strain and its derivatives used routinely in our laboratory (8, 60) and also extensively for studies of fluoroquinolone resistance (19) is a derivative of 8325 and carries an 11-bp deletion in rsbU. Because of this mutation, ISP794 possesses impaired transcriptional responses mediated by the alternative stress sigma factor σB. A functional σB is thought to play a role in glycopeptide resistance (5, 7), but its role in the early steps of emergence of drug resistance have not been previously examined. Therefore, to test what role, if any, σB played in the emergence of glycopeptide resistance in this strain background, we constructed three isogenic derivatives of ISP794: AR850, AR851, and AR852, which were characterized by deletion of the entire four-gene rsbUVW-sigB operon, disruption of the sigB alone, or restoration of rsbU+, respectively. Each strain was tested in multiple independent assays as described above. The results are shown in Table 5.
TABLE 5.
Effect of alternative sigma factor sigB on the emergence of glycopeptide resistance
| Strain | Description | Relevant genotype | CFU Σtot (n) | FOEa (T2/T0) | MIC (μg/ml)b |
|
|---|---|---|---|---|---|---|
| Teicoplanin | Vancomycin | |||||
| ISP794 | ΔrsbU | 1,065 (3) | 1.1 × 10−6 | 1-2 | 1-2 | |
| AR850 | ISP794, ΔsigB operon | ΔrsbUVW sigB::ermB | 280 (3) | 3.5 × 10−7 | 2 | 1 |
| AR851 | ISP794, ΔsigB | sigB::Tn551 | 633 (3) | 8.0 × 10−7 | 1 | 1 |
| AR852 | ISP794, rsbU+ | (rsbUVW-sigB)+, tetL nearby | >3,000 (3)c | >3.0 × 10−5d | 2 | 2 |
The frequency of emergence (FOE) was determined as described in Table 4, footnote a.
Defined in Table 3, footnote a.
Viable counts on agar containing 2 μg of teicoplanin/ml were too high to accurately measure using these conditions compared to ISP794. More than 1,000 CFU were estimated in each experiment.
The value reported is an estimate of the lower limit using CFU data obtained under identical conditions for the frequency determined with ISP794.
We observed that the deletion of the entire rsbUVW-sigB operon, or sigB alone, did not abolish the emergence of teicoplanin-resistant colonies. In contrast, when rsbU+ was restored in ISP794, we noted dramatic enhanced emergence frequency compared to ISP794 (ΔrsbU) conducted under identical conditions. We conclude that a functional σB markedly enhances the emergence of glycopeptide resistance in this strain background but that σB is not absolutely required for resistant colonies to arise.
DISCUSSION
In this study, we have identified the amino acids, VraS H156 and VraR D55, involved in phosphotransfer signaling in the VraRS TCS, a sentinel system for cell wall stress in S. aureus. Importantly, we have found that three distinct strains—ISP794, Newman, and COL, each genetically engineered to harbor the mutation encoding autophosphorylation-defective VraS H156A—are unable to generate first step low-level teicoplanin resistance mutants at a detectable frequency. The mutation also abolished detectable first-step low-level vancomycin-resistant mutants in strain ISP794 and severely reduced the frequency of mutants in Newman and COL strains. Disruption of VraS-mediated signaling in these strains also significantly altered the MICs of both teicoplanin and vancomycin as measured by broth macrodilution and also significantly altered the oxacillin MIC in the MRSA strain COL.
A broad range of antibiotics provoke cell wall stress in S. aureus and transcriptional upregulation of the vraSR four-gene operon is a feature commonly observed (22, 40, 77). Recent study provides a mechanistic explanation since VraR binds directly to its own promoter and thus autoregulates itself (4). Besides extensive evidence from transcriptome and sequence analysis of clinical isolates that highlight the importance of VraRS for glycopeptide resistance (9, 16, 22, 32, 34, 40, 46, 49, 52, 56, 73), additional mutations in the graRS TCS or tcaA disruption promote glycopeptide resistance (10, 15, 30, 45, 48, 53). Transcriptional upregulation of the WalKR TCS is also associated with altered glycopeptide resistance (31). Considering our inability to isolate first-step mutants using strains carrying the vraS(H156A) mutation, it is tempting to speculate that a functional VraRS phosphotransfer signaling system is necessary for all, or certainly a majority, of pathways leading to the emergence of glycopeptide resistance. It is important to stress, nevertheless, that the emergence assay that we describe in the present study used selection at very low drug concentrations, and therefore the role of VraRS has not been systematically assessed under other selection conditions using, for example, higher drug concentrations.
