

Abstract
The small-colony-variant (SCV) phenotype of Staphylococcus aureus has been associated with difficult-to-treat infections, reduced antimicrobial susceptibility, and intracellular persistence. This study represents a detailed intra- and extracellular investigation of a clinical wild-type (WT) S. aureus strain and its counterpart with an SCV phenotype both in vitro and in vivo, using the THP-1 cell line model and the mouse peritonitis model, respectively. Bacteria of both phenotypes infected the mouse peritoneum intra- and extracellularly. The SCV phenotype was less virulent and showed distinct bacterial clearance, a reduced multiplication capacity, and a reduced internalization ability. However, some of the SCV-infected mice were still culture positive up to 96 h postinfection, and bacteria of this phenotype could spread to the mouse kidney and furthermore revert to the more virulent WT phenotype in both the mouse peritoneum and kidney. The SCV phenotype is therefore, despite reduced virulence, an important player in S. aureus pathogenesis. In the THP-1 cell line model, both dicloxacillin (DCX) and linezolid (LZD) reduced the intracellular inocula of bacteria of both phenotypes by approximately 1 to 1.5 log10 in vitro, while DCX was considerably more effective against extracellular bacteria. In the mouse peritonitis model, DCX and LZD were also able to control both intra- and extracellular infections caused by either phenotype. Treatment with a single dose of DCX and LZD was, however, insufficient to clear the SCVs in the kidneys, and the risk of recurrent infection remained. This stresses the importance of an optimal dosing of the antibiotic when SCVs are present.
Staphylococcus aureus infections are characterized by high rates of complications, a slow response to antibiotic treatment, and a high incidence of infection recurrence (12, 14, 22, 39). The presence of small-colony variants (SCVs), a naturally occurring subpopulation of S. aureus, has been associated with these persistent and recurring infections (36, 37, 55, 56). SCVs are phenotypically characterized by a slow growth rate, often nonpigmented and nonhemolytic colonies 1/10 the size of those of the parent (wild-type [WT]) phenotype, with reduced coagulase activity, thus presenting a challenge to the microbiology laboratory (37, 52, 55).
While hemin- and/or menadione-auxotrophic SCVs have been recovered from patients with chronic osteomyelitis, persistent soft tissue infection, and various other recurrent infections, thymidine-dependent SCVs have been detected mainly from cystic fibrosis patients, especially after the prior use of trimethoprim-sulfamethoxazole (7, 24). Random mutations in the thymidylate synthase-encoding thyA gene which lead to an intracellular lack of dTMP have been identified as being responsible for the formation of the these SCVs (6, 13, 57). Of particular interest, hypermutability due to a defective DNA mismatch repair system was shown to favor the acquisition of these mutations (8). Since thymidine-auxotrophic SCVs are able to use thymidine and/or the metabolite dTMP from the environment, this subpopulation is able to bypass the antibiotic effect of folic acid antagonists and, consequently, are resistant to trimethoprim-sulfamethoxazole (57).
The characteristics of hemin- and/or menadione-auxotrophic SCVs, which can also be reverted to the parent phenotype by the supplementation of these two compounds, are associated with a defective electron transport system (37). Defining the precise genetic lesions in these clinical SCVs has proven difficult (37), but Lannergard et al. have recently identified mutations in menB (which has a putative function in menadione biosynthesis) in clinical menadione-auxotrophic SCVs (25).
The intracellular presence of S. aureus has been increasingly recognized (9, 15, 19, 20, 29, 30), and studies have shown that SCVs are more efficiently internalized and survive inside cells better than the parent WT (43, 50, 54). It has further been shown that the intracellular milieu induces and/or selects for the SCV phenotype (51). The intracellular persistence of bacteria complicates the antibiotic treatment of staphylococcal infections (41, 48, 49), as most antibiotics have an impaired intracellular activity against this organism (3, 11, 41) and especially against SCVs (33).
Besides the intracellular presence, bacteria of the SCV phenotype are often less susceptible to several antibiotics than bacteria of the WT phenotype because of (i) an interruption of the electron transport, which reduces the electron gradient across the bacterial membrane and affects the uptake of some antibiotics such as aminoglycosides, and/or (ii) their reduced growth rate for cell wall-active antibiotics such as the β-lactams (35, 55).
In the present study we compared the virulence of a clinical S. aureus menadione-dependent SCV isolate (OM1b) to that of its WT counterpart (OM1a) in mice. By using a recently described mouse peritonitis intracellular S. aureus model (41), the intra- and extracellular presence of the bacteria could be distinguished, and the leukocyte responses for both phenotypes could be determined. Furthermore, kidneys were sampled to detect the metastatic spread of bacteria. We also investigated the relapse tendency of bacteria with the SCV phenotype in both the mouse peritoneum and kidneys. In parallel, intra- and extracellular treatment studies with both dicloxacillin (DCX) and linezolid (LZD) were performed in vitro and in vivo for both phenotypes. The impact of antibiotic treatment (DCX and LZD) on the tendency to relapse in both the mouse peritoneum and kidneys was also investigated further.
(Part of this study was presented at the 18th European Congress of Clinical Microbiology and Infectious Diseases, Barcelona, Spain, April 2007.)
MATERIALS AND METHODS
Bacterial strains and antimicrobial agents.
A clinical menadione-auxotrophic SCV isolate (OM1b) and the corresponding clonally related wild-type (WT) isolate (OM1a), recovered from a patient with chronic osteomyelitis (53), were used for all studies. These strains were described previously (25). DCX and LZD were procured as the commercial products registered in Denmark for parenteral use (Diclocil [Bristol-Myers Squibb Company, New York, NY] and Zyvoxid [Pfizer Inc., New York, NY], respectively). Both products complied with the provisions of the European Pharmacopeia.
PFGE typing.
