

Abstract
Group A streptococcus (GAS) is an important human pathogen that causes a wide spectrum of diseases, ranging from mild throat and skin infections to severe invasive diseases such as necrotizing fasciitis and streptococcal toxic shock syndrome. Dextromethorphan (DM), a dextrorotatory morphinan and a widely used antitussive drug, has recently been reported to possess anti-inflammatory properties. In this study, we investigated the potential protective effect of DM in GAS infection using an air pouch infection mouse model. Our results showed that DM treatment increased the survival rate of GAS-infected mice. Bacterial numbers in the air pouch were lower in mice treated with DM than in those infected with GAS alone. The bacterial elimination efficacy was associated with increased cell viability and bactericidal activity of air-pouch-infiltrating cells. Moreover, DM treatment prevented bacterial dissemination in the blood and reduced serum levels of the proinflammatory cytokines interleukin-6 (IL-6), tumor necrosis factor alpha (TNF-α), and IL-1β and the chemokines monocyte chemotactic protein 1 (MCP-1), macrophage inflammatory protein 2 (MIP-2), and RANTES. In addition, GAS-induced mouse liver injury was reduced by DM treatment. Taken together, DM can increase bacterial killing and reduce inflammatory responses to prevent sepsis in GAS infection. The consideration of DM as an adjunct treatment in combination with antibiotics against bacterial infection warrants further study.
Group A streptococcus (GAS) is an important human pathogen that causes a wide spectrum of diseases, ranging from noninvasive infections such as pharyngitis, impetigo, and scarlet fever to invasive infections such as streptococcal toxic shock syndrome and necrotizing fasciitis. Immune-mediated postinfectious sequelae may also occur (2, 5, 6, 21, 27, 36-38). An epidemiological survey revealed that GAS causes high rates of morbidity and mortality in severe infections (3). Although there have been major advances in our understanding of the molecular mechanisms of host and GAS interactions, limited therapeutic progress has been made, and no vaccine is available (7, 21, 43).
Despite the availability of effective antimicrobial agents, an increase in the incidence of severe, invasive GAS infection has been noted worldwide in recent years. One of the possible reasons that GAS can persist in human populations is because it produces a number of virulence factors to either escape from host immunity or trigger hyperactive responses of the immune system (14, 34). Each virulence factor has a unique characteristic interaction with the host immune system and can facilitate colonization, invasion, immune evasion, and/or the long-term survival of GAS within the host (23, 28). GAS invasion may trigger apoptotic cell death in epithelial cells (22, 39), which is mediated by reactive oxygen species (ROS) production (1). Clinical studies indicate that patients suffering from severe invasive infection have higher levels of inflammatory cytokines than do those suffering from noninvasive infection (24-26, 29, 30). In addition to conventional antibacterial treatment, enhancing bacterial clearance and reducing excessive inflammatory responses may be of therapeutic benefit in invasive GAS infection.
Dextromethorphan (DM), an over-the-counter antitussive drug, is the d-isomer of the codeine analog levorphanol, a dextrorotatory morphinan. DM has been reported to be neuroprotective through acting as an N-methyl-d-aspartic acid (NMDA) receptor antagonist to prevent glutamate excitatory toxicity (4, 9). In addition, recent studies indicated that DM has an anti-inflammatory property. DM was shown to reduce the overactivation of microglia induced by lipopolysaccharide (LPS) to protect the degeneration of dopaminergic neurons (18, 44). Moreover, DM can improve endothelial function and attenuate vascular oxidative stress and inflammation markers in habitual smokers (16). DM can also reduce cytokine and superoxide production in macrophages via the inhibition of NADPH oxidase, leading to a decrease of atherosclerosis and neointima formation in mice (17). In an LPS-induced endotoxic shock mouse model, DM protected against hepatotoxicity by reducing the production of tumor necrosis factor alpha (TNF-α) and ROS (15). However, the effects of DM on Gram-positive bacterial infections have never been demonstrated.
In the present study, the potential for DM protection against Gram-positive GAS was evaluated in an air pouch infection mouse model. Our results showed that DM treatment reduced the mortality of GAS-infected mice. The protective effect of DM was correlated with enhancing bacterial clearance and reducing the systemic inflammatory response and organ injury.
MATERIALS AND METHODS
Mice.
BALB/c mice were obtained from Jackson Laboratories, Bar Harbor, ME, and maintained on standard laboratory food and water in the Laboratory Animal Center of National Cheng Kung University Medical College. Their progeny, ranging from 8 to 10 weeks of age, were used for the experiments. Housing, breeding, and experimental use of the animals were performed in strict accordance with the Experimental Animal Committee of National Cheng Kung University.
Bacteria.
GAS NZ131 (type M49, T14) was a gift from D. R. Martin, New Zealand Communicable Disease Center, Porirua, New Zealand. A fresh colony was inoculated into tryptic soy broth containing 0.5% yeast extract (TSBY) (Difco Laboratories, Detroit, MI) and cultured for 8 h at 37°C. The bacteria were harvested by centrifugation and resuspended in sterile phosphate-buffered saline (PBS). The bacterial density was determined by measuring the absorbance at 600 nm (A600) with a spectrophotometer (Beckmen Instruments, Somerset, NJ). The bacterial suspension was then diluted with PBS to 109 CFU/ml, using a standard growth curve to relate the measured A600 to the bacterial concentration.
Air pouch infection model.