Several recent studies also provide evidence for glycopeptide resistance arising as a consequence of genetic selection with antibiotics other than glycopeptides. For example, selection of S. aureus growth in the presence of imipenem or daptomycin has been shown to produce strains with altered glycopeptide resistance profiles (32, 33, 50). These results are also supported by clinical case reports suggesting that the mechanisms responsible for altered sensitivity to daptomycin and the emergence of hVISA are related (37, 71). In light of these findings, it is conceivable that disruption of VraRS TCS signaling could also predictably attenuate the emergence of glycopeptide cross-resistance evoked by a nonglycopeptide drug encounter.
Derivatives of S. aureus strain 8325, such as ISP794 used in our study, are known to have an 11-bp deletion in rsbU, and consequently such strains display reduced availability of free σB, an alternative stress sigma factor that has been shown to regulate more than 200 genes in response to a variety of stress inducers (6, 23, 39). Free σB is sequestered by the anti-sigma factor RsbW and is thought to be released in times of stress by the anti-sigma factor RsbV, provided it is dephosphorylated by the RsbV-specific phosphatase, RsbU (65). The absence of a functional RsbU in 8325 derived strains thus explains the constitutive sequestration of σB by RsbW. Previous studies (5, 7) have hinted that σB controls gene(s) required for glycopeptide resistance in S. aureus and that teicoplanin exposure stress can select for enhanced σB activity. One recently identified σB-regulated gene contributing to reduced glycopeptide sensitivity is spoVG, although its mechanism of action in S. aureus remains to be clarified (64).
In our study, we examined the role of σB specifically at the level of the emergence of first step teicoplanin resistance mutants and found that strains derived from ISP794 lacking sigB (encoding σB) or the entire rsbUVW-sigB operon were still able to generate teicoplanin-resistant mutants, thus formally demonstrating that a functional σB was not strictly necessary for the emergence of glycopeptide resistance in this strain background. In contrast, the restoration of wild-type rsbU in ISP794, and thus the concomitant restoration of fully functional σB stress response, led to markedly enhanced emergence of teicoplanin resistance. Our inability to isolate first-step teicoplanin mutants in an ISP794 rsbU+ strain carrying vraS(H156A) indicates that disrupting VraRS signaling can apparently override any contribution to the emergence of low-level teicoplanin resistance dependent upon σB in this strain background.
Using quantitative transcript analysis, we have shown that the vraS(H156A) mutation abolishes most, but not all transcription induction of the vraSR operon after brief exposure to a subinhibitory amount of oxacillin to provoke cell wall stress. Only a single major transcription start site upstream of SA1703, the first open reading frame of the four-gene operon, has been detected by primer extension (77) and confirmed independently by our laboratory using 5′ rapid amplification of cDNA ends (5′RACE; A. Renzoni, unpublished results). The vraRS operon is not known to be transcriptionally regulated by σB under any condition tested (6). We therefore believe that additional regulatory pathways exist for this promoter that are not governed by VraS-VraR phosphotransfer. Experiments to uncover these accessory regulatory factors are in progress.
The consequences of genetic disruption of vraS by insertional inactivation or by deletion of the entire vraSR coding region have been studied independently by several laboratories (9, 16, 22, 40, 51, 56, 77). These reports link the loss of function of VraRS to oxacillin resistance in C-MRSA, altered drug sensitivity profiles in a range of clinical MSSA and MRSA isolates, and disruption of VraR-dependent target gene activation such as pbp2. In addition, a growing inventory of clinical hVISA and GISA isolates that have been partially sequenced reveals numerous examples of missense mutations mapping in vraS, vraR, or SA1702 (16, 32-34, 52). How missense mutations such as VraS-I5N modify VraRS-dependent signaling and gene expression is unknown. A reasonable hypothesis is that such mutations result in enhanced activation of the VraRS signaling pathway, even in the absence of an external inducing signal such as drug stress. With the VraSR pathway set in an ON, or semi-ON state, the cells realign their cell wall biosynthetic machinery to survive in the presence of glycopeptide.
