

Abstract
We show that anti-IgM-induced cell death in a human B lymphoma cell line, B104, is associated with early intracellular acidification and cell shrinkage. In contrast, another human B cell lymphoma line, Daudi, less susceptible to B cell antigen receptor-mediated cell death, responded to anti-IgM with an early increase in intracellular pH (pHi). The anti-IgM-induced changes of pHi were associated with different levels of activation of the Na+/H+ exchanger isoform 1 (NHE1) as judged by its phosphorylation status. Prevention of anti-IgM-induced cell death in B104 cells by the calcineurin phosphatase inhibitor, cyclosporin A, abrogated both intracellular acidification and cell shrinkage and was associated with an increase in the phosphorylation level of NHE1 within the first 60 min of stimulation. This indicates a key role for calcineurin in regulating pHi and cell viability. The potential role of pHi in cell viability was confirmed in Daudi cells treated with an Na+/H+ exchanger inhibitor 5-(N,N-hexamethylene)amiloride. These observations indicate that the outcome of the anti-IgM treatment depends on NHE1-controlled pHi. We suggest that inactivation of the NHE1 in anti-IgM-stimulated cells results in intracellular acidification and subsequently triggers or amplifies cell death.
Keywords: Na+/H+ exchanger, B cell receptor
Crosslinking of the B cell antigen receptor (BCR) with anti-IgM antibodies initiates a cascade of events that culminates with growth arrest and cell death. Many studies have shown that the caspase cascade plays a central role in this process, with or without involvement of mitochondria (1–5). However, the lack of a known biochemical link between cell cycle regulation and apoptosis suggests that they could represent the endpoints of two independent pathways. Indeed, a recent report has shown that z-VAD-fmk, the universal caspase inhibitor, or Bcl-2 overexpression prevented apoptosis but not growth arrest induced by anti-IgM treatment of a murine B lymphoma (1).
The regulation of cytosolic pH during BCR crosslinking might affect both cell cycle progression and viability. Numerous studies have implicated intracellular pH (pHi) in certain forms of cell death in response to various extracellular stimuli. Thus, intracellular acidification below a critical level precedes or is associated with apoptosis in different cells and is triggered by Fas (6) or somatostatin receptors (7), growth factor deprivation (8–11), or chemical stress (12–14). Most of these processes involve an alteration in Na+/H+ exchange, suggesting that the signal for intracellular acidification might be the same as the signal for apoptosis. The family of Na+/H+ exchange (NHE) represents one of the most effective mechanisms for the regulation of pHi and cell volume in virtually all mammalian cells. The NHE1 isoform is ubiquitously expressed and is the predominant isoform expressed on nonepithelial cells (reviewed in ref. 15).
Here, we show a comparative study of the pHi modulation after BCR hypercrosslinking with a polyclonal anti-IgM in two different human B lymphoma cell lines, B104 and Daudi. The former is very susceptible to anti-IgM-induced cell death and often used as a model for the study of B cell tolerance (3, 16, 17). The latter is less susceptible to killing but enters G1 arrest via a pathway involving the p21WAF1-cyclin-dependent kinase inhibitor (18). The observations implicate early calcineurin-mediated dephosphorylation of NHE1 as a critical step in the induction of cytosol acidification during anti-IgM-induced cell death.†
Methods
Materials.
Purified goat IgG specific to human IgM was purchased from Chemicon. Rabbit anti-phosphoserine (Z-PS1) was purchased from Zymed. Rabbit anti-human NHE1 antiserum was a generous gift from Orson Moe (University of Texas Southwestern Medical Center). Nigericin and 5-(N,N-hexamethylene)amiloride (HMA) were obtained from Sigma, and 2′,7′-bis-carboxyethyl-5(6)carboxyfluorescein, acetoxymethyl ester (BCECF-AM) was purchased from Molecular Probes.
Cell Culture.
The human Burkitt's lymphoma cell lines Daudi, BL60, and Ramos were maintained in culture by serial passage in RPMI 1640 medium containing 25 mM Hepes, 10% heat-inactivated FBS, and 100 mM L-glutamine (complete medium). The cells were grown in a humidified atmosphere of 5% CO2 and air. The B104 cell line was grown in the same media supplemented with 5 × 10−5 M β-mercaptoethanol.