The air pouch infection model was previously described (12, 13). Since infections of the skin and soft tissue are common primary foci of severe GAS disease, and air pouch exudates and cells can be conveniently collected for assays, we adopted the subcutaneous air pouch model to mimic skin and soft tissue infections. Mice were anesthetized by ether inhalation and then injected subcutaneously with 2 ml of air to form an air pouch. The bacterial suspension (0.3 ml) was inoculated into the air pouch. DM (12.5 mg/kg of body weight; Sigma, St. Louis, MO) was injected intraperitoneally into mice 30 min before and 1, 12, and 24 h after GAS inoculation. Multiple administrations of DM were given to evaluate the protective effects based on previous studies of endotoxic shock and 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) Parkinson's disease mouse models (15, 44), and the dose of DM was chosen based on a previous report (15). Mice in the control group received an equal volume of saline. Mouse survival rates were monitored daily for 14 days.
Bacterial counts in the air pouch and blood.
After mice were inoculated with GAS for various time periods, bacteria in the air pouch were collected by the injection of 1 ml PBS into the air pouch and aspiration of the exudates. Blood was collected from the heart. To quantify bacterial numbers, air pouch exudates and blood samples were diluted 1:10, plated onto TSBY plates, and incubated at 37°C for 48 h.
Infiltrating cell number and viability in the air pouch.
After mice were inoculated with GAS for various time periods, infiltrating cells in the air pouch were collected by the injection of 1 ml PBS into the air pouch and aspiration of the exudates. Cell numbers in the air pouch were counted, and cell viability was determined by trypan blue staining. For apoptotic cell detection, infiltrating cells were fixed with 70% ethanol for 24 h. The apoptotic cells were detected by using propidium iodide (PI) staining and analyzed by flow cytometry (FACSCalibur; BD Biosciences, San Jose, CA).
Viable phagocytosed bacterial cell counts in mouse air pouch infiltrating cells and human neutrophils.
The numbers of viable phagocytosed bacteria in the infiltrating cells from air pouch exudates of GAS-infected mice or in neutrophils from human peripheral blood were determined. The infiltrating cells collected from air pouch exudates were treated with 5 μg/ml of penicillin and 50 μg/ml of gentamicin to remove extracellular GAS and then centrifuged with PBS twice. Ice-cold distilled water was added for 15 min for the lysis of infiltrating cells. To measure the total number of intracellular GAS, the supernatants were plated onto TSBY plates and incubated at 37°C for 48 h. The neutrophils from human peripheral blood were prepared by using dextran sedimentation and Ficoll-Hypaque density gradient separation, followed by the hypotonic lysis of erythrocytes. The neutrophils were infected with GAS (multiplicity of infection of 10) for 1 h with or without 1.5 h of DM pretreatment. In order to remove extracellular GAS, penicillin (5 μg/ml) and gentamicin (50 μg/ml) were added at 37°C for 1 h. The neutrophil lysate was plated onto TSBY plates to count the total number of intracellular GAS.
Phagocytosis.
Freshly harvested viable GAS was labeled by using the PKH67 green fluorescent cell linker minikit (Sigma-Aldrich, St. Louis, MO). Briefly, 5 × 108 bacteria were stained and washed according to the manufacturer's instructions and resuspended in 0.5 ml of PBS. Staining with this dye has no detrimental effect on bacterial viability and growth. Purified human neutrophils were pretreated with or without DM at 37°C for 1.5 h and then infected with fluorescein isothiocyanate (FITC)-labeled GAS at 37°C for 1 h. The phagocytic activity of neutrophils was determined by using flow cytometry.
Determination of cytokine and chemokine levels in sera.
Mouse sera were collected, and the concentrations of TNF-α, interleukin-1β (IL-1β), IL-6, monocyte chemotactic protein 1 (MCP-1), macrophage inflammatory protein 2 (MIP-2), and regulated upon activation in normal T cells expressed and secreted (RANTES) were measured by using enzyme-linked immunosorbent assay (ELISA) kits (R&D Systems, Minneapolis, MN) according to the manufacturer's instructions.
Histopathology.
Mouse liver tissues were fixed in 10% neutral-buffered formalin solution and then dehydrated in graded alcohol. The fixed tissues were embedded in paraffin and sliced into 4-mm-thick sections. The tissue sections were mounted onto glass slides and stained with hematoxylin and eosin.
Determination of AST levels in the sera.
Mouse sera were collected, and the concentrations of aspartate aminotransferase (AST) were detected by using the Vitros 950 chemistry analyzer at the Department of Pathology, National Cheng Kung University Hospital.
Statistical analysis.
The statistical analysis was done with Prism 4.0 software (GraphPad Software, San Diego, CA). A comparison of the survival curves was performed by the Mantel-Haenszel log rank test. Significant differences between treatment groups were determined by using the unpaired t test. a P value of <0.05 was considered significant.
RESULTS
DM increases survival of GAS-infected mice.
To evaluate the protective effect of DM in GAS infection, BALB/c mice were inoculated in the air pouch with a lethal dose of NZ131 (3 × 108 CFU) with or without DM treatment (12.5 mg/kg) 30 min before and 1, 12, and 24 h after bacterial infection. After 14 days, the survival rate of GAS-infected mice was only 10%. In contrast, the DM-treated, GAS-infected mice showed approximately 72% survival (Fig. 1).
FIG. 1.

Dextromethorphan increases the survival rate of GAS-infected mice. Dextromethorphan (12.5 mg/kg) was intraperitoneally administrated to mice 30 min before and 1, 12, and 24 h after NZ131 inoculation (3 × 108 CFU) in the air pouch. The mouse survival rate was determined for 14 days (***, P < 0.001).
