

Abstract
DNA ligases are indispensable enzymes playing a critical role in DNA replication, recombination, and repair in all living organisms. Bacterial NAD+-dependent DNA ligase (LigA) was evaluated for its potential as a broad-spectrum antibacterial target. A novel class of substituted adenosine analogs was discovered by target-based high-throughput screening (HTS), and these compounds were optimized to render them more effective and selective inhibitors of LigA. The adenosine analogs inhibited the LigA activities of Escherichia coli, Haemophilus influenzae, Mycoplasma pneumoniae, Streptococcus pneumoniae, and Staphylococcus aureus, with inhibitory activities in the nanomolar range. They were selective for bacterial NAD+-dependent DNA ligases, showing no inhibitory activity against ATP-dependent human DNA ligase 1 or bacteriophage T4 ligase. Enzyme kinetic measurements demonstrated that the compounds bind competitively with NAD+. X-ray crystallography demonstrated that the adenosine analogs bind in the AMP-binding pocket of the LigA adenylation domain. Antibacterial activity was observed against pathogenic Gram-positive and atypical bacteria, such as S. aureus, S. pneumoniae, Streptococcus pyogenes, and M. pneumoniae, as well as against Gram-negative pathogens, such as H. influenzae and Moraxella catarrhalis. The mode of action was verified using recombinant strains with altered LigA expression, an Okazaki fragment accumulation assay, and the isolation of resistant strains with ligA mutations. In vivo efficacy was demonstrated in a murine S. aureus thigh infection model and a murine S. pneumoniae lung infection model. Treatment with the adenosine analogs reduced the bacterial burden (expressed in CFU) in the corresponding infected organ tissue as much as 1,000-fold, thus validating LigA as a target for antibacterial therapy.
DNA ligases join adjacent 3′-hydroxyl and 5′-phosphoryl termini to form a phosphodiester bond in duplex DNA (22, 38). DNA ligases function in DNA replication, by joining Okazaki fragments on the lagging strand of DNA, and are involved in several DNA repair pathways (e.g., nucleotide excision repair). DNA ligation proceeds in three nucleotidyl transfer steps. The first step involves the formation of a covalent DNA ligase-adenylate intermediate. In the second step, AMP is transferred from DNA ligase to the 5′ phosphate of nicked DNA through a pyrophosphate bond. In the third step, a phosphodiester bond is formed to join adjacent polynucleotides, and AMP is released (22, 38).
DNA ligases are grouped into two families based on the substrate (NAD+ or ATP) used to form the DNA ligase-adenylate intermediate. The bacterial NAD+-dependent DNA ligases belong to a distinct, highly conserved phylogenetic cluster of enzymes. In contrast, the ATP-dependent DNA ligases of mammals, fungi, and viruses form a loosely defined cluster of associated enzymes (32, 41). The bacterial NAD+-dependent DNA ligases are essential for viability in all Gram-positive and Gram-negative bacteria tested to date, including Bacillus subtilis, Staphylococcus aureus, Streptococcus pneumoniae, Salmonella enterica serovar Typhimurium, and Escherichia coli (12, 18, 23, 31, 33).
NAD+-dependent DNA ligase has attracted interest as a prospective broad-spectrum antibacterial target because it is indispensable for DNA replication, conserved among bacterial pathogens, and distinctly different from the eukaryotic DNA ligases (5, 6, 9, 10, 15, 23, 25). X-ray crystal structures have facilitated a better understanding of substrate binding and the catalytic mechanism of DNA ligases (14, 16, 28; S. Lahiri and S. D. Mills, U.S. patent application 2008/0262811). DNA ligase structures have also stimulated efforts to design small-molecule inhibitors of LigA activity. Novel inhibitors of LigA with selective antibacterial activity have been reported, but none of the studies have validated the target of the inhibitors in vivo by demonstrating efficacy in animal models of infection (6, 23).
Here we describe the discovery of novel substituted adenosine analogs that are selective inhibitors of NAD+-dependent DNA ligases from a broad spectrum of pathogenic bacteria. In vitro mode-of-inhibition and mode-of-action studies, as well as in vivo efficacy data, support the potential therapeutic value of LigA as an antibacterial target. To our knowledge, this is the first example of in vivo validation of LigA as an antibacterial target.
(This material was presented in part at the 49th Interscience Conference on Antimicrobial Agents and Chemotherapy, San Francisco, CA, 11 to 15 September 2009 [8, 13, 30, 39].)
MATERIALS AND METHODS
Strains and compounds.
The strains, plasmids, and DNA primers used in this study are described in Table 1. All commercially available antibiotics were obtained from Sigma-Aldrich, Inc. (St. Louis, MO). Adenosine analogs (Fig. 1) were prepared at AstraZeneca R&D Boston as previously described (M. Cavero-Tomas, M. Gowravaram, H. Huynh, H. Ni, and S. Stokes, 20 April 2006, international patent application WO/2006/040558). T4 DNA ligase was purchased from New England BioLabs (Ipswich, MA).
TABLE 1.
Strains, plasmids, and primers
| Strain, plasmid, or primer | Genotype, phenotype, use, or sequencea | Source or reference(s) |
|---|---|---|
| Strains | ||
| E. coli | ||
| W3110; ATCC 27325 | ATCC, Manassas, VA | |
| ARC523 | W3110 ΔtolC::Tn10 | AstraZeneca culture collection |
| MG1655; ATCC 700926 | F− λ− IlvG−rfb-50 rph-1 | ATCC, Manassas, VA |
| TOP10 | Invitrogen Corp., Carlsbad, CA | |
| BL21(DE3) | Invitrogen Corp., Carlsbad, CA | |
| B | Used to propagate T4 DNA according to ATCC protocol (11303-B4) | ATCC, Manassas, VA |
| H. influenzae | ATCC, Manassas, VA | |
| Rd KW20; ATCC 51907 | Wild-type parent | |
| ARM158 | Rd KW20 ΔacrB::cat; Cmr | AstraZeneca culture collection |
| HIN100 | Rd KW20 ΔacrB::aph3-A; Kmr | This study |
| HIN101 | Rd KW20 ΔacrB::aph3-A ΔompP1::cat; Kmr Cmr | This study |
| HIN102 | Rd KW20 ΔacrB::aph3-A ΔompP1::ligA + cat; Kmr Cmr | This study |
| S. pneumoniae | ||
| D39; NCTC 7466 | 2 | |
| Rx1 | Unencapsulated transformable derivative of R36 | 34 |
| R6; ATCC BAA-255 | Unencapsulated transformable derivative of R36 | 17 |
| SPN100 | Rx1 ΔgalU::ermR; Ermr | This study |
| SPN101 | Rx1 ΔgalU::T4 lig + ermR; Ermr | This study |
| SPN102 | Rx1 ΔgalU::T4 lig + ermR ΔligA::aph3-A; Ermr Kmr | This study |
| SPN103 | Spontaneous D39 mutant, resistant to compound 4; amino acid substitution in LigA (L75F) | This study |
| S. aureus RN4220 | Restriction-deficient mutant of strain 8325-4 | 20 |
| M. pneumoniae | ||
| M129; ATCC 29342 | ATCC, Manassas, VA | |
| FH; NCTC 10119 | NCTC, Salisbury, United Kingdom | |
| Candida albicans B2630 | 35 | |
| H. sapiens A549; ATCC CCL-185 | Human lung carcinoma cell line | ATCC, Manassas, VA |
| Plasmids and DNA | ||
| E. coli bacteriophage T4 | ATCC 11303-B4 | ATCC, Manassas, VA |
| H. sapiens cDNA | Human Universal QUICK-Clone cDNA; catalog no. 7109-1; lot no. 1050696 | Clontech, Mountain View, CA |