Pulsed-field gel electrophoresis (PFGE) analysis was performed according to the HARMONY protocol (32), using the CHEF Mapper system. In brief, purified DNA was digested with the SmaI restriction enzyme, and the electrophoretic conditions were as follows: initial switch time of 5.0 s, final switch time of 60 s, run time of 23 h, gradient of 6 V/cm, angle of 120°, and running temperature of 14°C. The PFGE profiles were visually inspected for clonal relationships, and only identical profiles were interpreted as being clones.
In vitro susceptibility studies.
MICs were determined with Mueller-Hinton broth (Becton Dickinson, Cockeysville, MD) by using a standard microtiter tray method according to recommendations provided by the Clinical and Laboratory Standards Institute (16), as described previously (44). Readings were made after 24 h for bacteria with the WT phenotype and after 24 h and 48 h for bacteria with the SCV phenotype. When bacteria with the SCV phenotype were grown in Mueller-Hinton broth overnight, no reversion to the WT phenotype was observed.
In vitro extracellular concentration-kill studies.
Extracellular concentration-kill studies were performed for bacteria of both phenotypes (OM1a and OM1b) as described previously (3). In brief, bacteria exhibiting exponential growth were diluted in Mueller-Hinton broth to a density of 106 CFU/ml. DCX or LZD was added at concentrations from 0.001- to 1,000-fold the MIC, and the tubes were incubated at 35°C with shaking. Samples for CFU quantification were withdrawn before the antibiotic admixture and after 24 h of incubation.
In vitro intracellular concentration-kill studies with human THP-1 monocytes.
In vitro studies of the intracellular activities of DCX and LZD against bacteria of both phenotypes (OM1a and OM1b) were performed with THP-1 myelomonocytic cells (47) as described previously (26, 33).
In brief, (i) bacteria were opsonized with 10% nondecomplemented human serum (Lonza, Walkersville, MD) for 45 min; (ii) phagocytosis was performed during 1 h at a 4:1 bacterium/macrophage ratio, and nonphagocytosed bacteria were subsequently eliminated by incubating the cells in phosphate-buffered saline (PBS) containing gentamicin (50 mg/liter) (MICs were 0.25 and 1.0 mg/liter for bacteria with the WT and SCV phenotypes, respectively) for 45 min; (iii) after gentamicin washout, cells were resuspended in RPMI 1640 medium (Invitrogen Corporation, Paisley, Scotland) supplemented with 10% fetal calf serum; and (iv) DCX or LZD was added at the desired concentrations for tests, or gentamicin (GEN) (1× MIC) was added as a control (to prevent the extracellular growth of bacteria [2]). Samples (100 μl) for CFU quantification were withdrawn before the antibiotic admixture (time zero) and after 24 h of incubation; cells were collected by centrifugation, washed twice in PBS, and finally lysed in sterile water before the CFU quantification. The lower detection limit was 100 CFU/ml. The large dilution of the cellular material made during collection and spread onto the plates ensured the absence of any carryover effects of the antibiotic. In parallel to the CFU determination, the quantity of cell protein in the samples was determined as described previously (27, 44). All results from these studies are shown as CFU per mg of cell protein. The effects of DCX and LZD on the cell viability were assessed by the trypan blue exclusion test to calculate the ratio of live cells to dead cells. The antibiotic influence was tested for 24 h.
Mouse peritonitis model.
The mouse peritonitis model, performed as described previously, was used for all in vivo infection studies (17, 41). In short, outbred female NMRI mice (Harlan Netherlands, Horst, Netherlands) were used for all studies. Mice were inoculated 2 h before antibiotic treatment by the injection of a total of 2.5 × 107 CFU intraperitoneally (i.p.), if not stated otherwise (injection volume of 0.5 ml). Antibiotic treatments were administered subcutaneously (s.c.). Mice were euthanized before peritoneal sample collection; a peritoneal flush was performed by injecting 2.0 ml Hanks balanced salt solution (HBSS) (catalog no. H-9269; Sigma-Aldrich Inc., St. Louis, MO) i.p. The peritoneal fluid, containing murine cells and bacteria, was then collected in EDTA-coated tubes. White blood cell (WBC) counts in the peritoneal fluid were performed as described previously (41). Kidneys were also sampled in specific experiments to assess the metastatic spread of bacteria via the blood, since these samples are easy to collect intact and to homogenize for quantitative bacterial counts. Both kidneys were placed into one tube, and saline was added at 1 ml to facilitate the homogenization procedure. The tissue was homogenized with a TissueLyser (Qiagen, Ballerup, Denmark) and used for colony counts. All animal experiments were approved by the Danish Animal Experimentation Inspectorate (license no. 2004/561-835).
Separation of intra- and extracellular S. aureus cells in peritoneal fluid.
The separation assay was performed as a modified version of a procedure described previously (41). The collected peritoneal fluid from one mouse was diluted 1:1 with HBSS. The total CFU count of the diluted sample was determined before any further procedures were performed. The diluted sample was then divided into two equal fractions of ∼1.5 ml each (fractions A and B). For extracellular CFU quantification, fraction A was centrifuged at 300 × g at room temperature for 10 min, and the extracellular CFU in the supernatant was quantified. For intracellular CFU quantification, lysostaphin (catalog no. L-7386; Sigma-Aldrich Inc.) was added to fraction B to a final concentration of 15 mg/liter, and the fraction was incubated for 15 min at room temperature. These conditions ensured a complete killing of the extracellular bacteria contaminating the preparation, as shown previously (41). The lysostaphin was removed by centrifugation (300 × g), and the fraction was prepared for CFU quantification as described previously (41).
Data analysis.