DM reduces bacterial counts in the air pouch and prevents bacteremia.
Bacterial numbers in the air pouch were determined to verify the protective effect of DM on GAS infection. Bacterial numbers in the air pouch were not significantly different between DM-treated and untreated mice 24 h after infection. By 48 h, the DM-treated group had lower bacterial numbers in the air pouch than did the untreated group (Fig. 2). Bacterial dissemination was further determined by the presence of bacteria in the blood. The percentage of positive blood cultures from untreated mice was 33% after 48 h of infection, and there was no positive blood culture from live DM-treated mice (Table 1). In addition, four of the untreated mice and two of the DM-treated mice died by 48 h. These results suggested that DM administration reduced the bacterial load in the local infection site and prevented bacterial dissemination to the circulation.
FIG. 2.

Bacterial numbers in the air pouch exudates from GAS-infected mice with or without dextromethorphan treatment. Dextromethorphan (12.5 mg/kg) was intraperitoneally administrated to mice 30 min before and 1, 12, and 24 h after NZ131 inoculation (3 × 108 CFU) in the air pouch. Mice were sacrificed 24 h (n = 10 per group of GAS with saline and n = 9 per group of GAS with DM) and 48 h (n = 11 per group of GAS alone and n = 12 per group of GAS with DM) after NZ131 infection, and the air pouch exudates were collected. The bacterial numbers were determined by plate counting (*, P < 0.05).
TABLE 1.
Bacterial dissemination in the blood 48 h after GAS infection with or without dextromethorphan treatment
| Treatment | No. of mice with bacteria in blood culture | No. of mice without bacteria in blood culture | No. of dead mice | No. of mice with bacteria in blood culture/total no. of mice |
|---|---|---|---|---|
| Saline | 4a | 8 | 4 | 4/12 |
| DM | 0 | 14 | 2 | 0/14 |
Bacterial numbers in the blood of these four mice were 2.1 × 103, 1.0 × 103, 2.0 × 102, and 6.0 × 101 CFU/ml, respectively.
DM increases infiltrating cell viability, which is associated with reduced cell apoptosis.
The mechanism of the DM-mediated antibacterial effect was then studied. When DM was added to bacterial cultures, no bactericidal effect on or inhibition of bacterial growth was observed, which indicated that the protection was not due to a direct antibacterial activity of DM (data not shown). Furthermore, DM had no effect on streptococcal pyrogenic exotoxin B (SPE B) production, which is an important virulence factor of GAS (data not shown). We previously observed the recruitment of phagocytes into mouse air pouches infected with NZ131 (13). We herein detected the effect of DM on infiltrating cell numbers, which is related to bacterial clearance. The results showed that DM treatment did not influence the numbers of infiltrating cells after 24 or 48 h of infection with NZ131 (Fig. 3 A). We further checked for the viability of these infiltrating cells. Although the viability of infiltrating cells continually dropped (at 24 h, 81.4% ± 1.7% and 87.4% ± 1.0% for the untreated and DM-treated groups, respectively; at 48 h, 17.8% ± 11.1% and 47.5% ± 7.5% for the untreated and DM-treated groups, respectively), the cell viability remained higher in DM-treated mice than in untreated mice by 48 h after infection (Fig. 3B). At 24 h, there was also a statistically significant difference between untreated and DM-treated groups (P = 0.0061). In this infection model, neutrophils were the predominant cell type of infiltrating cells detected in air pouch exudates (13; data not shown). A previously reported study indicated that GAS infection rapidly induced the apoptosis of neutrophils (11). We found that DM treatment reduced the percentages of apoptosis of infiltrating cells (Fig. 3C and D).
FIG. 3.

Dextromethorphan increases the viability and reduces the apoptosis of infiltrating cells in the air pouch of GAS-infected mice. Mice were inoculated with 3 × 108 CFU of NZ131 with or without the administration of DM 30 min before and 1, 12, and 24 h after NZ131 infection. The air pouch exudates were collected 24 and 48 h (A and B) or 48 h (C and D) after NZ131 infection. The infiltrating cell numbers (A), cell viability determined by trypan blue staining (B) (at 24 h, n = 9 per group without or with DM treatment; at 48 h, n = 5 per group of GAS with saline and n = 7 per group of GAS with DM), and apoptosis determined by PI staining (C and D) were determined. A set of representative histograms is shown (C), and means ± standard deviations (SD) of air pouch exudates from six mice per group are shown (D) (*, P < 0.05; **, P < 0.01).
DM reduces viable bacterial numbers in infiltrating cells.
We next determined the numbers of viable bacteria in air pouch-infiltrating cells. At 36 h after NZ131 infection, the infiltrating cell numbers and viabilities were similar in the groups with and those without DM treatment (Fig. 4 A and B). Nevertheless, counts of viable phagocytosed bacteria at 36 h postinfection were lower for the DM-treated group than for the untreated group (Fig. 4C). Low bacterial numbers in air pouches of DM-treated mice were observed at 36 h after infection compared with untreated mice (with averages of 1.25 × 109 ± 0.44 × 109 bacteria for the untreated group versus 0.49 × 109 ± 0.19 × 109 bacteria for the DM-treated group) (see Table S1 in the supplemental material). At 36 h after NZ131 inoculation, the number of positive blood cultures was 4 of 5 live untreated mice (80%) and 0 of 7 live DM-treated mice. Two of the untreated mice and one of DM-treated mice died by 36 h after NZ131 infection (see Table S1 in the supplemental material). Bacterial counts in various organs were determined, and the results showed that all except one mouse in the untreated mouse group had detectable bacteria present in the kidney, and only one DM-treated mouse had a low bacterial count present in the kidney. For the liver and spleen, all untreated mice but not DM-treated mice had detectable bacteria in these organs (see Table S1 in the supplemental material). This suggested a more effective bacterial clearance ability at the local infection site with DM treatment than without DM treatment.