| pGEM-T | TA cloning vector; Apr | Promega Corp., Madison, WI |
| pCR4-TOPO | TOPO TA cloning vector; Kmr | Invitrogen Corp., Carlsbad, CA |
| pET21b | Protein expression vector | EMD4Biosciences, San Diego, CA |
| pET30 | Protein expression vector | EMD4Biosciences, San Diego, CA |
| pET28 | Protein expression vector | EMD4Biosciences, San Diego, CA |
| pUC18K | Source of nonpolar aph3-A gene in pUC18; Apr Kmr | 24 |
| pASK4 | Source of 1,238-bp cat cassette in pGEM-T: Apr Cmr | 36, 40 |
| pASK5 | Plasmid used for site-directed integration and expression of selected genes at H. influenzaeompP1 (HI0401) locus of Rd KW20; Apr Cmr | 36 |
| pBA467 | Plasmid used for site-directed integration and expression of selected genes at galU (spr1903) locus of S. pneumoniae R6; Apr Ermr | This study |
| pWY375 | pGEM-T containing H. influenzaeacrB (HI0895) deletion and insertion of 840 bp of nonpolar aph3-A cassette; Apr Kmr | This study |
| pLP109 | pASK5 containing H. influenzae ligA (HI1100) subcloned as NdeI-SalI fragment from pHIN-LigA; Apr Cmr | This study |
| pWY473 | pBA467 containing T4 lig subcloned as NdeI-SalI fragment from pT4-lig; Apr Ermr | This study |
| pLP112 | pGEM-T containing S. pneumoniaeligA (spr1024) deletion and insertion of 840 bp of nonpolar aph3-A cassette; Apr Kmr | This study |
| pT4-lig | pCR4-TOPO containing T4 lig; Kmr | This study |
| pHIN-LigA | H. influenzae LigA expression plasmid; pET30a; Kmr | This study |
| pSPN-LigA | S. pneumoniae LigA expression plasmid; pET30a; Kmr | This study |
| pECO-LigA | E. coli LigA expression plasmid; pET30a; Kmr | This study |
| pMPN-LigA | M. pneumoniae LigA expression plasmid; pET30a; Kmr | This study |
| pSAU-LigA | S. aureus LigA expression plasmid; pET30a; Kmr | This study |
| pHSA-LIGI | H. sapiens LIGI expression plasmid (N-terminal truncation and T7 tag); pET21b; Apr | This study |
| Primers | ||
| LigA protein expression | ||
| E. coli ligA-F (NdeI) | GCATTGATGGTGCCATATGGAATCAATCG | MG1655 |
| E. coli ligA-R (SalI) | CGTATTGGCTATTTCAGTCGACTGC | MG1655 |
| H. influenzae ligA-F (NdeI) | CCGAGAATCATATGACAAATATTCAAACTCAAC | KW20 |
| H. influenzae ligA-R (SalI) | GCCTGTCGACCCTGTCACACCAAGCGTCGG | KW20 |
| M. pneumoniae ligA-F (NdeI) | CCTACATATGGCAAAGGTCGCACAAATTCG | M129 |
| M. pneumoniae ligA-R (SalI) | GCATGTCGACTTAGGTCCAAATTGGTTC | M129 |
| S. aureus ligA-F (NcoI) | GGATTAAGCCATGGCTGATTTATCG | RN4220 |
| S. aureus ligA-R (SalI) | CGACGTCGACCTCTAACTATTTAATTC | RN4220 |
| S. pneumoniae ligA-F (NdeI) | GGTCTTATGAATAAACATATGAATGAGTTAGTCGCTTTGC | R6 |
| S. pneumoniae ligA-R (SalI) | CTCTGTCGACAAACGATCCATTACAAACTTTCTAGCC | R6 |
| Expression of other ligases | ||
| T4 lig-F (NdeI) | GCGCATATGATTCTTAAAATTCTGAACG | Bacteriophage T4 |
| T4 lig-R (SalI) | CTTGTCGACTCATAGACCAGTTACCTC | Bacteriophage T4 |
| H. sapiens lig-F (SacI) | GAGGAGAGCTCGGCTCCAGGAAAGGAGGGAGCTGCTGAGG | H. sapiens |
| H. sapiens lig-R (SalI) | GATCGTCGACTTAGTAGGTATCTTCAGGGTCAGAGCCTGAGTCC | H. sapiens |
Restriction enzyme sites in primer sequences are in boldface italics.
FIG. 1.

Chemical structures of the adenosine analogs discussed in this study. The 2-position of the adenine ring and the 5′ position of the ribose are indicated for compound 1.
Recombinant DNA methods.
The recombinant DNA methods and reagents used have been described previously (36). The DNAs of all PCR-generated clones and selected PCR-generated DNA fragments were sequenced using an ABI Prism genetic analyzer, model 3100, after the preparation of ABI Prism BigDye Terminator (version 3.1) cycle sequencing reactions according to the manufacturer's instructions (Life Technologies Corp., Carlsbad, CA). The resulting DNA sequence chromatographs were assembled and analyzed with Sequencher software (version 4.7; Gene Codes Corporation, Ann Arbor, MI). The strains, plasmids, and primers used for the overexpression of DNA ligases and for the characterization of the mode of action can be found in Table 1. The conditions for growth, cell preparation, and transformation are described in the supplemental material.
Cloning and expression of bacterial NAD+-dependent DNA ligase isozymes.
DNA fragments containing the ligA gene were PCR amplified using primers and template genomic DNA isolated from Haemophilus influenzae Rd KW20, S. pneumoniae R6, S. aureus RN4220, Mycoplasma pneumoniae M129, and E. coli MG1655 (Table 1). The ligA-containing PCR fragments were ligated into pGEM-T (Promega, Madison, WI) according to the manufacturer's instructions. The cloned DNA containing ligA from each strain listed above was sequenced and analyzed relative to the published gene sequence for each strain. The ligA-containing DNA from each organism was subcloned into pET30a or pET28b (EMD4Biosciences, San Diego, CA). LigA isozymes were overexpressed in E. coli BL21(DE3) (Invitrogen, Carlsbad, CA) for purification and activity assay development. The primers, template DNA, and plasmids used to make the overexpression plasmids are listed in Table 1.
Cloning and expression of Homo sapiens DNA ligase 1.
The Homo sapiens DNA ligase 1 gene, encoding amino acid residues 250 to 919 of the full-length protein, was PCR amplified from H. sapiens cDNA (Clontech, Mountain View, CA), and was cloned into pGEM-T as described previously (19). Once the sequence of the cloned DNA was verified, the truncated H. sapiens DNA ligase 1 gene was subcloned as a SacI-to-SalI fragment into pET21b to create an in-frame N-terminal fusion with the T7 tag for expression in E. coli BL21(DE3). The overexpressed T7-tagged protein was extracted and purified. The primers, template DNA, and plasmids used to make this overexpression plasmid are listed in Table 1.
Construction of strains for LigA mode-of-action studies.
H. influenzae strains for assessment of the mode of action against LigA were constructed in an efflux-negative (ΔacrB::aph3-A) mutant of strain KW20 (HIN100). H. influenzae HIN101 (ΔacrB::aph3-A ΔompP1::cat) was constructed to serve as an isogenic control strain, and H. influenzae HIN102 (ΔacrB::aph3-A ΔompP1::ligA + cat) was constructed to overexpress LigA as the experimental strain for assessment of the mode of action in H. influenzae. In HIN102, the ligA gene was under the transcriptional control of the strong, constitutively active ompP1 promoter (26, 27, 36). The supplemental material describes the construction of HIN101 and HIN102 (see Fig. S1).
Likewise, S. pneumoniae SPN100 (ΔgalU::ermR) was constructed to serve as an isogenic control strain, and S. pneumoniae SPN102 (ΔgalU::T4 lig + ermR ΔligA::aph3-A) was constructed to express T4 ligase in a ΔligA::aph3-A genetic background, as the experimental strain for assessment of the mode of action against S. pneumoniae LigA. The supplemental material describes the construction of SPN100 and SPN102 (see Fig. S2).
Purification of DNA ligases from H. influenzae, S. pneumoniae, E. coli, S. aureus, M. pneumoniae, and H. sapiens.
The conditions for the expression and purification of DNA ligase isozymes are described in the supplemental material.
DNA ligase assay.