Detection of pHi.
pHi was measured by flow cytometry by using the pH-sensitive fluorescent probe BCECF-AM. Approximately 5 × 106 cells/ml were incubated for 30 min in 7 μM BCECF-AM in Hanks' balanced salt solution at 37°C with gentle shaking. Then, cells were washed, resuspended in the same buffer at room temperature, and analyzed on a FACScan with an excitation at 488 nm. The relative pHi of individual cells was displayed as a two-dimensional dot-plot according to their fluorescence intensity at 520 nm (base) on x axis vs. 640 nm (acid) on y axis. The pHi values were obtained from the green/red fluorescence ratio values. The calibration curve was constructed by incubating BCECF-AM-loaded cells in different high K buffers in a range of pH from 6.2 to 8.0 in the presence of the K+ ionophore nigericin at 5 μM. The pH calibration buffers were prepared by mixing appropriate proportions of K buffers (135 mM KH2PO4/20 mM NaCl and 110 mM K2HPO4/20 mM NaCl) at room temperature to give a range of pH 6.2 to 8.0.
Cell Viability and Cell Cycle Analysis.
Cell viability was detected by propidium iodide (PI) exclusion and analyzed by flow cytometry by using a FACScan (Becton Dickinson). For cell cycle analysis, approximately 5 × 106 cells were pelleted, resuspended in 0.2 ml of PBS, and fixed by the addition of 1 ml of ice-cold 70% ethanol in PBS. Fixed cells were pelleted, gently resuspended in PBS, and incubated overnight at 4°C, in the dark, with 100 μg/ml RNase A and 40 μg/ml PI. In total, 10,000 events were collected and analyzed by using paint-a-gate software (Becton Dickinson); apoptotic cells were identified by their sub-G1 DNA content. Cell death was also quantified by simultaneous detection of phosphatidylserine (PS) exposure and cell permeability by using FITC-Annexin V (BD-PharMingen) and PI. Cells (5 × 105) were suspended in 100 μl of binding buffer (10 mM Hepes, pH 7.4/140 mM NaCl/2.5 mM CaCl2) together with 5 μl of FITC-Annexin V and 10 μl of PI (20 μg/ml), incubated for 15 min at room temperature in the dark, and analyzed within 1 h by flow cytometry.
Immunoprecipitation and Western Blotting.
Cells (107) were washed once in PBS and then lysed in 1 ml of ice-cold immunoprecipitation buffer (50 mM Hepes, pH 7.4/150 mM NaCl/3 mM KCl/5 mM EDTA/1 mM iodoacetamide/25 mM sodium pyrophosphate/0.1 mM sodium orthovanadate/1 mM PMSF/10 μg/ml each of aprotinin and leupeptin/1% Triton X-100). The pellets were removed by centrifugation of the cell lysate at 14,000 × g for 30 min at 4°C. The supernatants were cleared by incubation for 1 h at 4°C with 20 μl of protein A-agarose (Santa Cruz Biotechnology) coated with 1 μg of normal rabbit IgG. A 1:500 dilution of nonimmune or immune rabbit serum was added to the supernatant. After overnight incubation at 4°C, the immune complexes were collected by addition of 20 μl of protein A-agarose and incubation for 1 h at 4°C. The agarose beads were washed four times with the cold precipitation buffer, and the immunoprecipitated proteins were dissolved in 2× Laemmli sample buffer and boiled for 5 min. The samples were resolved on a 7.5% SDS/PAGE under reducing conditions and then transferred to a poly(vinylidene difluoride) membrane (Bio-Rad). The membranes were blocked in Tris-buffered saline containing 2% BSA and 0.1% Tween-20 and incubated overnight at 4°C with either 1 μg/ml anti-phosphoserine antibody or rabbit serum anti-NHE1 (1:1,500). The blots were developed by the enhanced chemiluminescent (ECL) detection system by using a 1:2,500 dilution of horseradish peroxidase-conjugated goat-anti rabbit IgG (Amersham Pharmacia).
Results
Anti-IgM-Induced Cell Death of B104 Cells Is Preceded by a Decrease in pHi.
B104 cells are very susceptible to killing by anti-IgM, as reported (3, 15, 16), whereas Daudi cells show less cell death, as judged by the loss of their membrane integrity after 24 h of treatment (Fig. 1). The inhibitory effect was not associated with DNA fragmentation because a significant number of cells with subdiploid levels was not observed. However, Daudi cells showed cell cycle arrest at the G1/S interphase, whereas B104 cells showed cell cycle arrest at the G2/M interphase (Fig. 1).
Figure 1.

The effect of anti-IgM on the viability and cell cycle distribution of B104 and Daudi cells. Cells (5 × 105) were incubated for 24 h in the presence or absence of 20 μg/ml anti-IgM, stained with PI before and after permeabilization, and analyzed by flow cytometry. The percentage of dead cells was detected by PI exclusion (upper left corners), and the proportion of cells in each phase of the cell cycle and in sub-G1 phase was calculated according to the DNA content. This is a representative experiment of three experiments performed.