FIG. 4.
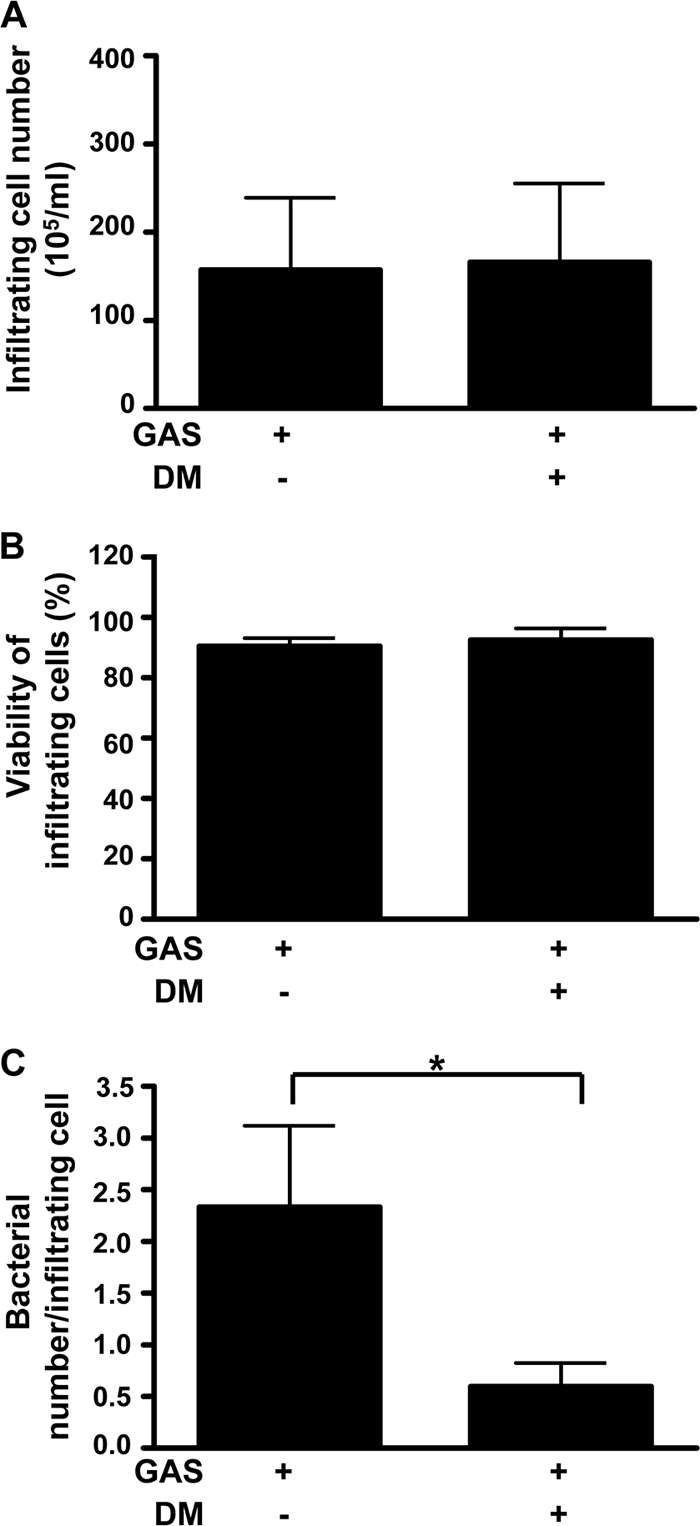
Dextromethorphan reduces viable bacterial numbers in infiltrating cells in the air pouch of GAS-infected mice. Mice were inoculated with 3 × 108 CFU of NZ131 with or without the administration of DM 30 min before and 1, 12, and 24 h after NZ131 infection (n = 5 per group of GAS with saline and n = 7 per group of GAS with DM). The air pouch exudates were collected 36 h after NZ131 infection. The infiltrating cell numbers (A), cell viability (B), and bacterial numbers in infiltrating cells (C) were determined (*, P < 0.05). The experiments were performed three times, and one set of representative data is shown.
Because neutrophils were the predominant type of infiltrating cells, we therefore used human neutrophils to check for the effects of DM. Purified human neutrophils were pretreated with or without DM for 1.5 h and then incubated with FITC-labeled NZ131 for 1 h, followed by flow cytometric analysis. The results showed that DM increased the phagocytic capability of neutrophils (Fig. 5 A). In a bactericidal assay, human neutrophils were infected with NZ131 for 1 h with or without 1.5 h of DM pretreatment, followed by an additional 1 h of incubation with antibiotics to remove unengulfed bacteria, and the numbers of viable intracellular bacteria were determined. The results showed that DM actually increased the bactericidal activity of human neutrophils (Fig. 5B). This is consistent with our findings with the mouse model showing more efficient bacterial clearance by neutrophils with DM treatment.
FIG. 5.