A fluorescence resonance energy transfer (FRET) assay was developed for measuring the DNA ligase activity of H. influenzae LigA by using a format similar to that previously described (5, 9). The DNA substrate in the reaction mixture was created by annealing three synthetic oligonucleotides (Eurofins MWG Operon, Huntsville, AL). The longest oligonucleotide (32-mer) was modified with a fluorescent carboxyfluorescein (FAM) label at its 5′ end. The shortest oligonucleotide (11-mer) was modified with a 5′ phosphate and a tetramethyl carboxyrhodamine (TAMRA) acceptor label at its 3′ end. The third oligonucleotide (18-mer) was unlabeled. The sequences for the three oligonucleotides were as follows: for the 32-mer, 5′-FAM-TGGATTGATGACTGGGCTGAATACTGACCTTA-3′; for the 11-mer (complementary to the 5′ end of the 32-mer with a single mismatch), 5′-PO4-CAGTCATCTAT-TAMRA-3′; and for the 18-mer (complementary to the 3′ end of the 32-mer), 5′-TAAGGTCAGTATTCAGCC-3′.
The three oligonucleotides were hybridized to form a DNA duplex with a nick in one strand and fluorophores on the 5′ and 3′ ends of opposite strands. Ligation of the nick in this substrate DNA resulted in the formation of a ligated full-length DNA duplex. The ligated product was resistant to denaturation by 4 M urea at pH 10, whereas the unligated substrate was denatured. In the ligated product, the fluorophores were in close proximity, such that upon excitation of FAM, FRET occurred from the FAM donor to the TAMRA acceptor, resulting in enhanced TAMRA fluorescence and reduced FAM fluorescence (e.g., quenching). The extent to which FRET persisted after denaturation reported the fraction of the DNA substrate that was ligated.
Assays for H. influenzae LigA were performed in 96-well black polystyrene flat-bottom plates in 100-μl reaction mixtures containing the following: 20% glycerol, 30 mM potassium chloride, 30 mM ammonium sulfate, 10 mM dithiothreitol, 1 mM EDTA, 0.002% Brij 35, 50 mM morpholinepropanesulfonic acid (MOPS; pH 7.5), 100 nM bovine serum albumin, 1 μM NAD+, 40 nM DNA substrate, 16 mM magnesium chloride, and 0.15 nM H. influenzae LigA. The assay reaction mixtures were incubated at room temperature for approximately 20 min before the reactions were terminated by the addition of 30 μl Quench reagent (8 M urea, 1 M Trizma base, 20 mM EDTA in water) to denature any remaining unligated substrate DNA. Plates were read in a Tecan Ultra plate reader monitoring the ratio of fluorescence intensities at two emission wavelengths (the TAMRA acceptor at 595 nm and the FAM donor at 535 nm) upon excitation of the FAM donor at 485 nm. The larger the ratio of 595- to 535-nm emission values, the larger the fraction of ligated DNA in the reaction.
Data were initially expressed as a ratio of the 595-nm emission values to the 535-nm emission values, and percentages of inhibition were calculated using 0.2% dimethyl sulfoxide (no compound) as the 0% inhibition control and 50 mM EDTA-containing reaction mixtures as 100% inhibition controls. For the high-throughput screening (HTS) campaign, the assay described above was performed in a 384-well format (30 μl) with a single test compound per well at a final concentration of 10 μM. For hit follow-up and subsequent structure-activity relationship (SAR) analysis, compound potency was based on measurements of the 50% inhibitory concentration (IC50) in reactions performed as described above but in the presence of 10 different compound concentrations.
Assays for the other bacterial LigA isozymes, human DNA ligase 1, and T4 DNA ligase were performed as described above for H. influenzae LigA, with the following changes in order to optimize enzyme performance and normalize substrate concentrations relative to the individual isozymes' Km values. S. aureus LigA reaction mixtures contained 0.15 nM enzyme, 72 nM DNA, and 94 μM NAD+. S. pneumoniae LigA reaction mixtures contained 0.1 nM enzyme, 24 nM DNA, and 6.8 μM NAD+. E. coli LigA reaction mixtures contained 0.1 nM enzyme, 22 nM DNA, and 5.3 μM NAD+, with potassium chloride and ammonium sulfate concentrations adjusted to 40 mM and 5 mM, respectively. M. pneumoniae LigA reaction mixtures contained 0.15 nM enzyme, 24 nM DNA, and 0.13 μM NAD+, with potassium chloride and ammonium sulfate concentrations adjusted to 5 mM each. Human ligase 1 reaction mixtures contained 0.5 nM enzyme, 50 nM DNA, and 3 μM ATP (replacing NAD+), with no potassium chloride and 12 mM ammonium sulfate. T4 bacteriophage DNA ligase reaction mixtures contained 2 U enzyme, 100 nM DNA, and 100 μM ATP (replacing NAD+), with potassium chloride and ammonium sulfate concentrations adjusted to 30 mM each.
To determine the apparent Ki of compound 1, the H. influenzae LigA FRET assay was performed in the presence of increasing concentrations of the compound (0 to 2.6 μM) in reaction mixtures with a range of concentrations of the NAD+ substrate (0.5 to 3.5 μM) and a fixed concentration (60 nM) of the DNA substrate. Nonlinear regression analysis of the resulting data was performed using GraFit (Erithacus Software Limited, Surrey, United Kingdom).
X-ray cocrystallography of the H. influenzae LigA adenylation domain.
The X-ray cocrystal structure of an H. influenzae LigA adenylation domain fragment (amino acid residues 1 to 324) containing AMP covalently bound to a conserved lysine residue (K116) and NAD+ bound to adenylation subdomain 1a has been reported previously (13; Lahiri and Mills, U.S. patent application 2008/0262811).
Susceptibility and cytotoxicity assays.
MICs were determined using broth microdilution and in vitro killing kinetics according to the guidelines of the Clinical and Laboratory Standards Institute (formerly NCCLS) (11, 29).
Cytotoxicity was assessed in the human lung carcinoma cell line A549 by using the CellTiter 96 AQueous One Solution cell proliferation assay with MTS [3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, inner salt] from Promega Corp. (Madison, WI). A549 cells were grown at 37°C under 5% CO2 in RPMI 1640 medium supplemented with 10% fetal bovine serum and 1 mM glutamine (Invitrogen, Carlsbad, CA). Cytotoxicity was quantified by determining the lowest compound concentration at which transmission was increased by 50%. Puromycin and amphotericin B served as controls.
Isolation of resistant mutants.
Mutants of S. pneumoniae D39 were isolated as described previously (7). Briefly, 107 to 108 CFU was spread onto blood agar plates (BAPs) containing 2-fold serial dilutions of compound 4 and was incubated for 24 h at 37°C. Mutants were isolated from BAPs that contained the compound at concentrations 4 times the agar dilution MIC or more and were purified on BAPs with the same compound concentration. Mutants were stored in growth medium containing 25% (vol/vol) glycerol at −80°C.
Okazaki fragment accumulation assay.
The procedure for determination of cellular Okazaki fragment accumulation in H. influenzae was adapted from the method described by Dermody et al. for E. coli (12). A description of the adapted method is in the supplemental material.
Murine infection models.
The animals used for the murine infection models were maintained in accordance with the criteria of the American Association for Accreditation of Laboratory Animal Care. All animal studies were carried out according to protocols approved by the Institutional Animal Care and Use Committee at AstraZeneca R&D Boston.
(i) Neutropenic mouse thigh model.