Within 8 h, anti-IgM treatment of B104 cells resulted in the appearance of a subpopulation of cells with acidic pHi as detected by flow cytometric analysis of cells loaded with the pH-sensitive fluorogen BCECF-AM (Fig. 2A) without evidence of cell death. By 8 h, the proportion of cells in the acidic population increased from 6.5% in the starting population to almost 35% (Fig. 2A). In the gated acidic population, the pHi averaged 6.65 ± 0.09, whereas the pHi of the cells in the major population remained at 7.15 ± 0.06. By contrast, treatment of Daudi cells induced only a minor increase in the proportion of acidic cells but an increase in pHi of the main population from 7.1 in untreated cells to 7.3 in treated cells. At 24 h, Daudi cells returned slowly to a pH with 0.15 units below the baseline level, a decrease we attributed to the G1 arrest, as shown (10, 19–21).
Figure 2.

The effect of anti-IgM stimulation on the pHi and size of B104 vs. Daudi cells. Cells (5 × 105) treated with 20 μg/ml anti-IgM for the indicated times were loaded with BCECF-AM and analyzed by flow cytometry. Their distribution, according to the pHi, was determined from the two-dimensional scatter plot of the fluorescence intensities at 520 nm (FL1, base) and 640 nm (FL3, acid) (A), and cell size determined from the forward (cell size) vs. side light scatter (cell granularity) plots (B). The shift of cells toward the FL3 axis indicates a decrease in their pHi. The gated events indicate the acidic subpopulation (pHi 6.65 ± 0.09). The percentage of cells in the gated area is indicated in the upper left corners. These are the results of a representative experiment of three experiments performed.
The proportion of cells with acidic pHi coincided with the proportion of smaller cells as judged by forward light scatter analyzed at the same time as the pHi, indicating that acidification and cell shrinkage were temporally related (Fig. 2B). Taken together, these findings provide suggestive evidence that a decrease in pHi is an early event in signaling.
Because B104 cells are EBV− and Daudi cells are EBV+, we investigated two EBV− B lymphoma cell lines, BL-60 and Ramos, that have different sensitivities to anti-IgM-mediated cell death. The scale of susceptibility to anti-IgM-mediated cell death was B104>BL60>Daudi>Ramos, it paralleled the scale of intracellular acidification (data not shown), and it was not related to Epstein–Barr virus status.
Role of NHE1 in Anti-IgM-Mediated Cell Death.
Regulation of pHi in mammalian cells largely
depends on the activity of NHE and the
HCO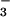 /Cl− exchanger.
Because experiments carried out in bicarbonate-free media gave similar
results (data not shown), this ruled out a contribution of the
HCO
/Cl− exchanger.
Because experiments carried out in bicarbonate-free media gave similar
results (data not shown), this ruled out a contribution of the
HCO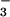 /Cl− exchange to
the anti-IgM-mediated change in pHi and indicated
that NHE could be the major player in regulating
pHi and cell volume.
/Cl− exchange to
the anti-IgM-mediated change in pHi and indicated
that NHE could be the major player in regulating
pHi and cell volume.
There are six known isoforms of NHE of which isoform 1 is the ubiquitous membrane transporter (15). The modulation of pHi by NHE depends on its affinity for cytosolic H+, which is enhanced mainly by Ser/Thr phosphorylation at multiple sites in the carboxyl-terminal cytoplasmic domain (22). Therefore, the acidification in B104 cells might arise from the loss of pHi regulation, possibly as a result of dephosphorylation by an intracellular phosphatase. In contrast, the slight intracellular alkalinization of Daudi cells in response to anti-IgM treatment might result from NHE1 activation. The phosphorylation levels of the NHE1 immunoprecipitates obtained from B104 cells stimulated with anti-IgM decreased within minutes. This was manifested by an increase in mobility on SDS/PAGE, without a change in the protein level (Fig. 3). The decrease in phosphorylation of NHE1 was reversed by preincubation of cells for 30 min with 200 nM cyclosporin A (CsA), a specific inhibitor of calcineurin phosphatase (Fig. 3). By contrast, Daudi cells underwent a rapid increase in the phosphorylation level of NHE1. and this increase remained constant within the same time interval (Fig. 4). Moreover, when 95% of the anti-IgM-induced death in B104 cells by 24 h was prevented by preincubation with 200 nM CsA, as reported (3, 17), the acidification was totally reversed by 8 h (Fig. 5). In an earlier study, Ishigami et al. (17) suggested that the protective effect of CsA on anti-IgM-induced death in B104 cells is not related to modulation of Ca2+ mobilization but to an event that occurs within 15–30 min after anti-IgM stimulation. The abrogation of anti-IgM-induced cell death in B104 cells by CsA and prevention of NHE1 dephosphorylation suggests that the signal for acidification, mediated by NHE1, and the signal for cell death converge at an early point.