Data from concentration-kill studies were analyzed by using the sigmoidal dose-response equation using a slope factor of 1 (three-parameter logistic equation), which allowed the calculation of the Emin, Emax, EC50, Cstatic, and goodness of fit (R2). Emin corresponds to the bacterial growth (in log10 CFU units) relative to that of the original inoculum (measured just before antibiotic admixture), as extrapolated for an infinitely low concentration of antibiotic; Emax corresponds to the decrease of bacterial counts (in log10 CFU) relative to that of the original inoculum, as extrapolated for an infinitely high concentration of antibiotic. EC50 indicates the concentration needed to obtain a change in bacterial counts halfway between the Emin and the Emax. Cstatic was defined as the concentration needed for a static effect (no apparent change in the number of CFU relative to that in the original inoculum). The best-fit values for the Emax and EC50 were compared between intra- and extracellular data or between phenotypes by using the extra-sum-of-squares F test. Data for the mouse treatment groups were compared by using the Kruskal-Wallis test followed by Dunn's multiple-comparison test when more than 2 groups were compared; otherwise, the Mann-Whitney test was applied. A P value of <0.05 was considered significant. All curve fittings and statistical analyses were performed by using GraphPad Prism 5.0 (GraphPad Prism Software, San Diego, CA).
RESULTS
In vitro THP-1 cell model.
The intracellular growth of bacteria was assessed at 24 h in the presence of gentamicin (1× the MIC) to control extracellular growth and showed a difference of about 1 log10 CFU between the WT and the SCV strains intracellularly (1.96 ± 0.46 log10 CFU versus 1.10 ± 0.42 log10 CFU, respectively), as described previously by others studies dealing with SCV isolates (see reference 33 for details about inoculum sizes and proportions of internalized bacteria, which were similar for both phenotypes). This intracellular growth was similar to that described previously for the WT phenotype of another SCV strain but larger than that of its SCV counterpart (0.10 to 0.21 log10 CFU only) (33). However, the level of intracellular growth was consistently lower than that in broth (3-log10 and 2-log10 increases in 24 h for WT and SCV bacteria, respectively).
Six-hour infection course study in the in vivo mouse peritonitis model.
Mice were inoculated with the SCV or the WT, and the peritoneal fluid was sampled 2 and 6 h after inoculation (n = 12) and immediately processed for WBC counts and for the intra- and extracellular separation assay. Data are shown in Fig. 1. After 2 h of infection the intracellular amount of SCV bacteria was significantly (P = 0.0022) lower than the amount of WT bacteria. No significant difference was observed between the two phenotypes extracellularly or when the two compartments were observed combined (total CFU count) (Fig. 1A). After 6 h of infection a significantly smaller amount of SCV bacteria was observed for all three fractions than the amount of WT bacteria, totally (P = 0.0023), extracellularly (P = 0.0337), and intracellularly (P = 0.0022) (Fig. 1A). The leukocyte responses were generally equal between the two phenotypes (Fig. 1B). However, a small significant difference was observed for the lymphocyte count after 6 h of infection (P = 0.013). The clinical appearances of the mice were very different depending on the bacterial phenotype, as the mice infected with the WT showed clear signs of sickness (e.g., piloerection, wasp waist, cold skin, stiff movements with a lack of curiosity, closed eyes, and forced ventilation), while the SCV-infected mice appeared healthy.
FIG. 1.

Illustrations of the increase in numbers of CFU (A) and white blood cells (B) in the peritoneal fluid from NMRI mice after 2 and 6 h of infection with S. aureus strains with the wild-type (WT) and the small-colony-variant (SCV) phenotypes, respectively. (A) Increase in CFU in total and when separated by intra- and extracellular growth. (B) Amount of granulocytes and lymphocytes in the mouse peritoneum at 2 and 6 h postinfection. Each set of data corresponds to 12 animals. Significant difference are marked by asterisks (* = 0.05 > P > 0.01; ** = 0.01 > P > 0.001).
Twenty-four hour infection course study and impact of inoculum in the in vivo mouse peritonitis model.
Since the amount of bacteria injected into the peritoneum might affect the infection course and, therefore, the virulence of the SCV phenotype, mice were infected with a low (106 CFU/ml) or a high (108 CFU/ml) inoculum (n = 6) of bacteria of the SCV phenotype. The results are shown in Fig. 2. The amount of bacteria in the peritoneal fluid was different after 2 h with respect to the high and low inocula, at (5.58 ± 1.01) × 106 CFU/ml and (9.88 ± 1.59) × 104 CFU/ml, respectively. However, the infection course was not influenced by the inoculum used, as shown in Fig. 2A. The leukocyte responses were also equal between the high- and low-inoculum-infected mice (Fig. 2B). All mice appeared healthy after 24 h of infection.
FIG. 2.