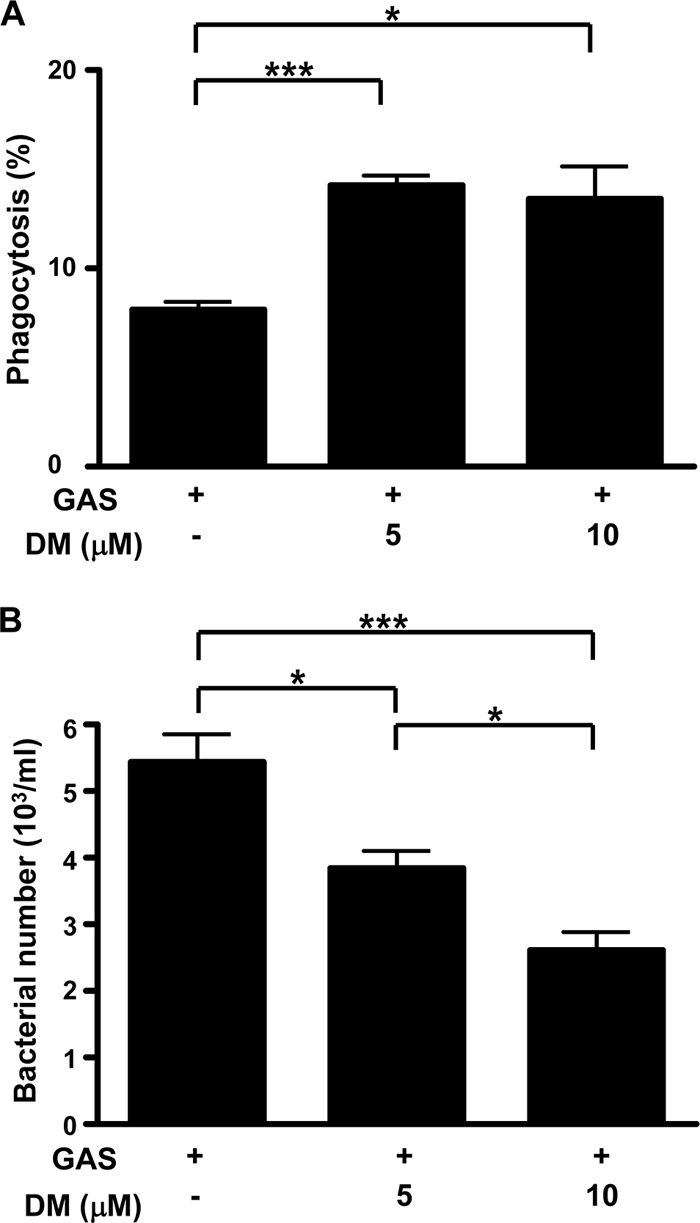
Phagocytic and bactericidal activities of human neutrophils are increased when pretreated with dextromethorphan. Human neutrophils were cultured with FITC-labeled NZ131 at 37°C for 1 h without or with 5 and 10 μM DM pretreatment. The phagocytic activity (A) and bacterial numbers in the cells (B) were determined for triplicate cultures (*, P < 0.05; ***, P < 0.001). The experiments were performed twice, and one set of representative data is shown.
DM reduces GAS-induced systemic inflammation and organ damage.
A systemic inflammatory response and liver injury were determined to confirm the protection of DM on GAS infection. Previous studies reported that invasive GAS infection caused an excess production of inflammatory cytokines, which correlated with disease severity and organ failure (24, 25). We compared the serum levels of inflammatory cytokines in GAS-infected mice with and those without DM treatment at 48 h after bacterial inoculation. By that time, GAS dissemination to the systemic organs was observed. Lower levels of proinflammatory cytokines (i.e., IL-6, TNF-α, and IL-1β) (Fig. 6 A) and chemokines (i.e., MCP-1, MIP-2, and RANTES) (Fig. 6B) were detected in the sera of DM-treated mice than in the sera of untreated mice following GAS infection. In our previous study, liver histopathological changes occurred in GAS-infected mice (12). The pathology of hepatocytes was fatty degeneration in which the cytoplasm was filled with foamy vacuoles of fat. Histological examination of liver tissue obtained 48 h after GAS infection showed hepatocyte damage after GAS infection (see Fig. S1B and S1E in the supplemental material), which was inhibited by DM treatment (see Fig. S1C and S1F in the supplemental material). To further confirm the protective effect of DM against liver injury, we found that the serum levels of AST were lower in DM-treated mice than in mice treated with GAS alone (Fig. 7).
FIG. 6.

Dextromethorphan reduces inflammatory cytokine and chemokine levels in sera of GAS-infected mice. Mice were inoculated with 3 × 108 CFU of NZ131 with or without the administration of DM 30 min before and 1, 12, and 24 h after NZ131 infection (n = 6 per group of GAS with saline and n = 7 per group of GAS with DM). The sera were collected 48 h after NZ131 infection, and the amounts of inflammatory cytokines (A) and chemokines (B) were determined by ELISA (*, P < 0.05; **, P < 0.01).
FIG. 7.