Figure 3.

The levels of NHE1 phosphorylation in anti-IgM-treated B104 cells. B104 cells (5 × 105/ml), pretreated or not with 200 ng/ml CsA for 30 min, were stimulated with 20 μg/ml anti-IgM for the indicated times. Immunoprecipitates isolated from cell lysates with either normal rabbit serum (NRS) or rabbit anti-NHE1 serum were subjected to a 7.5% SDS/PAGE, transferred to poly(vinylidene difluoride), and immunoblotted with either rabbit anti-phosphoserine antibody or rabbit anti-NHE1 serum. The blots were developed by the ECL method. This is a representative experiment of three experiments performed.
Figure 4.

The levels of NHE1 phosphorylation in anti-IgM-treated Daudi cells. Cells were incubated with 20 μg/ml anti-IgM for various times as indicated. NHE1 immunoprecipitates and control precipitates were electrophoresed on a 7.5% SDS/PAGE, transferred to poly(vinylidene difluoride), and immunoblotted as described in Fig. 3. This is a representative experiment of three experiments performed.
Figure 5.

The effect of CsA on anti-IgM-induced acidification of B104 cells. B104 cells were incubated with 20 μg/ml anti-IgM for the indicated time, and the pH was detected by flow cytometry as described in Fig. 3. The percentage of cells within the acidic subpopulation (gated area) is indicated in the upper left corners. This is a representative experiment of three experiments performed.
Inhibition of NHE1 Affects Cell Growth and Viability.
The finding that B104 cells that undergo NHE1-controlled acidification after treatment with anti-IgM die suggests that a sustained fall in pHi per se below a critical level may play an active role in the induction of cell death. Therefore, inhibition of NHE in cells actively generating H+ would result in intracellular acidification. To test this hypothesis, we studied the effect of NHE1 inhibition in Daudi cells with the amiloride analog, HMA. Treatment of Daudi cells with 50 μM HMA for up to 24 h led to a dramatic decrease in pHi and was paralleled by an increase in PS exposure and a loss of membrane integrity, confirming a correlation between intracellular acidification and cell death (Fig. 6). The decrease in pHi was reflected by an increase in the proportion of the acidic subpopulation with a pHi below 6.7 and by the decrease of pHi in the main population to 6.97 ± 0.05. The apparent decrease in the proportion of cells with acidic pHi at 24 h was caused by the increase in the proportion of necrotic cells (PI-permeable cells), which are unable to efficiently incorporate the pH-sensitive fluorogen BCECF-AM (Fig. 6). Analysis of the cell cycle distribution of cells treated for 24 h with 50 μM HMA showed an increase in the proportion of cells in G1 and G2/M at the expense of cells in the S phase with no significant increase in cells with DNA fragmentation (sub-G1 fraction; data not shown).
Figure 6.

The effect of HMA on pHi, PS exposure, and membrane integrity in Daudi cells. Cells (5 × 105/ml) were incubated for the indicated times with 50 μM HMA. At the indicated times, half of the cells were loaded with BCECF-AM and analyzed for the level of pHi (A), and the other half was analyzed for PS exposure/PI exclusion by staining with FITC-Annexin V (FL1) and PI (FL3) (B). The numbers in the upper left corner in A represent the proportion of acidic cells. Number of negative, single, and double Annexin-V/PI positive cells is indicated in each quadrant of B. These are the results of a representative experiment of three experiments performed.
Discussion
The anti-IgM-mediated induction of cell death in B104 cells was preceded in the first 8 h by a decrease in both the pH and cell volume. However, these cells did not undergo DNA fragmentation or present morphological features of apoptosis after 24 h of treatment. In contrast, anti-IgM stimulation of Daudi cells led to an early pHi increase followed by a slow decrease by 24 h to 0.15 units below the control level, when cells are mainly in G1 arrest with little cell death. These data suggest that anti-IgM-induced cell death can be mediated by an early change of pHi and cell volume.