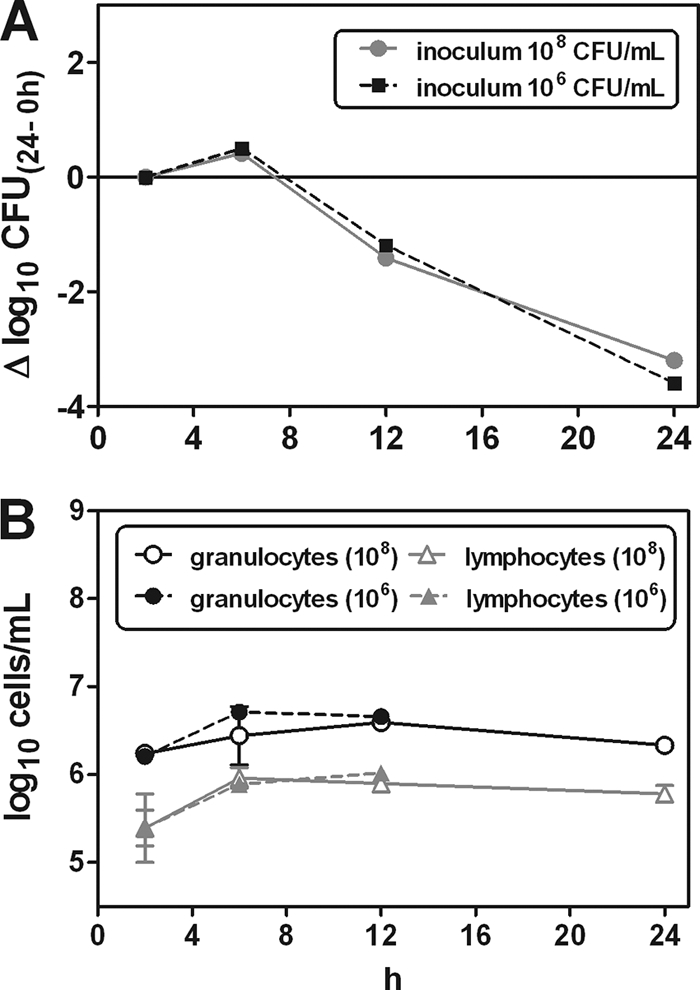
(A) Infection course for S. aureus bacteria with the small-colony-variant (SCV) phenotype in the mouse peritoneum. Mice were inoculated intraperitoneally (i.p.) with a high inoculum of 108 CFU/ml and a low inoculum of 106 CFU/ml. After 2 h of infection the bacterial loads in mice inoculated with the high and low inocula were (5.58 ± 1.01) × 106 CFU/ml and (9.88 ± 1.59) × 104 CFU/ml, respectively (n = 3). Baseline corrections were made according to the 2-h colony count. (B) Amount of white blood cells measured in parallel with the CFU estimations. The amounts of granulocytes and lymphocytes are also shown in relation to the initial inoculum size; the high inoculum was 108 CFU/ml, and the low inoculum was 106 CFU/ml. Data are provided as means and bars denoting the standard deviations (SD) of data from 3 mice per point.
Ninety-six hour infection course study in the in vivo mouse peritonitis model.
A 96-h study was performed for the SCV phenotype in order to detect the incidence of clearance and relapse of the infection in both the mouse peritoneum and the kidneys. The kidneys were used for the detection of metastatic spread. Mice were inoculated i.p. with bacteria of the SCV phenotype, and peritoneal fluid and kidneys were sampled at 2, 6, 24, 48, 72, and 96 h, respectively, after infection onset and processed for CFU determinations and PFGE analysis. In counting the CFU, morphological differentiation was used to distinguish between the SCV phenotype and WT phenotypes that possibly resulted from the reversion of the SCVs. The clonality of the bacteria sampled in the peritoneum and in the kidneys (both SCV and the reverted WT bacteria) was verified by PFGE. Results are displayed in Fig. 3.
FIG. 3.

Ninety-six-hour infection course study of the small colony variant (SCV) in the mouse peritoneum and kidney. Mice were inoculated i.p. with bacteria of the SCV phenotype only (n = 8). Morphological discrimination between bacteria of the SCV phenotype (original small nonpigmented colonies) and those of the reverted wild-type (WT) phenotype (large pigmented colonies) was performed for each sample between 2 h and 96 h postinoculation. Each sample is therefore representative of both an SCV colony count (original phenotype) and a WT colony count (reverted WT phenotype).
(i) Peritoneal fluid.
Figure 3A shows that the amount of SCVs decreased for the first 48 h. However, a positive culture of the peritoneal fluid was maintained for some of the mice between 48 and 96 h. For one mouse sampled at 72 h and one sampled at 96 h, the culture indicated a relapse of the infection, as the amount of bacteria was higher than that detected at 48 h. Even though the mice were infected only with bacteria of the SCV phenotype, reversion to a WT phenotype was already detected after 24 h of infection. The incidence of revertants decreased from 24 h to 48 h but phenotypically WT colonies were still present at 96 h postinfection.
(ii) Kidneys.
SCVs appeared in the mouse kidney at as early as 2 h postinfection (Fig. 3B). Although some of the mice seemed to clear the infection in the kidneys, most mice had positive cultures at 96 h postinfection, and some had the same number or a greater number of bacteria at 96 h than that detected in the mice sampled at 6 h postinfection. Revertants with WT phenotypes were present in the kidneys at 24 h to 96 h postinfection.
In vitro concentration-kill studies in broth (extracellular) and THP-1 cells (intracellular).
Intra- and extracellular concentration-kill studies with DCX and LZD were performed at 24 h for both strains OM1a (WT) and OM1b (SCV) in vitro by using the THP-1 cell model. These studies enabled the detection of possible pharmacodynamic differences between the two phenotypes both intra- and extracellularly. The results for the intra- and the extracellular concentration-kill studies are shown graphically in Fig. 4 and 5, and the pertinent regression parameters are listed in Table 1.
FIG. 4.

In vitro concentration-response curves for dicloxacillin (DCX) against extracellular and intracellular S. aureus bacteria. The activities against bacteria of both the small-colony-variant (SCV) phenotype and the wild-type (WT) phenotype were measured. The graphs show the activity as changes in the numbers of CFU at 24 h compared to the numbers in the initial inoculum (Δlog10 CFU) (means ± SD; n = 3). The changes in numbers of CFU were measured as CFU/ml of broth (extracellular bacteria) and as CFU per mg of cell protein (intracellular bacteria). The original inocula for the extracellular study (time zero) were (1.24 ± 0.12) × 106 CFU/ml for bacteria of the SCV phenotype and (1.82 ± 0.11) × 106 CFU/ml for bacteria of the WT phenotype (n = 3). The original (postphagocytosis) inocula for the intracellular study (time zero) were (5.65 ± 1.10) × 106 CFU/mg protein for bacteria of the SCV phenotype and (4.47 ± 0.57) × 106 CFU/mg protein for bacteria of the WT phenotype (n = 3). The sigmoidal function was used. Goodness-of-fit and regression parameters are shown in Table 1. (Upper panel) Comparison of intra- and extracellular activities for each strain versus the extracellular weight concentration (mg/liter). (Lower panel) Comparison of the extracellular (left) and intracellular (right) activities for the two strains versus equipotent extracellular concentrations (multiples of MICs).