Six-week-old specific-pathogen-free female CD-1 Swiss mice (Charles River Laboratories, MA) weighing 18 to 22 g were used for all studies. Mice were rendered neutropenic by intraperitoneal injection of cyclophosphamide (Sigma-Aldrich, St. Louis, MO) 4 days (150 mg/kg of body weight) and 1 day (100 mg/kg) before experimental infection (1). Two hours prior to infection, mice received a single oral administration of aminobenzotriazole (ABT) at 100 mg/kg to inhibit cytochrome P450 (CYP450) activity (4). A frozen stock of S. aureus ARC516 was thawed and diluted to a concentration of 7 × 106 CFU/ml. Mice were infected to achieve a target inoculum of 5 × 105 CFU/thigh. Groups of five animals each received an intraperitoneal injection of 5, 15, 30, or 45 mg/kg of body weight of compound 4, prepared with 0.75% hydroxypropylmethyl cellulose (HPMC), on a q.i.d., q3 (4 times a day, every 3 h) regimen starting 2 h after infection. An additional group of 10 mice received the vehicle alone (0.75% HPMC) on the same regimen. Efficacy was determined 24 h after the start of treatment. Thigh tissue was homogenized with an Omni TH homogenizer (Omni International, Warrenton, VA), and 100 μl of homogenate was serially diluted in tryptic soy broth and was plated onto tryptic soy agar plates for CFU determination (21). Plates were incubated at 37°C overnight.
(ii) Immunocompetent mouse lung model.
Six-week-old specific-pathogen-free female CD-1 Swiss mice (Charles River Laboratories, MA) weighing 18 to 22 g were used for all studies. Two hours prior to infection, mice received a single oral administration of 100 mg/kg ABT as described above. A frozen stock of S. pneumoniae isolate D39 was thawed and diluted to a concentration of 2 × 106 CFU/ml. Animals were anesthetized by isoflurane inhalation and were then infected with 105 CFU by delivery of 50 μl of diluted culture directly into the trachea of each mouse. Groups of 10 mice received an intraperitoneal injection of 2, 15, or 45 mg/kg of body weight of compound 4, prepared with 0.75% HPMC, on a q.i.d., q3 regimen starting 18 h after infection. An additional group of 10 mice received the vehicle (0.75% HPMC) on the same regimen. Efficacy was determined 24 h after the start of treatment (3). Lung tissue was homogenized and diluted as described above. Dilutions were spread onto blood agar plates for CFU determination. Plates were incubated overnight at 37°C under 5% CO2.
Protein structure accession number.
The coordinates of a refined cocrystallographic structure of compound 1 bound to the H. influenzae LigA adenylation domain, and the associated methods for protein preparation, have been deposited in the Protein Data Bank (PDB) under accession code 3PN1.
RESULTS
Discovery of LigA inhibitors and progression to antibacterial activity.
An HTS campaign was carried out to identify inhibitors of H. influenzae LigA by using a FRET-based DNA ligation assay and compounds from the AstraZeneca corporate library. After confirmation of IC50s against H. influenzae LigA and compound filtering based on physicochemical properties such as ClogP and solubility, 517 compounds were selected for testing for selectivity and spectrum of inhibition. IC50s against NAD+-dependent DNA ligases from E. coli, S. aureus, S. pneumoniae, and M. pneumoniae, in addition to the enzyme from H. influenzae, were determined. Selectivity for NAD+-dependent DNA ligases was determined by measuring the IC50s of selected hits against the ATP-dependent ligases human DNA ligase 1 and bacteriophage T4 ligase.
Several distinct chemotypes were identified from the HTS campaign, including a cluster of adenosine analogs, exemplified by compound 1 (Fig. 1). Compound 1 had an IC50 of 0.5 μM against H. influenzae LigA and displayed broad-spectrum biochemical potency for all LigA isozymes tested (Table 2) but was inactive (IC50, >200 μM) against human DNA ligase 1 and T4 bacteriophage ligase. Compound 1 also possessed favorable physical properties for an initial hit from HTS, including equilibrium solubility greater than 4 mM at pH 7.4, a logD7.4 (logD measured at pH 7.4) value of 1.24, and a free fraction (fu) in human plasma greater than 0.3.
TABLE 2.
Inhibition of bacterial DNA ligase enzymes by the adenosine analogsa
| Compound | IC50 (μM) for DNA ligase from: |
||||
|---|---|---|---|---|---|
| H. influenzae | E. coli | S. pneumoniae | S. aureus | M. pneumoniae | |
| 1 | 0.507 | 1.27 | 0.136 | 0.081 | 0.075 |
| 2 | 0.076 | 0.158 | 0.052 | 0.063 | 0.021 |
| 3 | 0.112 | 0.370 | 0.076 | 0.117 | 0.019 |
| 4 | 0.106 | 0.320 | 0.039 | 0.158 | <0.010 |
| 5 | 0.082 | 0.180 | 0.038 | 0.060 | 0.062 |
The chemical structures of the adenosine analogs are shown in Fig. 1.
Kinetics studies using the H. influenzae LigA FRET assay demonstrated that compound 1 was competitive with NAD+ with an apparent Ki of 110 nM (Fig. 2).
FIG. 2.
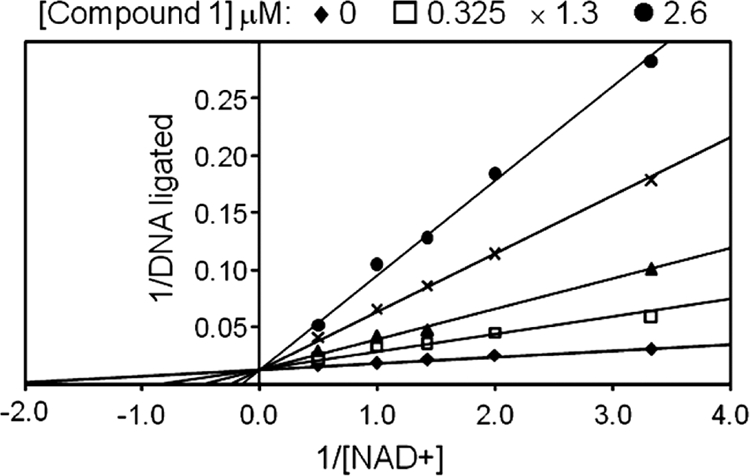
Enzyme mode of inhibition for compound 1. The activity of H. influenzae LigA was measured using a FRET assay in the presence of rising concentrations of compound 1 (0 to 2.6 μM) and NAD+ (0.5 to 3.5 μM), with a fixed concentration (60 nM) of the nicked DNA substrate. As represented in the double-reciprocal plot, the resulting data are consistent with competitive binding between compound 1 and NAD+. The apparent Ki for compound 1 was determined to be 110 nM by nonlinear regression analysis of the data.
The binding mode for compound 1 in the X-ray crystal structure of the adenylation domain fragment of H. influenzae LigA is shown in Fig. 3A. The crystallographic data revealed interactions between the adenine ring and the side chains of Glu-114 and Lys-291, as well as the backbone of Pro-115 and Leu-117, and also revealed an interaction between the 3′ hydroxyl of the ribose ring and the side chain of Glu-174. The X-ray crystal structure provided the foundation for an iterative structure-based medicinal chemistry effort to improve biochemical potency and achieve antibacterial activity, ultimately leading to compounds for in vivo efficacy studies.
FIG. 3.

(A) X-ray cocrystal structure of compound 1 bound to the AMP-binding site of H. influenzae LigA. Predicted hydrogen bonds between the inhibitor and the enzyme are shown as dashed lines. Enzyme residues proposed to make key interactions with the compound are labeled. (B) Surface representation of the hydrophobic tunnel of the binding pocket containing compound 1. The surface of L82 (corresponding to L75 in S. pneumoniae LigA) is shown in cyan.
Adenosine analogs that inhibited the activities of DNA ligase enzymes from a broad spectrum of bacterial species were synthesized; they are exemplified by compounds 2 to 5, with IC50s ranging from <0.010 μM to 1.27 μM (Table 2). Structure-activity relationships pertaining to the potency of enzyme inhibition led to changes at the 2-position of the adenine ring and the 5′ position on the ribose ring (Fig. 1). The cocrystal structure of compound 1 bound to the H. influenzae LigA AMP-binding pocket helped to define a hydrophobic tunnel predicted to accommodate the butanethiol at the 2-position of the adenine ring (Fig. 3B). This tunnel was explored in order to increase biochemical potency. The butanethiol at the 2-position of compound 1 was changed to a cycloalkoxy substituent in compounds 2 to 5, and the 5′-hydroxyl of compound 1 was replaced by either a fluorine (compounds 2 and 4) or a hydride (compounds 3 and 5). The resulting potencies of inhibition increased for enzymes from Gram-negative organisms and were essentially equipotent for enzymes from Gram-positive species. Any changes to the nitrogens of the adenine ring resulted in reduced potency; this is in agreement with the proposed binding mode in which hydrogen bonds to the residues mentioned above are important for the binding of the adenosine analogs to the active site of LigA.