There are numerous examples in which apoptosis induced by signaling mechanisms or chemical stress is associated with intracellular acidification. Thus, intracellular acidification precedes apoptosis induced in anti-Fas-treated Jurkat cells (6) or in somatostatin-treated MCF-7 cells (7). Acidification-mediated apoptosis has also been reported in murine T cell lines deprived of the IL-2 (8, 9) or murine pre-B (10) or premast cells (11) deprived of IL-3. Intracellular acidification has been shown to correlate with apoptosis induced by different chemical stimuli, such as ionomycin (23), etoposide (12), cycloheximide (13), lovastatin (24), and camptothecin (14).
We have considered two mechanisms that might be responsible for the
above effects:
Na+/H+ exchangers
and Na-dependent and independent
Cl−/HCO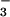 exchangers.
However, anti-IgM-mediated intracellular acidification of B104 cells in
the absence of sodium bicarbonate ruled out a contribution of
bicarbonate exchangers.
Na+/H+ exchange is
the most effective mechanism for maintaining pHi
homeostasis in mammalian cells under physiological conditions.
Intracellular alkalinization results from H+
extrusion and Na+ uptake, which also induce the
uptake of water, leading to an increase in cell volume (15). A second
function of NHE is therefore the regulation of cell volume. NHE1 is a
phosphoprotein expressed on virtually all tissues and cells, in
contrast to other isoforms whose expression is restricted to the
gastrointestinal tract and kidney. The balance between the kinase and
phosphatase activity is the major controller of its activity. Thus, the
different pHi resulting from anti-IgM treatment
on the two cell lines was associated with different levels of NHE1
phosphorylation as judged by the level of phosphoserine detected in the
NHE1 immunoprecipitated from cells stimulated up to 60 min.
exchangers.
However, anti-IgM-mediated intracellular acidification of B104 cells in
the absence of sodium bicarbonate ruled out a contribution of
bicarbonate exchangers.
Na+/H+ exchange is
the most effective mechanism for maintaining pHi
homeostasis in mammalian cells under physiological conditions.
Intracellular alkalinization results from H+
extrusion and Na+ uptake, which also induce the
uptake of water, leading to an increase in cell volume (15). A second
function of NHE is therefore the regulation of cell volume. NHE1 is a
phosphoprotein expressed on virtually all tissues and cells, in
contrast to other isoforms whose expression is restricted to the
gastrointestinal tract and kidney. The balance between the kinase and
phosphatase activity is the major controller of its activity. Thus, the
different pHi resulting from anti-IgM treatment
on the two cell lines was associated with different levels of NHE1
phosphorylation as judged by the level of phosphoserine detected in the
NHE1 immunoprecipitated from cells stimulated up to 60 min.
A wide variety of signals including growth factors, phorbol esthers, and phosphatase inhibitors induce NHE1 phosphorylation and enhance its affinity for cytosolic H+ leading to alkalinization (22). At least three serine/threonine kinases can account for the activation of NHE1: protein kinase C (25, 26), calmodulin kinase II (27), and mitogen-activated protein kinase (28–30). Theoretically, all of these kinases might account for the NHE1 phosphorylation in anti-IgM-treated cells. Thus, activation of the src family kinases after BCR crosslinking leads to phospholipase Cγ-mediated hydrolysis of phosphoinositide bisphosphate into inositol triphosphate and diacylglycerol leading to Ca2+-dependent activation of calmodulin kinase II and calcineurin, and activation of protein kinase C isoenzymes, respectively. Alternatively, activation of the Ras or Rho pathway via activated syk tyrosine kinase leads to activation of mitogen-activated protein kinases. Regardless of the effector kinase, the resulting alkalinization is critical for cell survival.
Here, we propose that cytosolic alkalinization or prevention of acidification has a protective effect in anti-IgM-mediated cell death in B lymphoma cells. The protective effect of the increased pHi has been demonstrated in both receptor- and chemotherapy-mediated apoptosis. Thus, pHi elevation by imidazole or chloroquine prevents Fas-mediated apoptosis in Jurkat cells (6), whereas NHE-mediated intracellular alkalinization plays an important role in PMA, IL-3, granulocyte macrophage colony-stimulating factor (31), and stem cell factor (32) suppression of apoptosis in interleukin-3-dependent cell lines. Induction of multidrug resistance by expression of P-glycoprotein on tumor cells was correlated with an increased pHi, which prevented cytosolic drug accumulation (33). Reversal of the protective effect conferred by P-glycoprotein with specific inhibitors (e.g., verapamil) was associated with a reduced pHi and an increased sensitivity to apoptotic stimuli.