Our study clearly predicts that pharmacologically blocking VraS signaling by interfering with its sensor histidine kinase function will significantly block the emergence of glycopeptide resistance or restore fully, or partially, sensitivity to glycopeptide antibiotics in strains showing preexisting reduced sensitivity to these drugs. The idea of using kinase inhibitors to target bacterial TCS systems has been suggested for many years, especially in light of the fact that TCS systems are not found in humans (2, 25, 58, 62). Our genetic study reported here therefore provides a firm foundation for this strategy and further predicts that the efficacy of existing antistaphylococcal cell wall-active antibiotics might be prolonged if such kinase inhibitor compounds were to be ultimately identified and coadministered.
Evidence presented to date thus underscores the importance of the vraRS operon at the crossroads mediating S. aureus response to cell wall stress and cell wall-active antibiotics. The ensemble of downstream events that are regulated by VraR-dependent transcription may be quite extensive judging by the number of genes suspected to be regulated by this system. Additional response pathways mediated by other TCS systems (AgrAB, GraRS, and WalKR) and global regulators such as σB would appear to contribute to the establishment of endogenous glycopeptide resistance. By targeting the initial molecular features of the VraRS signaling response pathway at the onset of this multifactorial regulatory cascade, we strongly believe that emergence of glycopeptide resistance could be contained.
Acknowledgments
This study was supported by Swiss National Science Foundation grant 3100A0-120428 (to W.L.K.), a Novartis Consumer Health Foundation postdoctoral fellowship grant (to A.R.), and an F. Hoffmann-La Roche M.D.-Ph.D. training grant (to D.O.A.). Portions of this study were financed by a student exchange fellowship from the Italian Ministry of Education and Research (E.G.).
We thank Markus Bischoff, Brigitte Berger-Bachi, Reinhold Bruckner, and Ambrose Cheung for generously providing strains and plasmids and members of the laboratory for their encouraging support and helpful comments on the manuscript.
Footnotes
Published ahead of print on 20 December 2010.
REFERENCES
- 1.Baba, T., T. Bae, O. Schneewind, F. Takeuchi, and K. Hiramatsu. 2008. Genome sequence of Staphylococcus aureus strain Newman and comparative analysis of staphylococcal genomes: polymorphism and evolution of two major pathogenicity islands. J. Bacteriol. 190:300-310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barrett, J. F., and J. A. Hoch. 1998. Two-component signal transduction as a target for microbial anti-infective therapy. Antimicrob. Agents Chemother. 42:1529-1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Belcheva, A., and D. Golemi-Kotra. 2008. A close-up view of the VraSR two-component system: a mediator of Staphylococcus aureus response to cell wall damage. J. Biol. Chem. 283:12354-12364. [DOI] [PubMed] [Google Scholar]
- 4.Belcheva, A., V. Verma, and D. Golemi-Kotra. 2009. DNA-binding activity of the vancomycin resistance associated regulator protein VraR and the role of phosphorylation in transcriptional regulation of the vraSR operon. Biochemistry 48:5592-5601. [DOI] [PubMed] [Google Scholar]
- 5.Bischoff, M., and B. Berger-Bachi. 2001. Teicoplanin stress-selected mutations increasing σB activity in Staphylococcus aureus. Antimicrob. Agents Chemother. 45:1714-1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bischoff, M., et al. 2004. Microarray-based analysis of the Staphylococcus aureus σB regulon. J. Bacteriol. 186:4085-4099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bischoff, M., et al. 2001. Involvement of multiple genetic loci in Staphylococcus aureus teicoplanin resistance. FEMS Microbiol. Lett. 194:77-82. [DOI] [PubMed] [Google Scholar]