FIG. 5.

In vitro concentration-response curves for linezolid (LZD) against extracellular and intracellular S. aureus. Activities against bacteria of both the small-colony-variant (SCV) phenotype and the wild-type (WT) phenotype were measured. The graphs show the activity as changes in the numbers of CFU at 24 h (Δlog10 CFU) (means ± SD; n = 3) compared to numbers in the initial inoculum. This was measured as CFU/ml of broth (extracellular bacteria) and as CFU per mg of cell protein (intracellular bacteria). The original inocula for the extracellular study (time zero) were (1.49 ± 0.33) × 106 CFU/ml for bacteria of the SCV phenotype and (1.58 ± 0.60) × 106 CFU/ml for bacteria of the WT phenotype (n = 3). The original (postphagocytosis) inocula for the intracellular study (time zero) were (5.57 ± 2.33) × 106 CFU/mg protein for bacteria of the SCV phenotype and (5.69 ± 1.68) × 106 CFU/mg protein for bacteria of the WT phenotype (n = 3). The sigmoidal function was used. Goodness-of-fit and regression parameters are shown in Table 1. (Upper panel) Comparison of intra- and extracellular activities for each strain versus the extracellular weight concentration (mg/liter). (Lower panel) Comparison of the extracellular (left) and intracellular (right) activities for the two strains versus equipotent extracellular concentrations (multiples of MICs).
TABLE 1.
In vitro intra- and extracellular regression data for 24-h concentration-kill studies of DCX and LZD against S. aureus bacteria with the SCV and WT phenotypes (OM1b and OM1a, respectively)
| Antibiotic | Phenotype | MIC (mg/liter) | Extracellular value (95% confidence interval)a |
R2 | Intracellular value (95% confidence interval)b |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Emaxc (Δlog10 CFU) | EC50d (mg/liter) | EC50 (multiple of MIC) | Cstatice (mg/liter) | Cstatic (multiple of MIC) | Emaxc (Δlog10 CFU) | EC50d (mg/liter) | EC50 (multiple of MIC) | Cstatice (mg/liter) | Cstatic (multiple of MIC) | R2 | ||||
| DCX | WT | 0.25 | −4.14 (−5.31-−2.97) | 0.20 (0.09-0.48) | 0.81 (0.35-1.90) | 0.18 (0.10-0.31) | 0.70 (0.40-1.25) | 0.92 | −1.01 (−1.94-−0.08) | 0.11 (0.03-0.40) | 0.26 (0.16-0.42) | 0.37 (0.14−1.45) | 1.49 (0.56-5.79) | 0.87 |
| SCV | 0.06 | −2.99 (−3.34-−2.64) | 0.11 (0.07-0.15) | 1.73 (1.22-2.44) | 0.08 (0.06-0.10) | 1.29 (1.03-1.65) | 0.99 | −1.59 (−1.85-−1.32) | 0.02 (0.01-0.02) | 0.45 (0.13-1.59) | 0.014 (0.01-0.02) | 0.23 (0.17-0.31) | 0.97 | |
| LZD | WT | 1.0 | −1.54 (−2.52-−0.55) | 1.49 (0.42-5.27) | 1.49 (0.42-5.27) | 3.46 (1.34-7.38) | 3.46 (1.34-7.38) | 0.86 | −0.98 (−1.46-−0.50) | 1.06 (0.55-2.01) | 1.06 (0.55-2.01) | 3.31 (2.03-5.60) | 3.31 (2.03-5.60) | 0.95 |
| SCV | 1.0 | −1.38 (−1.38-−0.92) | 3.57 (1.81-7.04) | 3.57 (1.81-7.04) | 5.92 (7.45-3.17) | 5.92 (7.45-3.17) | 0.96 | −1.47 (−1.81-−1.12) | 0.20 (0.07-1.37) | 0.20 (0.07-1.37) | 0.25 (0.17-0.36 | 0.25 (0.17-0.36 | 0.95 | |
For DCX, the original inocula (time zero) were (1.82 ± 0.11) × 106 CFU/ml for the WT phenotype and (1.24 ± 0.12) × 106 CFU/ml for the SCV phenotype (n = 3). For LZD, the original inocula (time zero) were (1.58 ± 0.60) × 106 CFU/ml for the WT phenotype and (1.49 ± 0.33) × 106 CFU/ml for the SCV phenotype (n = 3).
For DCX, the original (postphagocytosis) inocula (time zero) were (4.47 ± 0.51) × 106 CFU/mg protein for the WT phenotype and (5.65 ± 1.11) × 106 CFU/mg protein for the SCV phenotype (n = 3). For LZD, the original (postphagocytosis) inocula (time zero) were (5.69 ± 1.68) × 106 CFU/mg protein for the WT phenotype and (4.57 ± 2.33) × 106 CFU/mg protein for the SCV phenotype (n = 3).
Emax, decrease in the numbers of CFU (on a log10 scale) at 24 h from the numbers in the corresponding original inoculum, as extrapolated for an infinitely large antibiotic concentration (obtained from the Hill equation).
EC50, concentrations causing a reduction of the inoculum halfway between the increase in bacterial counts (in log10 CFU) for an infinitely low antibiotic concentration (Emin) and Emax values, as obtained from the Hill equation.
Cstatic, concentrations resulting in no apparent bacterial growth (number of CFU identical to that of the original inoculum), as determined by graphical intrapolation.
(i) Results obtained for DCX.