We postulate that acidification in B104 cells treated with anti-IgM is induced as a result of the NHE1 dephosphorylation by calcineurin phosphatase. This is suggested by the finding that the prevention of anti-IgM-induced cell death in B104 cells by CsA was associated with the total abrogation of the early intracellular acidification and increase in NHE1 phosphorylation. This suggests that the mechanism(s) by which CsA can render cells resistant to BCR-mediated negative signaling involves inactivation of calcineurin, which in turn directly or indirectly controls NHE1 activation. Calcineurin can achieve this by direct dephosphorylation of or binding to NHE1. In addition to its extensively described role in mediating apoptosis in T cells by dephosphorylation and activation of the nuclear factor of activated T cells (NF-AT) (34), recent evidence supports a role for calcineurin in transcription-independent apoptosis. Thus, calcineurin has been implicated in dephosphorylation of the pro-apoptotic protein BAD, leading to mitochondria-mediated apoptosis in neuronal cells (35). This pro-apoptotic role could be partially inhibited by the specific inhibitors CsA and FK506 (36). The second possibility is suggested by the fact that a constitutively phosphorylated calcineurin B homolog protein can associate with and inhibit NHE1 (37).
The present results suggest that a dysfunction/inhibition of the NHE1, resulting in intracellular acidification after anti-IgM treatment of B lymphoma cells, could favor apoptosis or could represent an important cause of cellular damage. The first possibility is suggested by the fact that various pathways are directly influenced by pHi. Thus, up-regulation (38) or dimerization (39) of Bax proteins as well as caspase activation (10) are triggered by an acidic pHi. It has recently been shown that the cytosolic pH of the multidrug-resistant MCF-7 human breast cancer cells is 0.4 units higher than that of the drug-sensitive ones (40). In contrast, an acidic intracellular environment can sensitize cells to chemotherapy. In this context, the potential chemosensitization of tumor cells via modulation of pHi by therapeutic signaling antibodies (e.g., anti-CD20 or anti-HER2) has profound implications for combination antibody/chemotherapy and may partly explain their synergism reported in a series of experimental models and clinical trials (41–43).
In support for the second possibility, we found that inhibition of NHE1 in all of the B cell lines used in this study led to the induction of apoptosis, as judged by PS externalization and lost of membrane integrity. However, the levels of PS exposure and cell death induced by inhibition of NHE1 were not similar, suggesting that cells have different basal levels of Na+/H+ exchange activity.
NHE is an important regulator of the acidification that may occur during growth of a tumor in vivo. Because of insufficient vascular supply, many malignant cells have a high glycolytic activity that generates large amounts of H+ in the form of lactic acid. Therefore, they need to extrude H+ at the same rate it is synthesized to survive. This very active proton extrusion may account for the neutral or slightly alkaline cytosolic pH and the very acidic extracellular pH of tumors (44). This consideration suggests that NHE can be a therapeutic target, as recently reported (45).
As we mentioned above, cell death mediated by anti-IgM in B104 cells does not involve DNA fragmentation. However, the two manifestations of cell death, apoptosis and necrosis, coexist in many situations, and their balance depends on the ATP level produced by mitochondrial respiration and glycolysis. We propose that in addition to ATP-dependent mechanisms, modulation of pHi and cell volume by ATP-independent mechanisms may contribute to the nature, intensity, and rate of cell death. Hence, signaling receptors or pharmacological agents can initiate a plethora of cell death pathways that may not be restricted to classical apoptosis, as judged by activation of caspase cascade, DNA fragmentation, and typical morphological features.
Acknowledgments
We are deeply appreciative to Dr. Mitsufumi Mayumi and Dr. Ed Clark for providing the B104 cell line, Dr. Peter Krammer for the BL60 cell line, and Dr. Orson Moe for the rabbit anti-NHE1 antiserum. We thank Dr. Xiaodong Wang for his critical reading of the manuscript. This research was supported by American Cancer Society Grant RPG-99-337-01-CIM.
Abbreviations
- BCR
B cell antigen receptor
- pHi
intracellular pH
- CsA
cyclosporin A
- NHE
Na+/H+ exchanger
- HMA
5-(N,N-hexamethylene)amiloride
- BCECF-AM
2′,7′bis-carboxyethyl-5(6)carboxyfluorescein, acetoxymethyl ester
- PI
propidium iodide
- PS
phosphatidylserine
Footnotes
This is paper no. 9 of a series.