Compound 1 exhibited modest antibacterial activity, with MIC values of 32 μg/ml against S. pneumoniae and activity against an efflux mutant of H. influenzae lacking the AcrB subunit of its main efflux pump (37). The lack of activity against the parental H. influenzae strain suggested that compound 1 was subject to active efflux out of the cell, poor penetration into the cell, or a combination of efflux and poor penetration. Antibacterial activities for compounds 2 to 5 were significantly improved against the Gram-positive pathogens S. pneumoniae and S. aureus, atypical M. pneumoniae, and mutants of the Gram-negative pathogens H. influenzae and E. coli that lacked components of the major AcrAB-TolC efflux pumps (Table 3). Although these compounds displayed modest antibacterial activity (MICs, 8 to 64 μg/ml) against the wild-type H. influenzae strain, they were inactive against the wild-type E. coli strain. Compound 5 displayed potent, broad-spectrum MIC values of 8, 4, 1, 1, and 2 μg/ml against wild-type H. influenzae, Moraxella catarrhalis, S. pneumoniae, Streptococcus pyogenes, and S. aureus, respectively. Additional examples of adenosine analogs, with corresponding IC50s and MICs, can be found in Table S1 in the supplemental material.
TABLE 3.
Antimicrobial activities of adenosine analogs against selected pathogenic bacteria
| Compound | MIC (μg/ml) for: |
||||||
|---|---|---|---|---|---|---|---|
|
H. influenzae |
E. coli |
S. pneumoniae D39 | S. aureus ARC516 | M. pneumoniae FH | |||
| KW20 | ARM158a | W3110 | ARC523b | ||||
| 1 | >64 | 32 | >64 | >64 | 32 | >64 | NDc |
| 2 | 8 | 2 | >64 | 4 | 2 | 8 | 1 |
| 3 | 16 | 4 | >64 | 8 | 4 | 16 | 2 |
| 4 | 64 | 2 | >64 | 4 | 1 | 2 | 0.2 |
| 5 | 8 | 2 | >64 | 4 | 1 | 2 | ND |
A KW20 efflux mutant with an acrB deletion.
A W3110 efflux mutant with a Tn10 insertion in tolC.
ND, not determined.
Selectivity for the bacterial target over eukaryotic targets was maintained for these compounds, as demonstrated by IC50s of >200 μM against human DNA ligase and bacteriophage T4 ligase. General cytotoxicity was not observed for these compounds, as shown by 50% effective concentrations (EC50s) of >64 μg/ml for the lysis of intact sheep red blood cells, as well as MIC values of >64 μg/ml for the proliferation of the human cell line A549 and the fungus Candida albicans.
Compounds 2 to 5 maintained favorable physical properties (solubility, >1 mM; logD7.4, <2; fu in human plasma, >0.3) such that they were amenable to in vivo dosing and efficacy.
Cellular characterization of LigA inhibitors and confirmation of a DNA ligase-specific mode of action.
The killing kinetics of compound 4 at multiples of the MIC was assessed against S. pneumoniae D39. When compound 4 was added at concentrations equal to 8 and 32 times the MIC, a 3-log10 drop in viability (CFU) was observed within 7 h, demonstrating that the adenosine analogs are rapidly bactericidal to S. pneumoniae (Fig. 4).
FIG. 4.
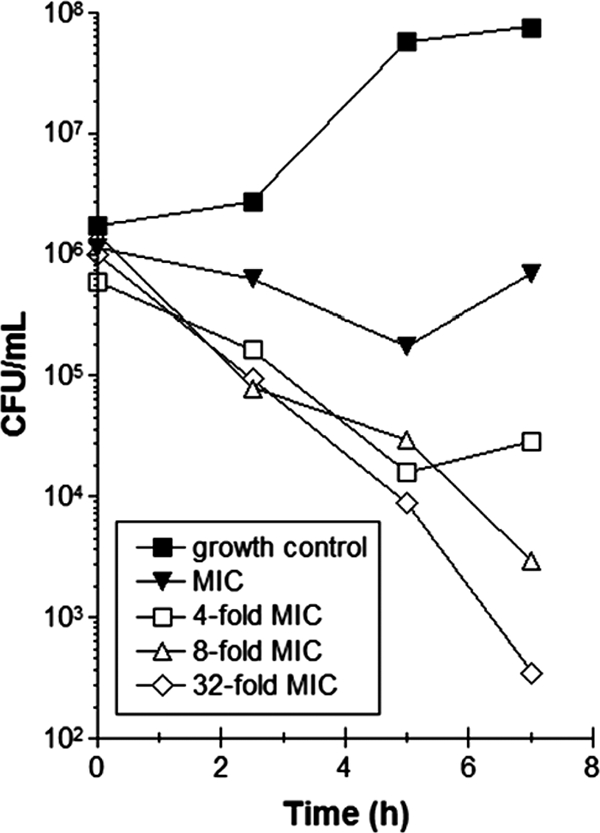
Time-kill study. Shown are the effects of increasing concentrations of compound 4 (multiples of the MIC) on the viability of S. pneumoniae (measured as CFU/ml).
In order to determine whether DNA ligase was inhibited in bacteria, intracellular levels of Okazaki fragments in H. influenzae were measured by fractionating DNA pools on sucrose gradients upon exposure to adenosine analogs (compounds 2 and 4). DNA extracted from H. influenzae grown in the absence of antibiotics led to a fractionated sucrose gradient profile with a single broad peak (spanning fractions 9 to 12) representing ligated genomic DNA (Fig. 5A to C, open circles). Incubation with ciprofloxacin (CIP) also led to the detection of a single peak, corresponding to ligated DNA, that became less abundant as the CIP concentration was increased to a level above the MIC (Fig. 5A). In this case, CIP predictably inhibited thymidine incorporation but did not cause an accumulation of Okazaki fragments. Incubation with adenosine analogs at concentrations close to their MICs against H. influenzae ARM158 resulted in the accumulation of low-molecular-weight DNA fragments consistent with Okazaki fragments (12). These data strongly suggested that the adenosine analogs inhibit DNA ligase in H. influenzae.
FIG. 5.
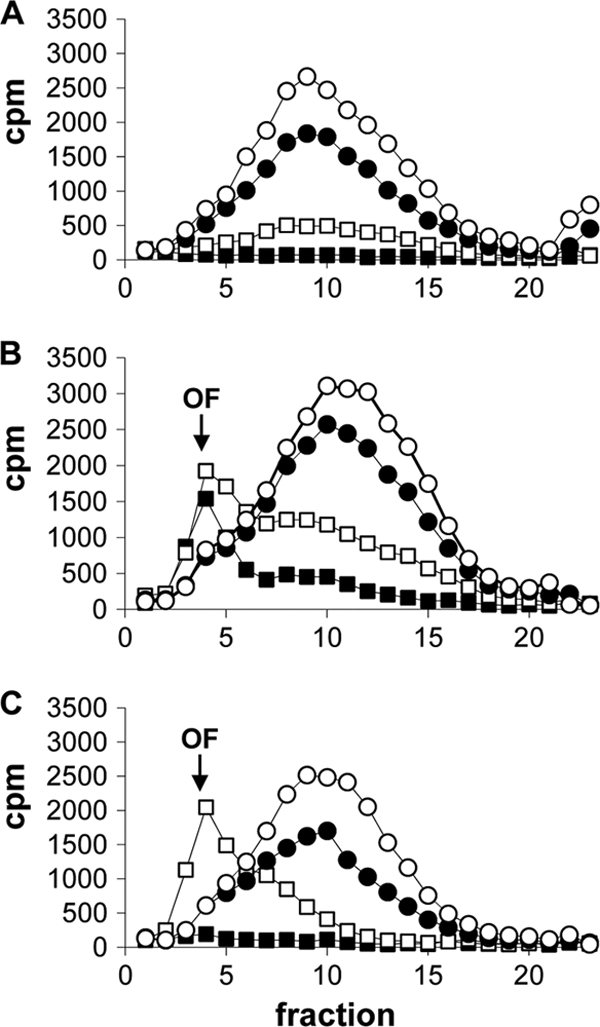
Accumulation of Okazaki fragments (OF) in [methyl-3H]thymidine-labeled DNA isolated from H. influenzae ARM158 cells treated with increasing concentrations of CIP (0, 2, 8, and 31 ng/ml) (A), compound 2 (0, 16, 250, and 1,000 ng/ml) (B), or compound 4 (0, 63, 500, and 2,000 ng/ml) (C). Symbols represent the absence of the compound (open circles) or increasing concentrations from lowest to highest (filled circles, open squares, and filled squares, respectively). The amount of radioactivity (cpm) was measured in 23 fractions (x axis) obtained from alkaline sucrose gradients. Okazaki fragments typically appeared in fraction 4, whereas high-molecular-weight DNA peaked in fractions 9 to 12.