The maximal intracellular relative efficacy of DCX (Emax) was significantly impaired for both phenotypes (P = 0.0002 for the WT and P < 0.0001 for the SCV) compared to the extracellular efficacy (Fig. 4.). Despite this reduced intracellular efficacy, DCX showed a reduction of 1 to 1.5 log10 CFU compared to the CFU in the original inoculum intracellularly. There was no significant difference between the two phenotypes according to their intracellular maximal relative efficacies (Emax values). However, the extracellular maximal relative efficacy obtained for the SCV phenotype was slightly lower (fewer negative Emax values) than that obtained for the WT phenotype (P = 0.0476). When considering the relative potency (EC50) of DCX, equal values were obtained intra- and extracellularly for the WT phenotype. The relative potency of DCX, however, was significantly better (lower EC50) intracellularly than extracellularly for the SCV phenotype (P < 0.0001). Furthermore, when the relative potencies between phenotypes were compared, in multiples of the MIC value, equal values were observed both intra- and extracellularly.
(ii) Results obtained for LZD.
For both phenotypes, the maximal relative efficacies (Emax) were equal intra- and extracellularly (Fig. 5). Furthermore, there was no significant difference between the two phenotypes according to either the intracellular or extracellular Emax values. The reductions obtained were 1 to 1.5 log10 CFU (Table 1). When considering the relative potency of LZD (EC50) against bacteria of the WT phenotype, equal values were obtained intra- and extracellularly. However, for the SCV phenotype, the relative potency of LZD was significantly higher (lower EC50) intracellularly than extracellularly (P < 0.0001). Furthermore, when the relative potencies between phenotypes were compared, equal values were obtained extracellularly, but different ones were observed intracellularly (P = 0.0002), as LZD was more potent against bacteria of the SCV phenotype than against bacteria of the WT phenotype.
Single-dose 24-h intra- and extracellular time-kill studies in the in vivo mouse peritonitis model.
Intra- and extracellular time-kill studies (24-h exposure) with DCX and LZD were performed for both phenotypes in vivo. Mice were treated with a single s.c. dose of DCX (60 mg/kg of body weight) or LZD (17 mg/kg) 2 h after inoculation (n = 3). The peritoneal fluid was sampled just before (time zero) and 4 h and 24 h after treatment onset and immediately processed for the intra- and extracellular separation assay and CFU determinations. The results are displayed in Fig. 6. If the WT-inoculated mice remained untreated, they displayed signs of irreversible sickness after 6 h of infection, motivating the exclusion of this group from the 24-h time point.
FIG. 6.

Single-dose 24-h intra- and extracellular time-kill studies of dicloxacillin (DCX) and linezolid (LZD) against bacteria of the small-colony-variant (SCV) phenotype and the wild-type phenotype in the mouse peritoneum. Mice were inoculated i.p. with bacteria of the SCV phenotype and the WT phenotype. Mice were then treated s.c. with a single dose of DCX (60 mg/kg) or LZD (17 mg/kg). The graphs show activity as changes in numbers of CFU (Δlog10 CFU) (means ± SD; n = 3). The effect was estimated both in total and when differentiated into intra- and extracellular fractions.
(i) Results obtained for DCX.
Considering the WT phenotype of OM1a, the treatment with DCX was clearly effective, as the mice could survive for 24 h, in contrast to the untreated mice, and the effect increased with an increased time of exposure. A different picture appeared, however, for the OM1b (SCV phenotype)-infected mice; after 4 h of treatment, the difference between the treated and untreated mice was significant, but after 24 h, no significant difference was observed between the treated and untreated mice due to the extensive clearance observed for bacteria of the SCV phenotype.
(ii) Results obtained for LZD.
Considering the WT phenotype, the treatment with LZD was effective, as the mice could survive for 24 h compared to the untreated mice, and the effect increased with increasing times of exposure both intra- and extracellularly. For the SCV phenotype-infected mice, differences between treated and untreated mice after 4 h of treatment were significant for the extracellular compartment but not the intracellular compartment. After 24 h, however, no significant difference was observed between the treated and untreated mice either extra- or intracellularly due to the extensive bacterial clearance observed for the SCV phenotype.
Single-dose 96-h infection studies in the in vivo mouse peritonitis model.
A 96-h treatment study was performed for the SCV phenotype in order to detect the influence of a single dose of DCX or LZD on the relapse of infection in both the mouse peritoneum and the kidneys. Mice were inoculated i.p. with bacteria of the SCV phenotype and were treated with one single s.c. dose of DCX (60 mg/kg) or LZD (17 mg/kg) 2 h after inoculation (n = 8). The peritoneal fluid and the kidneys were sampled 2, 24, and 96 h postinfection, respectively, for CFU determinations. For CFU detection, morphological differentiations were made between the SCV phenotype and the WT phenotype, possibly due to reversion, and the genotype was verified by PFGE. Results are displayed in Fig. 7.
FIG. 7.

Single-dose 96-h infection study of dicloxacillin (DCX) and linezolid (LZD) against bacteria of the small-colony-variant (SCV) phenotype in the mouse peritoneum and kidney. Mice were inoculated i.p. with bacteria of the SCV phenotype only (n = 8). Mice were then treated s.c. with a single dose of DCX (60 mg/kg) or LZD (17 mg/kg). Morphological discrimination between bacteria of the SCV phenotype (original small nonpigmented colonies) and bacteria of the reverted wild-type (WT) phenotype (large pigmented colonies) was performed for each sample 24 and 96 h after inoculations. Each sample is therefore representative of both an SCV colony count and a WT colony count.
(i) Peritoneal fluid.
A significant clearance of bacteria of the SCV phenotype was observed for the first 24 h, and there was no significant difference between any of the groups (Fig. 7A). No revertants to WT phenotypes were observed for either group at 24 h postinfection. At 96 h, all the mice had bacteria cleared from their peritoneum completely. One mouse, however, had positive cultures of the WT phenotype at 96 h postinfection.
(ii) Kidneys.