References
- 1.Bras A, Ruiz-Vela A, Gonzalez de Buitrago G, Martinez A C. FASEB J. 1999;13:931–944. doi: 10.1096/fasebj.13.8.931. [DOI] [PubMed] [Google Scholar]
- 2.Berard M, Mondiere P, Casamayor-Palleja M, Hennino A, Bella C, DeFrance T. J Immunol. 1999;163:4655–4662. [PubMed] [Google Scholar]
- 3.Chen W, Wang H G, Srinivasula S M, Alnemri E S, Cooper N R. J Immunol. 1999;163:2483–2491. [PubMed] [Google Scholar]
- 4.Graves J D, Draves K E, Craxton A, Krebs E G, Clark E A. J Immunol. 1998;161:168–174. [PubMed] [Google Scholar]
- 5.Lens S M, den Drijver B F, Potgens A J, Tesselaar K, van Oers M H, van Lier R A. J Immunol. 1998;160:6083–6092. [PubMed] [Google Scholar]
- 6.Gottlieb R A, Nordberg J, Skowronski E, Babior B M. Proc Natl Acad Sci USA. 1996;93:654–658. doi: 10.1073/pnas.93.2.654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liu D, Martino G, Thangaraju M, Sharma M, Halwani F, Shen S H, Patel Y C, Srikant C B. J Biol Chem. 2000;275:9244–9250. doi: 10.1074/jbc.275.13.9244. [DOI] [PubMed] [Google Scholar]
- 8.Li J, Eastman A. J Biol Chem. 1995;270:3203–3211. doi: 10.1074/jbc.270.7.3203. [DOI] [PubMed] [Google Scholar]
- 9.Rebollo A, Gomez J, Martinez de Aragon A, Lastres P, Silva A, Perez-Sala D. Exp Cell Res. 1995;218:581–585. doi: 10.1006/excr.1995.1195. [DOI] [PubMed] [Google Scholar]
- 10.Furlong I J, Ascaso R, Lopez Rivas A, Collins M K. J Cell Sci. 1997;110:653–661. doi: 10.1242/jcs.110.5.653. [DOI] [PubMed] [Google Scholar]
- 11.Chen Q, Benson R S, Whetton A D, Brant S R, Donowitz M, Montrose M H, Dive C, Watson A J. J Cell Sci. 1997;110:379–387. doi: 10.1242/jcs.110.3.379. [DOI] [PubMed] [Google Scholar]
- 12.Barry M A, Reynolds J E, Eastman A. Cancer Res. 1993;53:2349–2357. [PubMed] [Google Scholar]
- 13.Gottlieb R A, Nordberg J, Skowronski E, Babior B M. Proc Natl Acad Sci USA. 1996;93:654–658. doi: 10.1073/pnas.93.2.654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Goossens J F, Henichart J P, Dassonneville L, Facompre M, Bailly C. Eur J Pharmacol Sci. 2000;10:125–131. doi: 10.1016/s0928-0987(99)00091-3. [DOI] [PubMed] [Google Scholar]
- 15.Orlowski J, Grinstein S. J Biol Chem. 1997;272:22373–22376. doi: 10.1074/jbc.272.36.22373. [DOI] [PubMed] [Google Scholar]
- 16.Kim K M, Yoshimura T, Watanabe H, Ishigami T, Nambu M, Hata D, Higaki Y, Sasaki M, Tsutsui T, Mayumi M, et al. J Immunol. 1991;146:819–825. [PubMed] [Google Scholar]
- 17.Ishigami T, Kim K M, Horiguchi Y, Higaki Y, Hata D, Heike T, Katamura K, Mayumi M. J Immunol. 1992;148:360–368. [PubMed] [Google Scholar]
- 18.Marches R, Scheuermann R H, Uhr J W. Cancer Res. 1999;58:691–697. [PubMed] [Google Scholar]
- 19.Gerson D F, Kiefer H. J Cell Physiol. 1982;112:1–4. doi: 10.1002/jcp.1041120102. [DOI] [PubMed] [Google Scholar]
- 20.Gerson D F, Kiefer H. J Cell Physiol. 1983;114:132–136. doi: 10.1002/jcp.1041140121. [DOI] [PubMed] [Google Scholar]
- 21.Ingber D E, Prusty D, Frangioni J V, Cragoe E J, Jr, Lechene C, Schwartz M A. J Cell Biol. 1990;110:1803–1811. doi: 10.1083/jcb.110.5.1803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wakabayashi S, Fafournoux P, Sardet C, Pouyssegur J. Proc Natl Acad Sci USA. 1992;89:2424–2428. doi: 10.1073/pnas.89.6.2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Barry M A, Eastman A. Biochem Biophys Res Commun. 1992;186:782–789. doi: 10.1016/0006-291x(92)90814-2. [DOI] [PubMed] [Google Scholar]