A strain of H. influenzae (HIN102) was constructed in which H. influenzae LigA expression was controlled by the ompP1 promoter. This promoter is much stronger than the native LigA promoter (27), and as a result, it significantly increased the expression level of H. influenzae LigA as determined by Western blot analysis (see Fig. S2 in the supplemental material). It was predicted that the elevated levels of LigA in HIN102 would lead to elevated MIC values only if the antibacterial activity of the adenosine analogs was mediated by inhibition of LigA. The control inhibitors levofloxacin (LVX) and linezolid (LZD) did not have elevated MICs for HIN102, whereas the MIC values of compounds 1 to 4 for HIN102 were at least 16-fold higher than those for the parent strain, HIN101 (Table 4). These data demonstrate that the antibacterial activity of the adenosine analogs was mediated by inhibition of H. influenzae NAD+-dependent DNA ligase.
TABLE 4.
DNA ligase mode-of-action confirmation using recombinant expression strains
| Compounda | MICb |
|||||
|---|---|---|---|---|---|---|
|
H. influenzae |
S. pneumoniae |
|||||
| HIN101 | HIN102 | Fold increase | SPN100 | SPN102 | Fold increase | |
| LVX | 0.01 | 0.01 | 0.5 | 0.5 | ||
| LZD | 4 | 4 | 0.5 | 0.5 | ||
| 1 | 32 | 512 | 16 | ND | ND | |
| 2 | 4 | >64 | >16 | 2 | >64 | >32 |
| 3 | 8 | >64 | >8 | 2 | >64 | >32 |
| 4 | 2 | 32 | 16 | 1 | >64 | >64 |
LVX, levofloxacin; LZD, linezolid.
MICs for strains are given in micrograms per milliliter. ND, not determined.
Since there is precedent for antibacterial compounds with widely different modes of action against Gram-positive and Gram-negative pathogens (7), mode-of-action studies were also performed with S. pneumoniae. A strain of S. pneumoniae was constructed in which the S. pneumoniae ligA gene was replaced with the ATP-dependent T4 lig gene (SPN102). ATP-dependent T4 lig fully compensated for the loss of bacterial ligA in SPN102. No difference in growth was observed when SPN102 and the isogenic strain SPN100 were compared. By virtue of the replacement of S. pneumoniae ligA with T4 lig in SPN102, it was expected that compounds inhibiting growth by acting on the native S. pneumoniae LigA would have MICs higher than those for the isogenic strain SPN100, since the adenosine analogs had been shown to be inactive against T4 DNA ligase in vitro. Negative-control inhibitors (LVX and LZD) showed no change in the MIC for SPN102 relative to the MIC for the parent strain, SPN100. The MIC values of the adenosine analogs (compounds 1 to 4) were at least 64-fold higher for SPN102 than for SPN100. These data supported the premise that the antibacterial mode of action for the adenosine analog series was LigA inhibition (Table 4).
Finally, spontaneous resistant mutants of S. pneumoniae D39 were isolated on agar plates containing compound 4. Sequencing of the ligA gene from independently obtained mutants, including SPN103, revealed a mutation in the hydrophobic tunnel of the adenosine analog binding pocket (L75F) associated with an elevated MIC (64 μg/ml). The corresponding leucine residue in H. influenzae (L82) LigA is mapped (cyan surface) to the cocrystal structure shown in Fig. 3B. These data further supported LigA inhibition as the mode of action in S. pneumoniae.
In vivo efficacy.
Mice received a single oral administration of 100 mg/kg ABT 2 h prior to infection in order to maximize the potential exposure of bacteria to the compound in the efficacy experiments by reducing the high hepatic clearance of the adenosine analogs in rodents (data not shown). At the start of therapy, mice were colonized with 6.4 ± 0.1 log10 CFU/thigh of S. aureus ARC516 or 5.0 ± 0.07 log10 CFU/lung of S. pneumoniae D39. The organisms grew by 3.3 ± 0.5 log10 CFU/lung and 2.9 ± 0.2 log10 CFU/thigh after 24 h in untreated control mice in the respective models (Fig. 6). Escalating doses of compound 4 resulted in concentration-dependent killing of both strains in the respective tissue compartments. The highest doses studied reduced the organism burden by 1.8 ± 0.3 log10 S. aureus CFU/thigh and 2.9 ± 0.06 log10 S. pneumoniae CFU/lung from the inocula (input) administered to the mice at the beginning of the studies (Fig. 6). No adverse reactions were observed in the mice dosed with compound 4 in efficacy experiments.
FIG. 6.
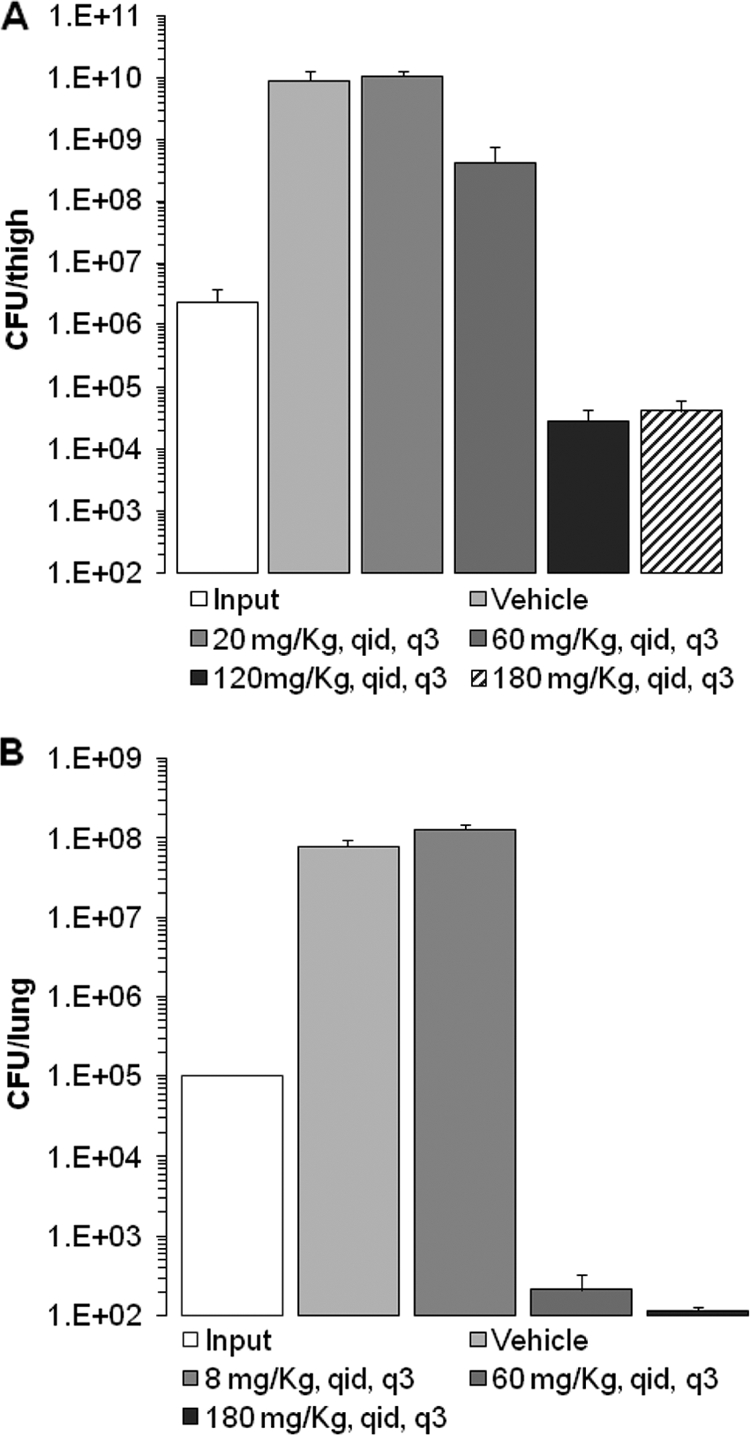
In vivo activity of compound 4 on infection. (A) S. aureus ARC516 in the thighs of neutropenic mice; (B) S. pneumoniae D39 in the lungs of immunocompetent mice. Results are plotted as means; the error bars represent the standard errors of the means. Dosing regimens are given in milligrams per kilogram per day. q.i.d., 4 times per day; q3, every 3 h.