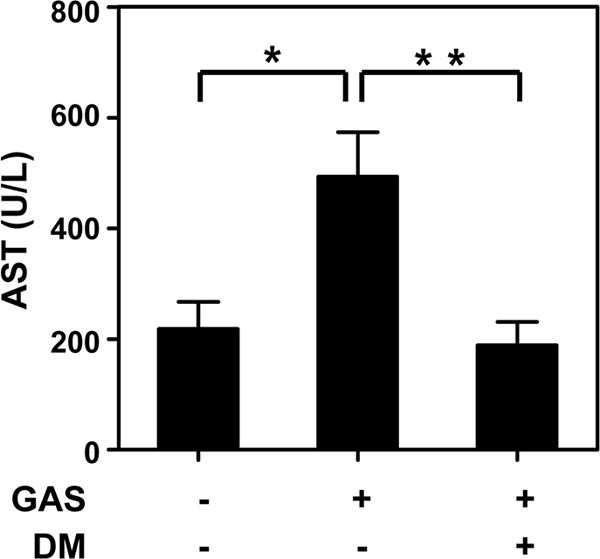
Dextromethorphan reduces AST levels in sera of GAS-infected mice. Mice were inoculated with 3 × 108 CFU of NZ131 with or without the administration of DM 30 min before and 1, 12, and 24 h after NZ131 infection. The sera were collected 48 h after NZ131 infection for AST measurements (n = 5 for the untreated group, n = 6 for the GAS-alone group, and n = 6 for GAS and DM group) (*, P < 0.05; **, P < 0.01).
DISCUSSION
Although sepsis is a major cause of death for severely infected patients, efficient treatment has been lacking. We therefore aimed to develop therapeutic interventions using small molecules for this disease. DM is a dextrorotatory morphinan and a widely used antitussive drug, which was recently found to possess anti-inflammatory properties and to provide protection in an LPS-induced endotoxic shock mouse model (15). This study further demonstrates the efficacy and novel mechanisms of DM in protecting mice against Gram-positive GAS-elicited sepsis. We believe that this live bacterial infection model represents a useful system for studying the clinical septic syndrome.
A previously reported study showed that the administration of carboxyfullerene was able to protect mice from GAS-induced death. Surveys of exudates of the air pouch of carboxyfullerene-treated mice revealed that the survival of infiltrating neutrophils was prolonged and that the bacteria were eliminated as a result of the enhanced bactericidal activity of neutrophils (41). We have also found a prolonged survival of neutrophils, which correlated with a DM-mediated inhibition of neutrophil apoptosis. In addition, DM also enhanced the bacterial clearance ability of neutrophils. However, unlike the effect of carboxyfullerene, which directly inhibits the in vitro growth of GAS, DM does not cause a direct effect on bacterial growth.
For the detection of DM effects on innate immunity, we found that DM inhibits cytokine and chemokine expression while increasing the phagocytic activity of neutrophils. At 36 h after GAS infection, infiltrating neutrophil numbers and viabilities were similar between DM-treated and untreated groups of mice. However, there were reduced bacterial counts in infiltrating neutrophils in the DM-treated group at that time. Because lower bacterial counts in the air pouches of DM-treated mice were observed after 36 h of infection with NZ131, we cannot exclude the possibility that the lower bacterial numbers in the air pouch at least partially accounted for the reduced bacterial counts in infiltrating neutrophils. Nevertheless, in vitro studies showed that DM increased the bacterial clearance activity of human neutrophils. This, taken together with the fact that no or few bacteria were detected in the blood and organs of DM-treated mice, supports the notion of a DM-enhanced bacterial killing efficacy.
At 48 h, the neutrophil viability was markedly decreased, which correlated with apoptotic cell death, whereas DM inhibited this effect. These results indicate that the enhanced bacterial clearance of neutrophils by DM is associated with at least two factors: one is the prolonged survival of infiltrating neutrophils, and the other is the increased phagocytic activity of neutrophils. Neutrophils undergo apoptosis after phagocytosis, a process also known as phagocytosis-induced cell death (PICD) (10). Many bacteria have been shown to trigger neutrophil apoptosis or PICD, including GAS. To survive and persist within the host, GAS has developed a number of strategies to circumvent the host immune system, such as virulence factors directed to prevent phagocytosis or enhance survival within phagocytic cells and induce neutrophil apoptosis (20, 35, 42). In our study, DM was found to inhibit the apoptosis of GAS-induced infiltrating neutrophils. However, the precise mechanisms of the DM-mediated inhibition of GAS-induced apoptosis and the increase in the phagocytic activity remain to be determined. Neutrophils destroy pathogens through a combination of mechanisms, such as ROS production and the accumulation of cytotoxic molecules in the phagosomes (10, 31, 32). However, previous studies showed that DM decreases ROS at least in part through the inhibition of NADPH oxidase (15, 17, 44). The neutrophil bacterial clearance ability enhanced by DM is therefore less likely to be attributable to ROS action. Although detailed mechanisms need to be further resolved, we speculate that DM-induced prolonged survival and the decreased apoptosis of neutrophils may be related to its inhibition of ROS production.
Elevated levels of inflammatory cytokines may be associated with morbidity in patients with severe invasive GAS infection. In our model, the results showed that the levels of proinflammatory cytokines and chemokines in mouse sera were increased after GAS infection. The increased levels of inflammatory cytokines in the sera of GAS-infected mice most likely reflect the sustained systemic infection occurring in these animals. DM has an anti-inflammatory effect through reducing the levels of ROS and proinflammatory cytokines, such as TNF-α and IL-6 (15, 18). In the present study, we showed that DM reduced GAS-induced inflammatory cytokines (TNF-α, IL-1β, and IL-6) and chemokines (MCP-1, MIP-2, and RANTES) in a mouse model. It was previously reported that GAS can induce NF-κB activation in epithelial cells (19, 40). Because ROS may regulate the activation of NF-κB (8), it remains to be clarified whether the reduced ROS caused by DM result in the inhibition of GAS-induced NF-κB activation.