- 8.Bisognano, C., et al. 2004. A RecA-LexA-dependent pathway mediates ciprofloxacin-induced fibronectin binding in Staphylococcus aureus. J. Biol. Chem. 279:9064-9071. [DOI] [PubMed] [Google Scholar]
- 9.Boyle-Vavra, S., S. Yin, and R. S. Daum. 2006. The VraS/VraR two-component regulatory system required for oxacillin resistance in community-acquired methicillin-resistant Staphylococcus aureus. FEMS Microbiol. Lett. 262:163-171. [DOI] [PubMed] [Google Scholar]
- 10.Brandenberger, M., M. Tschierske, P. Giachino, A. Wada, and B. Berger-Bachi. 2000. Inactivation of a novel three-cistronic operon tcaR-tcaA-tcaB increases teicoplanin resistance in Staphylococcus aureus. Biochim. Biophys. Acta 1523:135-139. [DOI] [PubMed] [Google Scholar]
- 11.Bruckner, R. 1997. Gene replacement in Staphylococcus carnosus and Staphylococcus xylosus. FEMS Microbiol. Lett. 151:1-8. [DOI] [PubMed] [Google Scholar]
- 12.Chambers, H. F., and F. R. Deleo. 2009. Waves of resistance: Staphylococcus aureus in the antibiotic era. Nat. Rev. Microbiol. 7:629-641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cheung, A. L., Y. T. Chien, and A. S. Bayer. 1999. Hyperproduction of alpha-hemolysin in a sigB mutant is associated with elevated SarA expression in Staphylococcus aureus. Infect. Immun. 67:1331-1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cheung, A. L., K. Schmidt, B. Bateman, and A. C. Manna. 2001. SarS, a SarA homolog repressible by agr, is an activator of protein A synthesis in Staphylococcus aureus. Infect. Immun. 69:2448-2455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cui, L., J. Q. Lian, H. M. Neoh, E. Reyes, and K. Hiramatsu. 2005. DNA microarray-based identification of genes associated with glycopeptide resistance in Staphylococcus aureus. Antimicrob. Agents Chemother. 49:3404-3413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cui, L., H. M. Neoh, M. Shoji, and K. Hiramatsu. 2009. Contribution of vraSR and graSR point mutations to vancomycin resistance in vancomycin-intermediate Staphylococcus aureus. Antimicrob. Agents Chemother. 53:1231-1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.DeLeo, F. R., and H. F. Chambers. 2009. Reemergence of antibiotic-resistant Staphylococcus aureus in the genomics era. J. Clin. Invest. 119:2464-2474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dubrac, S., P. Bisicchia, K. M. Devine, and T. Msadek. 2008. A matter of life and death: cell wall homeostasis and the WalKR (YycGF) essential signal transduction pathway. Mol. Microbiol. 70:1307-1322. [DOI] [PubMed] [Google Scholar]
- 19.Fournier, B., R. Aras, and D. C. Hooper. 2000. Expression of the multidrug resistance transporter NorA from Staphylococcus aureus is modified by a two-component regulatory system. J. Bacteriol. 182:664-671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Galperin, M. Y. 2006. Structural classification of bacterial response regulators: diversity of output domains and domain combinations. J. Bacteriol. 188:4169-4182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gao, R., and A. M. Stock. 2009. Biological insights from structures of two-component proteins. Annu. Rev. Microbiol. 63:133-154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gardete, S., S. W. Wu, S. Gill, and A. Tomasz. 2006. Role of VraSR in antibiotic resistance and antibiotic-induced stress response in Staphylococcus aureus. Antimicrob. Agents Chemother. 50:3424-3434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Giachino, P., S. Engelmann, and M. Bischoff. 2001. σB activity depends on RsbU in Staphylococcus aureus. J. Bacteriol. 183:1843-1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gill, S. R., et al. 2005. Insights on evolution of virulence and resistance from the complete genome analysis of an early methicillin-resistant Staphylococcus aureus strain and a biofilm-producing methicillin-resistant Staphylococcus epidermidis strain. J. Bacteriol. 187:2426-2438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Guarnieri, M. T., L. Zhang, J. Shen, and R. Zhao. 2008. The Hsp90 inhibitor radicicol interacts with the ATP-binding pocket of bacterial sensor kinase PhoQ. J. Mol. Biol. 379:82-93. [DOI] [PubMed] [Google Scholar]