A significant clearance of the SCVs was also observed for the kidneys for the first 24 h. The mice treated with DCX, however, had significantly lower bacterial counts in the kidneys than did the control groups after 24 h. The LZD-treated mice were not significantly different from the control group after 24 h. A larger amount of SCVs was detected in the kidneys at 96 h than at the beginning of the study, covering both potential bacterial growth and the accumulation of bacteria filtrated from the blood. At this time point, neither the DCX treatment group nor the LZD treatment group was found to be significantly different compared to the control group, and after a single dose they were unable to prevent the increase in colony counts from 24 to 96 h. The variability in both treatment groups, however, was noticeable, since there were mice with colony counts as high as those for the control group, while some mice had completely cleared the kidney infection. When the results obtained for the control groups in this experiment (Fig. 7) were compared with those obtained with the 96-h virulence study (Fig. 3), some differences appeared, as the colony counts and the incidence of revertants were higher for the infection course study in general (except for the kidney colony count at 96 h). This shows that the infection course with the SCV phenotype is highly unpredictable and associated with great variability.
DISCUSSION
The present study represents a detailed investigation of a clinical WT S. aureus strain and an isogenic isolate with the SCV phenotype. In comparison with data from a previously reported in vitro study (33), the data also cover in vivo intra- and extracellular colonization and infection course studies. With respect to treatment, we have focused on dicloxacillin and linezolid, two important antibiotics in the control of methicillin-susceptible and methicillin-resistant isolates, respectively.
Both phenotypes were able to induce infection both intra- and extracellularly in the mouse peritoneum and induced an equal immune response. However, the isolate with the SCV phenotype was less virulent (based on the appearance of the mice) and showed noticeable bacterial clearance and a reduced multiplicative capacity compared to the isolate with the WT phenotype. Bacteria of the SCV phenotype showed signs of reduced intracellular internalization compared to bacteria of the WT phenotype, as we observed significantly fewer intracellular SCV bacteria than WT bacteria after 2 h of infection but equal total colony counts and equal numbers of leukocytes at the infection site at the same time point. Therefore, the difference in the intracellular colony counts was not caused by a difference in the total amount of leukocytes or bacteria and might be better explained by a reduced internalization of bacteria of the SCV phenotype. The lower growth rate of bacteria of the SCV phenotype may also play a role. In spite of an extensive clearance of bacteria of the SCV phenotype, some of the mice were still culture positive at up to 96 h postinfection, and bacteria of this phenotype were able to spread via the blood to the mouse kidney (metastatic spread) and induced here a more persistent infection than that in the mouse peritoneum. A very important observation was the ability of bacteria of the SCV phenotype to revert to the more virulent WT phenotype in both the mouse peritoneum and kidney.
Most of our data support previous findings generated by different in vivo models. Quie previously showed a decrease in mortality and colonization with bacteria of the SCV phenotype compared to bacteria of the WT phenotype and observed a complete clearance of bacteria of both phenotypes after 14 days (38). Brouillette et al. also showed reduced tissue colonization and increased clearance of an SCV mutant (hemB mutant) compared to the isogenic parent (10). However, Pelletier et al. reported increased mortality associated with the SCV phenotype (34). Jonsson et al. showed a higher density of bacteria of the WT phenotype in both the kidneys and the joints than that of SCV mutant (hemB mutant) bacteria but more severe arthritis in mice infected with bacteria of the SCV phenotype (23). Finally, Sifri et al. showed that clinical SCVs as well as hemB- and menD-deficient mutants of S. aureus were greatly reduced in virulence in the Caenorhabditis elegans infection model (45).
Our detection of a reduced intracellular internalization of bacteria of the SCV phenotype is, however, surprising considering that previous studies have shown that bacteria of the SCV phenotype internalize and survive inside cells more efficiently than do bacteria of the WT phenotype (10, 43, 50, 54). However, our findings are strongly supported by data from a previous study by Sadowska et al. (40), who showed a reduced in vitro uptake of bacteria of the SCV phenotype compared to bacteria of the WT phenotype by granulocytes harvested from mouse peritoneum. This indicates that the uptake of bacteria of the SCV phenotype could be host cell dependent, as the above-mentioned studies (10, 43, 50, 54) used cell types other than mouse granulocytes. The uptake could also be strain dependent.
As shown by us and others, the SCV phenotype plays a significant role in S. aureus pathogenesis: although the SCV phenotype shows a low level of virulence, it induces latent and, in some cases, persistent infections. Combined with its ability to revert to the rapidly growing and virulent wild-type phenotype, it is plausible that this phenomenon explains the recurrence of S. aureus infections. It is therefore important to also focus on the eradication of bacteria of the SCV phenotype when considering the antibiotic treatment of S. aureus infections.
DCX and LZD are antibiotics commonly used for S. aureus infections (1, 18, 21, 28, 42, 46). As commonly observed for β-lactams (3, 5, 26, 33), DCX reduced the extracellular forms of S. aureus more efficiently than the intracellular forms in vitro for both the WT and the SCV phenotypes. The maximal bacterial reduction obtained intracellularly (1 to 1.5 log10 CFU) was surprisingly not significantly higher for the WT phenotype than for the SCV phenotype, contrary to previously reported observations for oxacillin (33). The different findings could, however, be explained by different growth rates of the test organism or a difference in auxotrophies of the SCVs (4). DCX appeared to be more potent against the intracellular form of the SCV phenotype than against the extracellular form when considering weight concentrations (mg/liter), which corresponds to previously reported findings (33). However, making allowance for the difference in MIC values between the two phenotypes (comparison in multiples of the MIC), DCX was as potent against bacteria of the SCV phenotype as it was against bacteria of the WT phenotype, showing that the MIC was the main driver of the intracellular expression of activity. The impaired intracellular activity of DCX compared to the extracellular activity was demonstrated in vivo for both phenotypes, although the extensive clearance of bacteria of the SCV phenotype obscured the real antimicrobial activity at the 24-h time point. The 96-h single-dose study indicated that DCX could, to some extent, prevent the accumulation of bacteria of the SCV phenotype in the kidneys but that a single dose, in most cases, was inadequate for complete clearance from the kidneys. These findings were supported by data from a previous study by Bates et al. (4), where oxacillin also failed to clear SCVs from the mouse kidney.