A previously reported study used an LPS-induced mouse model to investigate the DM effect (15). This is the first study investigate the effect of DM on bacterial infection in an animal model, using GAS as a Gram-positive bacterium for the study. The possible protective effect of DM against other bacteria remains to be investigated. Although infecting bacteria can be taken care of by antibiotics, the bacterial components released from dead bacteria, such as peptidoglycans, lipoproteins, lipoteichoic acids, and LPS, may be responsible for systemic inflammation and cause organ failure and sepsis. Our results showed that DM reduces the inflammatory response to protect mice against systemic GAS infection, which suggests that DM can be considered an adjunct agent for antimicrobial treatment. Combined with antimicrobial agents, the dosage of DM may be reduced comparably to the recommended doses as a cough suppressant (33), but this issue needs to be tested.
Supplementary Material
Acknowledgments
This work was supported by grant NHRI-EX95-9429SP from the National Health Research Institutes, Taiwan. This research was supported in part by the Intramural Research Program of the NIEHS, NIH.
We thank Robert Anderson for critical reading of the manuscript.
Footnotes
Published ahead of print on 3 January 2011.
Supplemental material for this article may be found at http://aac.asm.org/.
REFERENCES
- 1.Aikawa, C., et al. 2010. Reactive oxygen species induced by Streptococcus pyogenes invasion trigger apoptotic cell death in infected epithelial cells. Cell. Microbiol. 12:814-830. [DOI] [PubMed] [Google Scholar]
- 2.Bisno, A. L., M. O. Brito, and C. M. Collins. 2003. Molecular basis of group A streptococcal virulence. Lancet Infect. Dis. 3:191-200. [DOI] [PubMed] [Google Scholar]
- 3.Carapetis, J. R., A. C. Steer, E. K. Mulholland, and M. Weber. 2005. The global burden of group A streptococcal diseases. Lancet Infect. Dis. 5:685-694. [DOI] [PubMed] [Google Scholar]
- 4.Choi, D. W. 1987. Dextrorphan and dextromethorphan attenuate glutamate neurotoxicity. Brain Res. 403:333-336. [DOI] [PubMed] [Google Scholar]
- 5.Cleary, P. P. 2006. Streptococcus moves inward. Nat. Med. 12:384-386. [DOI] [PubMed] [Google Scholar]
- 6.Cunningham, M. W. 2000. Pathogenesis of group A streptococcal infections. Clin. Microbiol. Rev. 13:470-511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Currie, B. J. 2006. Group A streptococcal infections of the skin: molecular advances but limited therapeutic progress. Curr. Opin. Infect. Dis. 19:132-138. [DOI] [PubMed] [Google Scholar]
- 8.Gloire, G., S. Legrand-Poels, and J. Piette. 2006. NF-κB activation by reactive oxygen species: fifteen years later. Biochem. Pharmacol. 72:1493-1505. [DOI] [PubMed] [Google Scholar]
- 9.Haberecht, M. F., C. K. Mitchell, G. J. Lo, and D. A. Redburn. 1997. N-Methyl-D-aspartate-mediated glutamate toxicity in the developing rabbit retina. J. Neurosci. Res. 47:416-426. [PubMed] [Google Scholar]
- 10.Kennedy, A. D., and F. R. DeLeo. 2009. Neutrophil apoptosis and the resolution of infection. Immunol. Res. 43:25-61. [DOI] [PubMed] [Google Scholar]
- 11.Kobayashi, S. D., et al. 2003. Bacterial pathogens modulate an apoptosis differentiation program in human neutrophils. Proc. Natl. Acad. Sci. U. S. A. 100:10948-10953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kuo, C. F., et al. 2004. Histopathologic changes in kidney and liver correlate with streptococcal pyrogenic exotoxin B production in the mouse model of group A streptococcal infection. Microb. Pathog. 36:273-285. [DOI] [PubMed] [Google Scholar]
- 13.Kuo, C. F., et al. 1998. Role of streptococcal pyrogenic exotoxin B in the mouse model of group A streptococcal infection. Infect. Immun. 66:3931-3935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kurupati, P., et al. 2010. Chemokine-cleaving Streptococcus pyogenes protease SpyCEP is necessary and sufficient for bacterial dissemination within soft tissues and the respiratory tract. Mol. Microbiol. 76:1387-1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li, G., et al. 2005. Protective effect of dextromethorphan against endotoxic shock in mice. Biochem. Pharmacol. 69:233-240. [DOI] [PubMed] [Google Scholar]
- 16.Liu, P. Y., et al. 2008. Treatment with dextromethorphan improves endothelial function, inflammation and oxidative stress in male heavy smokers. J. Thromb. Haemost. 6:1685-1692. [DOI] [PubMed] [Google Scholar]
- 17.Liu, S. L., et al. 2009. Dextromethorphan reduces oxidative stress and inhibits atherosclerosis and neointima formation in mice. Cardiovasc. Res. 82:161-169. [DOI] [PubMed] [Google Scholar]
- 18.Liu, Y., et al. 2003. Dextromethorphan protects dopaminergic neurons against inflammation-mediated degeneration through inhibition of microglial activation. J. Pharmacol. Exp. Ther. 305:212-218. [DOI] [PubMed] [Google Scholar]
- 19.Medina, E., D. Anders, and G. S. Chhatwal. 2002. Induction of NF-κB nuclear translocation in human respiratory epithelial cells by group A streptococci. Microb. Pathog. 33:307-313. [DOI] [PubMed] [Google Scholar]