- 24.Perez-Sala D, Collado-Escobar D, Mollinedo F. J Biol Chem. 1995;270:6235–6242. doi: 10.1074/jbc.270.11.6235. [DOI] [PubMed] [Google Scholar]
- 25.Ober S, Pardee B. J Cell Physiol. 1987;132:311–317. doi: 10.1002/jcp.1041320216. [DOI] [PubMed] [Google Scholar]
- 26.Vicentini L M, Villereal M L. Life Sci. 1986;38:2269–2276. doi: 10.1016/0024-3205(86)90632-6. [DOI] [PubMed] [Google Scholar]
- 27.Fliegel L, Walsh M P, Singh D, Wong C, Barr A. Biochem J. 1992;282:139–145. doi: 10.1042/bj2820139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang H, Silva N L, Lucchesi P A, Haworth R, Wang K, Michalak M, Pelech S, Fliegel L. Biochemistry. 1997;36:9151–9158. doi: 10.1021/bi970802f. [DOI] [PubMed] [Google Scholar]
- 29.Aharonovitz O, Granot Y. J Biol Chem. 1996;271:16494–16499. doi: 10.1074/jbc.271.28.16494. [DOI] [PubMed] [Google Scholar]
- 30.Bianchini L, L'Allemain G, Pouyssegur J. J Biol Chem. 1997;272:271–279. doi: 10.1074/jbc.272.1.271. [DOI] [PubMed] [Google Scholar]
- 31.Rajotte D, Haddad P, Haman A, Cragoe E J, Jr, Hoang T. J Biol Chem. 1992;267:9980–9987. [PubMed] [Google Scholar]
- 32.Caceres-Cortes J, Rajotte D, Dumouchel J, Haddad P, Hoang T. J Biol Chem. 1994;269:12084–12091. [PubMed] [Google Scholar]
- 33.Simon S, Roy D, Schindler M. Proc Natl Acad Sci USA. 1994;91:1128–1132. doi: 10.1073/pnas.91.3.1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Latinis K M, Norian L A, Eliason S L, Koretzky G A. J Biol Chem. 1997;272:31427–31434. doi: 10.1074/jbc.272.50.31427. [DOI] [PubMed] [Google Scholar]
- 35.Wang H G, Pathan N, Ethell I M, Krajewski S, Yamaguchi Y, Shibasaki F, McKeon F, Bobo T, Franke T F, Reed J C. Science. 1999;284:339–343. doi: 10.1126/science.284.5412.339. [DOI] [PubMed] [Google Scholar]
- 36.Asai A, Qiu J H, Narita Y, Chi S, Saito N, Shinoura N, Hamada H, Kuchino Y, Kirino T. J Biol Chem. 1999;274:34450–34458. doi: 10.1074/jbc.274.48.34450. [DOI] [PubMed] [Google Scholar]
- 37.Lin X, Barber D L. Proc Natl Acad Sci USA. 1996;93:12631–12636. doi: 10.1073/pnas.93.22.12631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Park H J, Lyons J C, Ohtsubo T, Song C W. Br J Cancer. 1999;80:1892–1897. doi: 10.1038/sj.bjc.6690617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xie Z, Schendel S, Matsuyama S, Reed J C. Biochemistry. 1998;37:6410–6418. doi: 10.1021/bi973052i. [DOI] [PubMed] [Google Scholar]
- 40.Altan N, Chen Y, Schindler M, Simon S M. J Exp Med. 1998;187:1583–1598. doi: 10.1084/jem.187.10.1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Czuczman M S, Grillo-Lopez A J, White C A, Saleh M, Gordon L, LoBuglio A F, Jonas C, Klippenstein D, Dallaire B, Varns C. J Clin Oncol. 1999;17:268–276. doi: 10.1200/JCO.1999.17.1.268. [DOI] [PubMed] [Google Scholar]
- 42.Pegram M D, Lipton A, Hayes D F, Weber B L, Baselga J M, Tripathy D, Baly D, Baughman S A, Twaddell T, Glaspy J A, Slamon D J. J Clin Oncol. 1998;16:2659–2671. doi: 10.1200/JCO.1998.16.8.2659. [DOI] [PubMed] [Google Scholar]
- 43.Baselga J, Norton L, Albanell J, Kim Y M, Mendelsohn J. Cancer Res. 1998;58:2825–2831. [PubMed] [Google Scholar]
- 44.Vaupel P, Kallinowski F, Okunieff P. Cancer Res. 1989;49:6449–6465. [PubMed] [Google Scholar]
- 45.Rich I N, Worthington-White D, Garden O A, Musk P. Blood. 2000;95:1427–1434. [PubMed] [Google Scholar]