DISCUSSION
DNA ligases are essential in all organisms. Bacterial NAD+-dependent DNA ligases are distinct from eukaryotic and viral ATP-dependent ligases and therefore represent a potentially selective target for the discovery and development of novel antibacterial agents (38).
The adenosine analog series discovered and further elaborated in this report is attractive from the standpoint of physical properties as well as potency. The adenosine analogs retained good physical properties (solubility, >1 mM; logD7.4, <2; fu in human plasma, >0.3) throughout the medicinal chemistry efforts to improve potency. Compounds 2 to 5 showed broad-spectrum enzyme inhibition of LigA with similar IC50s. In general, the adenosine analogs were more potent against LigA from Gram-positive species than against LigA from Gram-negative species. The boundaries of the hydrophobic tunnel (depicted in Fig. 3B) were explored and found to accommodate both large and small nonaromatic hydrophobic substituents at the 2-position of the adenine ring. The cyclic alkoxides appeared either to fit more favorably in the binding pocket or to be less flexible, and therefore more entropically favored, than the linear side chains and had improved IC50s. Compounds 1 to 5 were all selective for the bacterial DNA ligases over human and bacteriophage T4 DNA ligases.
Antibacterial activity was improved significantly by replacement of the 5′ hydroxyl with fluorine or hydride. This was presumably due to improved permeation of the bacterial cell envelope, resulting from the higher lipophilicity of compounds 2 to 5 than of compound 1. Optimization of the 2-position cycloalkoxy substituent led to compounds with improved antibacterial activity, especially against the Gram-positive organisms S. pneumoniae and S. aureus.
The adenosine analogs exhibited bactericidal activity. Inhibition of S. pneumoniae NAD+-dependent DNA ligase resulted in concentration-independent bactericidal activity, based on the time-kill experiment performed in this study (Fig. 4). Brötz-Oesterhelt et al. similarly demonstrated that pyridochromanone LigA inhibitors were rapidly bactericidal for S. aureus, although the activity was concentration dependent (6). Further, the antibacterial activity of the adenosine analogs was selective in that they displayed no cytotoxic activity against the fungus C. albicans or the human cell line A549.
Several lines of evidence indicated that the adenosine analogs represented by compounds 1 to 5 specifically inhibit the target enzyme, NAD+-dependent DNA ligase, in the bacteria. Addition of adenosine analogs to growth medium inhibited the incorporation of radiolabeled thymidine into DNA and promoted the accumulation of polynucleotide fragments of a size consistent with Okazaki fragments (Fig. 5). Overexpression of LigA in H. influenzae or replacement of ligA with T4 lig in S. pneumoniae desensitized the bacteria to the growth-inhibitory action of the adenosine analogs (Table 4). Furthermore, selection for spontaneous resistance to the adenosine analogs resulted in the consistent isolation of an S. pneumoniae mutant strain with a ligA target-based mutation (L75F) in the inhibitor-binding pocket of LigA. Modeling with the phenylalanine residue in place of leucine (L82F) in the H. influenzae structure predicts that the cycloalkoxy substituents at the 2-position of the adenine ring would be occluded or altered in binding (Fig. 3B; data not shown).
The in vivo efficacy of the adenosine analogs was evaluated in murine models of infection. Compound 4 was efficacious in a neutropenic-mouse model of S. aureus thigh infection and an immunocompetent-mouse model of S. pneumoniae lung infection. In both models of infection, the compounds caused a dose-dependent decrease in CFU over 24 h, with maximal responses occurring at 120 mg/kg/day and 60 mg/kg/day, respectively. We believe this is the first publication to demonstrate in vivo efficacy with a bacterial DNA ligase inhibitor. Ultimately, to attain clinically relevant in vivo efficacy with the adenosine analog series, further optimization of potency and in vivo pharmacokinetic properties is required.
In summary, the target-based drug discovery approach described was successful in identifying a novel series of adenosine analogs that exhibited selective inhibition of bacterial NAD+-dependent DNA ligase. The inhibition of cellular growth by the adenosine analogs was mediated by inhibition of NAD+-dependent DNA ligase. Furthermore, LigA was validated in vivo with the demonstration of efficacy in a murine S. aureus thigh infection model and a murine S. pneumoniae lung infection model. These data support the potential use of NAD+-dependent DNA ligase as a target for antibiotic therapy.
Supplementary Material
Acknowledgments
We thank the AstraZeneca R&D Boston Susceptibility Group for MIC testing and the Drug Metabolism and Pharmacokinetics (DMPK) Group for analyses associated with in vivo efficacy studies. We specifically thank Robert Albert, Teresa Conneely, Lena Grosser, Irene Karantzeni, Val Laganas, Ce Feng Liu, Kathy MacCormack, Bob McLaughlin, Art Patten, Hongming Wang, and Maria Uria-Nickelsen for their technical expertise in supporting this study. We thank Tom Dougherty and Boudewijn deJonge for critical reading of the manuscript.
Footnotes
Published ahead of print on 28 December 2010.
Supplemental material for this article may be found at http://aac.asm.org/.
REFERENCES
- 1.Andes, D., and W. A. Craig. 1998. In vivo activities of amoxicillin and amoxicillin-clavulanate against Streptococcus pneumoniae: application to breakpoint determinations. Antimicrob. Agents Chemother. 42:2375-2379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Avery, O. T., C. M. MacLeod, and M. McCarty. 1944. Studies on the chemical nature of the substance inducing transformation of pneumococcal types. J. Exp. Med. 79:137-157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Azoulay-Dupuis, E., et al. 1991. Antipneumococcal activity of ciprofloxacin, ofloxacin and temafloxacin in an experimental mouse pneumonia model at various stages of the disease. J. Infect. Dis. 163:319-324. [DOI] [PubMed] [Google Scholar]
- 4.Balani, S. K., et al. 2004. Effective dosing regimen of 1-aminobenzotriazole for inhibition of antipyrine clearance in guinea pigs and mice using serial sampling. Drug Metab. Dispos. 32:1092-1095. [DOI] [PubMed] [Google Scholar]
- 5.Benson, E. L., et al. 2004. A high-throughput resonance energy transfer assay for Staphylococcus aureus DNA ligase. Anal. Biochem. 324:298-300. [DOI] [PubMed] [Google Scholar]
- 6.Brötz-Oesterhelt, H., et al. 2003. Specific and potent inhibition of NAD+-dependent DNA ligase by pyridochromanones. J. Biol. Chem. 278:39435-39442. [DOI] [PubMed] [Google Scholar]
- 7.Buurman, E. T., K. D. Johnson, R. K. Kelly, and K. MacCormack. 2006. Different modes of action of naphthyridones in Gram-positive and Gram-negative bacteria. Antimicrob. Agents Chemother. 50:385-387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Buurman, E. T., et al. 2009. Adenosine analogs mediating antibacterial activity via inhibition of NAD-dependent DNA ligase, poster F1-1982. Abstr. 49th Intersci. Conf. Antimicrob. Agents Chemother.