- 20.Medina, E., O. Goldmann, A. W. Toppel, and G. S. Chhatwal. 2003. Survival of Streptococcus pyogenes within host phagocytic cells: a pathogenic mechanism for persistence and systemic invasion. J. Infect. Dis. 187:597-603. [DOI] [PubMed] [Google Scholar]
- 21.Musser, J. M., and F. R. DeLeo. 2005. Toward a genome-wide systems biology analysis of host-pathogen interaction in group A streptococcus. Am. J. Pathol. 167:1461-1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nakagawa, I., M. Nakata, S. Kawabata, and S. Hamada. 2001. Cytochrome c-mediated caspase-9 activation triggers apoptosis in Streptococcus pyogenes-infected epithelial cells. Cell. Microbiol. 3:395-405. [DOI] [PubMed] [Google Scholar]
- 23.Nizet, V. 2007. Understanding how leading bacterial pathogens subvert innate immunity to reveal novel therapeutic targets. J. Allergy Clin. Immunol. 120:13-22. [DOI] [PubMed] [Google Scholar]
- 24.Norrby-Teglund, A., et al. 2000. Host variation in cytokine responses to superantigens determine the severity of invasive group A streptococcal infection. Eur. J. Immunol. 30:3247-3255. [DOI] [PubMed] [Google Scholar]
- 25.Norrby-Teglund, A., K. Pauksens, M. Norgren, and S. E. Holm. 1995. Correlation between serum TNF alpha and IL6 levels and severity of group A streptococcal infections. Scand. J. Infect. Dis. 27:125-130. [DOI] [PubMed] [Google Scholar]
- 26.Norrby-Teglund, A., et al. 2001. Evidence for superantigen involvement in severe group A streptococcal tissue infection. J. Infect. Dis. 186:853-860. [DOI] [PubMed] [Google Scholar]
- 27.O'Connell, D. 2006. Bacterial pathogenesis: triggering GAS invasive disease. Nat. Rev. Microbiol. 4:644-645. [Google Scholar]
- 28.Olsen, R. J., S. A. Shelburne, and J. M. Musser. 2009. Molecular mechanisms underlying group A streptococcal pathogenesis. Cell. Microbiol. 11:1-12. [DOI] [PubMed] [Google Scholar]
- 29.Pahlman, L. I., et al. 2008. Soluble M1 protein of Streptococcus pyogenes triggers potent T cell activation. Cell. Microbiol. 10:404-414. [DOI] [PubMed] [Google Scholar]
- 30.Proft, T., and J. D. Fraser. 2003. Bacterial superantigens. Clin. Exp. Immunol. 133:299-306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Quinn, M. T., M. C. Ammons, and F. R. DeLeo. 2006. The expanding role of NADPH oxidases in health and disease: no longer just agents of death and destruction. Clin. Sci. (Lond.) 111:1-20. [DOI] [PubMed] [Google Scholar]
- 32.Rosen, H., and S. J. Klebanoff. 1979. Bactericidal activity of a superoxide anion-generating system. A model for the polymorphonuclear leukocyte. J. Exp. Med. 149:27-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Siu, A., and R. Drachtman. 2007. Dextromethorphan: a review of N-methyl-D-aspartate receptor antagonist in the management of pain. CNS Drug Rev. 13:96-106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Smeesters, P. R., D. J. McMillan, and K. S. Sriprakash. 2010. The streptococcal M protein: a highly versatile molecule. Trends Microbiol. 18:275-282. [DOI] [PubMed] [Google Scholar]
- 35.Staali, L., S. Bauer, M. Morgelin, L. Bjorck, and H. Tapper. 2006. Streptococcus pyogenes bacteria modulate membrane traffic in human neutrophils and selectively inhibit azurophilic granule fusion with phagosomes. Cell. Microbiol. 8:690-703. [DOI] [PubMed] [Google Scholar]
- 36.Stevens, D. L. 1999. The flesh-eating bacterium: what's next? J. Infect. Dis. 179:S366-S374. [DOI] [PubMed] [Google Scholar]
- 37.Stevens, D. L. 2000. Streptococcal toxic shock syndrome associated with necrotizing fasciitis. Annu. Rev. Med. 51:271-288. [DOI] [PubMed] [Google Scholar]
- 38.Tart, A. H., M. J. Walker, and J. M. Musser. 2007. New understanding of the group A streptococcus pathogenesis cycle. Trends Microbiol. 15:318-325. [DOI] [PubMed] [Google Scholar]
- 39.Tsai, P. J., Y. S. Lin, C. F. Kuo, H. Y. Lei, and J. J. Wu. 1999. Group A streptococcus induces apoptosis in human epithelial cells. Infect. Immun. 67:4334-4339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tsai, P. J., et al. 2006. Streptococcus pyogenes induces epithelial inflammatory responses through NF-κB/MAPK signaling pathways. Microbes Infect. 8:1440-1449. [DOI] [PubMed] [Google Scholar]
- 41.Tsao, N., et al. 2001. Inhibition of group A streptococcus infection by carboxyfullerene. Antimicrob. Agents Chemother. 45:1788-1793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Voyich, J. M., J. M. Musser, and F. R. DeLeo. 2004. Streptococcus pyogenes and human neutrophils: a paradigm for evasion of innate host defense by bacterial pathogens. Microbes Infect. 6:1117-1123. [DOI] [PubMed] [Google Scholar]
- 43.Zagursky, R. J., and A. S. Anderson. 2008. Application of genomics in bacterial vaccine discovery: a decade in review. Curr. Opin. Pharmacol. 8:632-638. [DOI] [PubMed] [Google Scholar]
- 44.Zhang, W., et al. 2004. Neuroprotective effect of dextromethorphan in the MPTP Parkinson's disease model: role of NADPH oxidase. FASEB J. 18:589-591. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.