- 26.Haapa, S., S. Taira, E. Heikkinen, and H. Savilahti. 1999. An efficient and accurate integration of mini-Mu transposons in vitro: a general methodology for functional genetic analysis and molecular biology applications. Nucleic Acids Res. 27:2777-2784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hiramatsu, K. 2001. Vancomycin-resistant Staphylococcus aureus: a new model of antibiotic resistance. Lancet Infect. Dis. 1:147-155. [DOI] [PubMed] [Google Scholar]
- 28.Hiramatsu, K., et al. 1997. Dissemination in Japanese hospitals of strains of Staphylococcus aureus heterogeneously resistant to vancomycin. Lancet 350:1670-1673. [DOI] [PubMed] [Google Scholar]
- 29.Howden, B. P., J. K. Davies, P. D. Johnson, T. P. Stinear, and M. L. Grayson. 2010. Reduced vancomycin susceptibility in Staphylococcus aureus, including vancomycin-intermediate and heterogeneous vancomycin-intermediate strains: resistance mechanisms, laboratory detection, and clinical implications. Clin. Microbiol. Rev. 23:99-139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Howden, B. P., et al. 2008. Genomic analysis reveals a point mutation in the two-component sensor gene graS that leads to intermediate vancomycin resistance in clinical Staphylococcus aureus. Antimicrob. Agents Chemother. 52:3755-3762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jansen, A., et al. 2007. Role of insertion elements and yycFG in the development of decreased susceptibility to vancomycin in Staphylococcus aureus. Int. J. Med. Microbiol. 297:205-215. [DOI] [PubMed] [Google Scholar]
- 32.Katayama, Y., H. Murakami-Kuroda, L. Cui, and K. Hiramatsu. 2009. Selection of heterogeneous vancomycin-intermediate Staphylococcus aureus by imipenem. Antimicrob. Agents Chemother. 53:3190-3196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kato, Y., T. Suzuki, T. Ida, and K. Maebashi. 2010. Genetic changes associated with glycopeptide resistance in Staphylococcus aureus: predominance of amino acid substitutions in YvqF/VraSR. J. Antimicrob. Chemother. 65:37-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kato, Y., et al. 2008. Microbiological and clinical study of methicillin-resistant Staphylococcus aureus (MRSA) carrying VraS mutation: changes in susceptibility to glycopeptides and clinical significance. Int. J. Antimicrob. Agents 31:64-70. [DOI] [PubMed] [Google Scholar]
- 35.Kelley, W. L., and C. Georgopoulos. 1997. Positive control of the two-component RcsC/B signal transduction network by DjlA: a member of the DnaJ family of molecular chaperones in Escherichia coli. Mol. Microbiol. 25:913-931. [DOI] [PubMed] [Google Scholar]
- 36.Kim, D., and S. Forst. 2001. Genomic analysis of the histidine kinase family in bacteria and archaea. Microbiology 147:1197-1212. [DOI] [PubMed] [Google Scholar]
- 37.Kirby, A., et al. 2009. In vivo development of heterogeneous glycopeptide-intermediate Staphylococcus aureus (hGISA), GISA, and daptomycin resistance in a patient with methicillin-resistant S. aureus endocarditis. J. Med. Microbiol. 58:376-380. [DOI] [PubMed] [Google Scholar]
- 38.Kreiswirth, B. N., et al. 1983. The toxic shock syndrome exotoxin structural gene is not detectably transmitted by a prophage. Nature 305:709-712. [DOI] [PubMed] [Google Scholar]
- 39.Kullik, I., P. Giachino, and T. Fuchs. 1998. Deletion of the alternative sigma factor sigmaB in Staphylococcus aureus reveals its function as a global regulator of virulence genes. J. Bacteriol. 180:4814-4820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kuroda, M., et al. 2003. Two-component system VraSR positively modulates the regulation of cell-wall biosynthesis pathway in Staphylococcus aureus. Mol. Microbiol. 49:807-821. [DOI] [PubMed] [Google Scholar]