- 9.Chen, X. C., et al. 2002. Development of a fluorescence resonance transfer assay for measuring the activity of Streptococcus pneumoniae DNA ligase, an enzyme essential for DNA replication, repair and recombination. Anal. Biochem. 309:232-240. [DOI] [PubMed] [Google Scholar]
- 10.Ciarrocchi, G., D. G. MacPhee, L. W. Deady, and L. Tilley. 1999. Specific inhibition of the eubacterial DNA ligase by arylamino compounds. Antimicrob. Agents Chemother. 43:2766-2772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Clinical and Laboratory Standards Institute. 2009. Methods for dilution antimicrobial susceptibility tests for bacteria that grow aerobically; approved standard, 9th ed. M07-A8, vol. 29, no. 2. Clinical and Laboratory Standards Institute, Wayne, PA.
- 12.Dermody, J. J., G. T. Robinson, and R. Sternglanz. 1979. Conditional-lethal deoxyribonucleic acid ligase mutant of Escherichia coli. J. Bacteriol. 139:701-704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Eakin, A. E., et al. 2009. Discovery of novel adenosine analogs that selectively inhibit bacterial NAD+-dependent DNA ligase, poster F1-1981. Abstr. 49th Intersci. Conf. Antimicrob. Agents Chemother.
- 14.Gajiwala, K. S., and C. Pinko. 2004. Structural rearrangement accompanying NAD+ synthesis within a bacterial DNA ligase crystal. Structure 12:1449-1459. [DOI] [PubMed] [Google Scholar]
- 15.Gul, S. R., et al. 2004. Staphylococcus aureus DNA ligase: characterization of its kinetics of catalysis and development of a high-throughput screening compatible chemiluminescent hybridization protection assay. Biochem. J. 383:551-559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Han, S., J. S. Chang, and M. Griffor. 2009. Structure of the adenylation domain of NAD+-dependent DNA ligase from Staphylococcus aureus. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 65:1078-1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hoskins, J., et al. 2001. Genome of the bacterium Streptococcus pneumoniae strain R6. J. Bacteriol. 183:5709-5717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kaczmarek, F. S., et al. 2001. Cloning and functional characterization of an NAD+-dependent DNA ligase from Staphylococcus aureus. J. Bacteriol. 183:3016-3024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kodama, K., D. E. Barnes, and T. Lindahl. 1991. In vitro mutagenesis and functional expression in Escherichia coli of a cDNA encoding the catalytic domain of human DNA ligase I. Nucleic Acids Res. 19:6093-6099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kreiswirth, B., et al. 1983. The toxic shock syndrome exotoxin structural gene is not detectably transmitted by a prophage. Nature 305:709-712. [DOI] [PubMed] [Google Scholar]
- 21.Leggett, J. E., S. Ebert, B. Fantin, and W. A. Craig. 1990. Comparative dose-effect relations at several dosing intervals for beta-lactam, aminoglycoside and quinolone antibiotics against Gram-negative bacilli in murine thigh-infection and pneumonitis models. Scand. J. Infect. Dis. Suppl. 74:179-184. [PubMed] [Google Scholar]
- 22.Lehman, I. R. 1974. DNA ligase: structure, mechanism and function. Science 186:790-797. [DOI] [PubMed] [Google Scholar]
- 23.Meier, T. I., et al. 2008. Identification and characterization of an inhibitor specific to bacterial NAD+-dependent DNA ligases. FEBS J. 275:5258-5271. [DOI] [PubMed] [Google Scholar]
- 24.Ménard, R., P. J. Sansonetti, and C. Parsot. 1993. Nonpolar mutagenesis of the ipa genes defines IpaB, IpaC, and IpaD as effectors of Shigella flexneri entry into epithelial cells. J. Bacteriol. 175:5899-5906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Miesel, L., et al. 2007. A high-throughput assay for the adenylation reaction of bacterial DNA ligase. Anal. Biochem. 366:9-17. [DOI] [PubMed] [Google Scholar]
- 26.Munson, R., Jr., and S. Grass. 1988. Purification, cloning, and sequence of outer membrane protein P1 of Haemophilus influenzae type b. Infect. Immun. 56:2235-2242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Munson, R., Jr., and A. Hunt. 1989. Isolation and characterization of a mutant of Haemophilus influenzae type b deficient in outer membrane protein P1. Infect. Immun. 57:1002-1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nandakumar, J., P. A. Nair, and S. Shuman. 2007. Last stop on the road to repair: structure of E. coli DNA ligase bound to nicked DNA-adenylate. Mol. Cell 26:257-271. [DOI] [PubMed] [Google Scholar]
- 29.National Committee for Clinical Laboratory Standards. 1999. Methods for determining bactericidal activity of antimicrobial agents; approved guideline. M26-A, vol. 19, no. 18. National Committee for Clinical Laboratory Standards, Wayne, PA.
- 30.Newman, J. V., T. O'Shea, L. Grosser, H. Hu, and S. D. Mills. 2009. Adenosine analogs: pharmacokinetics and in vivo efficacy in Gram-positive infection models, poster F1-1983. Abstr. 49th Intersci. Conf. Antimicrob. Agents Chemother.
- 31.Park, U. E., B. M. Olivera, K. T. Hughes, J. R. Roth, and D. R. Hillyard. 1989. DNA ligase and the pyridine nucleotide cycle in Salmonella typhimurium. J. Bacteriol. 171:2173-2180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pascal, J. M. 2008. DNA and RNA ligases: structural variations and shared mechanisms. Curr. Opin. Struct. Biol. 18:96-105. [DOI] [PubMed] [Google Scholar]
- 33.Petit, M. A., and S. D. Ehrlich. 2000. The NAD-dependent ligase encoded by yerG is an essential gene of Bacillus subtilis. Nucleic Acids Res. 28:4642-4648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ravin, A. W. 1959. Reciprocal capsular transformations of pneumococci. J. Bacteriol. 77:296-309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ryley, J. F., R. G. Wilson, and K. J. Barrett-Bee. 1984. Azole resistance in Candida albicans. Sabouraudia 22:53-63. [PubMed] [Google Scholar]
- 36.Saeed-Kothe, A., W. Yang, and S. D. Mills. 2004. Use of the riboflavin synthase gene (ribC) as a model for development of an essential gene disruption and complementation system for Haemophilus influenzae. Appl. Environ. Microbiol. 70:4136-4143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sánchez, L., W. Pan, M. Vinas, and H. Nikaido. 1997. The acrAB homolog of Haemophilus influenzae codes for a functional multidrug efflux pump. J. Bacteriol. 179:6855-6857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shuman, S. 2009. DNA ligases: progress and prospects. J. Biol. Chem. 284:17365-17369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stokes, S. S., et al. 2009. Adenosine analogs: synthesis and SAR of novel inhibitors of bacterial NAD+-dependent DNA ligase, poster F1-1980. Abstr. 49th Intersci. Conf. Antimicrob. Agents Chemother.
- 40.Whitby, P. W., D. J. Morton, and T. L. Stull. 1998. Construction of antibiotic resistance cassettes with multiple paired restriction sites for insertional mutagenesis of Haemophilus influenzae. FEMS Microbiol. Lett. 158:57-60. [DOI] [PubMed] [Google Scholar]
- 41.Wilkinson, A., J. Day, and R. Bowater. 2001. Bacterial DNA ligases. Mol. Microbiol. 40:1241-1248. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.