- 41.Kuroda, M., et al. 2001. Whole genome sequencing of methicillin-resistant Staphylococcus aureus. Lancet 357:1225-1240. [DOI] [PubMed] [Google Scholar]
- 42.Lee, C. Y., S. L. Buranen, and Z. H. Ye. 1991. Construction of single-copy integration vectors for Staphylococcus aureus. Gene 103:101-105. [DOI] [PubMed] [Google Scholar]
- 43.Liu, C., and H. F. Chambers. 2003. Staphylococcus aureus with heterogeneous resistance to vancomycin: epidemiology, clinical significance, and critical assessment of diagnostic methods. Antimicrob. Agents Chemother. 47:3040-3045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lowy, F. D. 1998. Staphylococcus aureus infections. N. Engl. J. Med. 339:520-532. [DOI] [PubMed] [Google Scholar]
- 45.Maki, H., N. McCallum, M. Bischoff, A. Wada, and B. Berger-Bachi. 2004. tcaA inactivation increases glycopeptide resistance in Staphylococcus aureus. Antimicrob. Agents Chemother. 48:1953-1959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.McAleese, F., et al. 2006. Overexpression of genes of the cell wall stimulon in clinical isolates of Staphylococcus aureus exhibiting vancomycin-intermediate S. aureus-type resistance to vancomycin. J. Bacteriol. 188:1120-1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.McCallum, N., B. Berger-Bachi, and M. M. Senn. 2010. Regulation of antibiotic resistance in Staphylococcus aureus. Int. J. Med. Microbiol. 300:118-129. [DOI] [PubMed] [Google Scholar]
- 48.McCallum, N., A. K. Brassinga, C. D. Sifri, and B. Berger-Bachi. 2007. Functional characterization of TcaA: minimal requirement for teicoplanin susceptibility and role in Caenorhabditis elegans virulence. Antimicrob. Agents Chemother. 51:3836-3843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.McCallum, N., G. Spehar, M. Bischoff, and B. Berger-Bachi. 2006. Strain dependence of the cell wall damage-induced stimulon in Staphylococcus aureus. Biochim. Biophys. Acta 1760:1475-1481. [DOI] [PubMed] [Google Scholar]
- 50.Mishra, N. N., et al. 2009. Analysis of cell membrane characteristics of in vitro-selected daptomycin-resistant strains of methicillin-resistant Staphylococcus aureus. Antimicrob. Agents Chemother. 53:2312-2318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Muthaiyan, A., J. A. Silverman, R. K. Jayaswal, and B. J. Wilkinson. 2008. Transcriptional profiling reveals that daptomycin induces the Staphylococcus aureus cell wall stress stimulon and genes responsive to membrane depolarization. Antimicrob. Agents Chemother. 52:980-990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mwangi, M. M., et al. 2007. Tracking the in vivo evolution of multidrug resistance in Staphylococcus aureus by whole-genome sequencing. Proc. Natl. Acad. Sci. U. S. A. 104:9451-9456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Neoh, H. M., et al. 2008. Mutated response regulator graR is responsible for phenotypic conversion of Staphylococcus aureus from heterogeneous vancomycin-intermediate resistance to vancomycin-intermediate resistance. Antimicrob. Agents Chemother. 52:45-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Parkinson, J. S. 1995. Genetic approaches for signaling pathways and proteins, p. 9-24. In J. A. Hoch (ed.), Two-component signal transduction. ASM Press, Washington, DC.
- 55.Perichon, B., and P. Courvalin. 2009. VanA-type vancomycin-resistant Staphylococcus aureus. Antimicrob. Agents Chemother. 53:4580-4587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pietiainen, M., et al. 2009. Transcriptome analysis of the responses of Staphylococcus aureus to antimicrobial peptides and characterization of the roles of vraDE and vraSR in antimicrobial resistance. BMC Genomics 10:429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pinho, M. G. 2008. Mechanisms of β-lactam and glycopeptides resistance in Staphylococcus aureus, p. 207-226. In J. A. Lindsay (ed.), Staphylococcus molecular genetics. Caister Academic Press, Norfolk, United Kingdom.
- 58.Qin, Z., et al. 2006. Structure-based discovery of inhibitors of the YycG histidine kinase: new chemical leads to combat Staphylococcus epidermidis infections. BMC Microbiol. 6:96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Renzoni, A., et al. 2004. Modulation of fibronectin adhesins and other virulence factors in a teicoplanin-resistant derivative of methicillin-resistant Staphylococcus aureus. Antimicrob. Agents Chemother. 48:2958-2965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Renzoni, A., et al. 2009. Identification by genomic and genetic analysis of two new genes playing a key role in intermediate glycopeptide resistance in Staphylococcus aureus. Antimicrob. Agents Chemother. 53:903-911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Renzoni, A., W. L. Kelley, P. Vaudaux, A. L. Cheung, and D. P. Lew. 2010. Exploring innate glycopeptide resistance mechanisms in Staphylococcus aureus. Trends Microbiol. 18:55-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Roychoudhury, S., et al. 1993. Inhibitors of two-component signal transduction systems: inhibition of alginate gene activation in Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. U. S. A. 90:965-969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sass, P., and G. Bierbaum. 2009. Native graS mutation supports the susceptibility of Staphylococcus aureus strain SG511 to antimicrobial peptides. Int. J. Med. Microbiol. 299:313-322. [DOI] [PubMed] [Google Scholar]
- 64.Schulthess, B., et al. 2009. Functional characterization of the sigmaB-dependent yabJ-spoVG operon in Staphylococcus aureus: role in methicillin and glycopeptide resistance. Antimicrob. Agents Chemother. 53:1832-1839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Senn, M. M., et al. 2005. Molecular analysis and organization of the sigmaB operon in Staphylococcus aureus. J. Bacteriol. 187:8006-8019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sieradzki, K., and A. Tomasz. 2006. Inhibition of the autolytic system by vancomycin causes mimicry of vancomycin-intermediate Staphylococcus aureus-type resistance, cell concentration dependence of the MIC, and antibiotic tolerance in vancomycin-susceptible S. aureus. Antimicrob. Agents Chemother. 50:527-533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Stahl, M. L., and P. A. Pattee. 1983. Confirmation of protoplast fusion-derived linkages in Staphylococcus aureus by transformation with protoplast DNA. J. Bacteriol. 154:406-412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Stauff, D. L., V. J. Torres, and E. P. Skaar. 2007. Signaling and DNA-binding activities of the Staphylococcus aureus HssR-HssS two-component system required for heme sensing. J. Biol. Chem. 282:26111-26121. [DOI] [PubMed] [Google Scholar]
- 69.Steidl, R., et al. 2008. Staphylococcus aureus cell wall stress stimulon gene-lacZ fusion strains: potential for use in screening for cell wall-active antimicrobials. Antimicrob. Agents Chemother. 52:2923-2925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sullivan, M. A., R. E. Yasbin, and F. E. Young. 1984. New shuttle vectors for Bacillus subtilis and Escherichia coli which allow rapid detection of inserted fragments. Gene 29:21-26. [DOI] [PubMed] [Google Scholar]
- 71.Tenover, F. C., et al. 2009. Characterisation of a Staphylococcus aureus strain with progressive loss of susceptibility to vancomycin and daptomycin during therapy. Int. J. Antimicrob. Agents 33:564-568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tu Quoc, P. H., et al. 2007. Isolation and characterization of biofilm formation-defective mutants of Staphylococcus aureus. Infect. Immun. 75:1079-1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Utaida, S., et al. 2003. Genome-wide transcriptional profiling of the response of Staphylococcus aureus to cell-wall-active antibiotics reveals a cell-wall-stress stimulon. Microbiology 149:2719-2732. [DOI] [PubMed] [Google Scholar]
- 74.Vaudaux, P., et al. 2002. Increased expression of clumping factor and fibronectin-binding proteins by hemB mutants of Staphylococcus aureus expressing small colony variant phenotypes. Infect. Immun. 70:5428-5437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Vaudaux, P., et al. 2010. Underestimation of vancomycin and teicoplanin MICs by broth microdilution leads to underdetection of glycopeptide-intermediate isolates of Staphylococcus aureus. Antimicrob. Agents Chemother. 54:3861-3870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Yanisch-Perron, C., J. Vieira, and J. Messing. 1985. Improved M13 phage cloning vectors and host strains: nucleotide sequences of the M13mp18 and pUC19 vectors. Gene 33:103-119. [DOI] [PubMed] [Google Scholar]
- 77.Yin, S., R. S. Daum, and S. Boyle-Vavra. 2006. VraSR two-component regulatory system and its role in induction of pbp2 and vraSR expression by cell wall antimicrobials in Staphylococcus aureus. Antimicrob. Agents Chemother. 50:336-343. [DOI] [PMC free article] [PubMed] [Google Scholar]