It is difficult to obtain a clear picture of the effect of β-lactams on the SCV phenotype from the results of previous studies due to a lack of standardization in this field. This concerns various factors such as strain type, culture conditions, auxotrophy, antibiotic susceptibility, animal and infection model, use of in vitro versus in vivo models, and knowledge of intracellular presence, and so on (4, 10, 31).
In vitro, LZD showed equal maximal efficacies intra- and extracellularly against bacteria of both phenotypes, which is contrary to the findings described previously by Nguyen et al. (33), who measured a maximal extracellular efficacy of LZD that was significantly better than its intracellular activity at 24 h (33) (although prolonging the incubation to 72 h significantly improved the maximal efficacy of LZD against intracellular SCVs). As mentioned above, this difference could be explained by the use of different test organisms in these studies. Similar to our findings, however, those authors found that LZD achieved the same maximal efficacy intracellularly against bacteria of the SCV phenotype as against bacteria of the WT phenotype. Considering the relative potency of LZD in vitro, we found that similar concentrations are needed for the intra- and extracellular forms of bacteria of the WT phenotype and for the extracellular form of bacteria of the SCV phenotype, but lower concentrations are sufficient to obtain intracellular activity against bacteria of the SCV phenotype. This indicates that LZD is most potent against the intracellular form of bacteria of the SCV phenotype, which was also found by the study by Nguyen et al. (33).
In the mouse peritonitis model, similar intra- and extracellular activities against bacteria of the WT phenotype were observed after 24 h of treatment. For the first 4 h of treatment, LZD was more effective against the extracellular SCVs than against the intracellular SCVs, but after 24 h of treatment, it was not possible to differentiate between treated and untreated mice or between the intra- and extracellular compartments due to the extensive bacterial clearance. The 96-h single-dose study indicated that, as with DCX, LZD could to some extent prevent the accumulation of bacteria of the SCV phenotype in the kidneys but that a single dose was inadequate for a complete clearance from the kidneys. No comparative in vivo studies investigating the activity of LZD against bacteria of the SCV phenotype were found.
In summary, both DCX and LZD reduced the amount of bacteria in the intracellular compartment by approximately 1 to 1.5 log10 CFU in vitro for the two phenotypes investigated. However, DCX was considerably less effective against these intracellular forms than against the extracellular ones, while LZD displayed equal, albeit weak, efficacies (in terms of bacterial eradication) in both situations. However, while DCX and LZD were able to control both intra- and extracellular infections caused by either phenotype in vivo, treatment with a single dose of either antibiotic was insufficient for the clearance of the SCVs in the kidney, and the risk of a recurrence of the infection was impending. This confirms the decreased efficacy shown with the in vitro model. Future studies should examine (i) whether this may be compensated for by prolonging the treatment duration so as to minimize the risk of relapses, (ii) to what extent the present conclusions can be generalized to other growing conditions and to other strains, and (iii) how the model could be extended to other cell types, such as epithelial and endothelial cells, or professional noninflammatory phagocytes, in which S. aureus is also known to survive.
Acknowledgments
We thank J. M. Andersen, L. Borggild, D. Truelsen, F. R. Pedersen, P. N. Nielsen, L. U. Kurland, and F. E. Jensen (Statens Serum Institut) for their dedicated technical assistance during the animal studies and Pierre Baudoux, M.-C. Cambier, and M. Vergauwen (Unité de Pharmacologie Cellulaire et Moléculaire) for guidance and technical help during studies with the cellular (THP-1) in vitro model.
F.V.B. and S.L. are Maître de Recherches and Chargée de Recherches of the Belgian Fonds de la Recherche Scientifiques (FRS-FNRS). This work was supported by the M. L. Jørgensen and Gunnar Hansen Foundation, Denmark, and by the STAPHAUR program for the Région Wallonne, Belgium (grant no. EP1A320501R052F/415735).
Footnotes
Published ahead of print on 31 January 2011.
REFERENCES
- 1.Bamberger, D. M., and S. E. Boyd. 2005. Management of Staphylococcus aureus infections. Am. Fam. Physician 72:2474-2481. [PubMed] [Google Scholar]
- 2.Barcia-Macay, M., S. Lemaire, M. P. Mingeot-Leclercq, P. M. Tulkens, and F. Van Bambeke. 2006. Evaluation of the extracellular and intracellular activities (human THP-1 macrophages) of telavancin versus vancomycin against methicillin-susceptible, methicillin-resistant, vancomycin-intermediate and vancomycin-resistant Staphylococcus aureus. J. Antimicrob. Chemother. 58:1177-1184. [DOI] [PubMed] [Google Scholar]
- 3.Barcia-Macay, M., C. Seral, M. P. Mingeot-Leclercq, P. M. Tulkens, and F. Van Bambeke. 2006. Pharmacodynamic evaluation of the intracellular activities of antibiotics against Staphylococcus aureus in a model of THP-1 macrophages. Antimicrob. Agents Chemother. 50:841-851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bates, D. M., et al. 2003. Staphylococcus aureus menD and hemB mutants are as infective as the parent strains, but the menadione biosynthetic mutant persists within the kidney. J. Infect. Dis. 187:1654-1661. [DOI] [PubMed] [Google Scholar]
- 5.Baudoux, P., et al. 2007. Combined effect of pH and concentration on the activities of gentamicin and oxacillin against Staphylococcus aureus in pharmacodynamic models of extracellular and intracellular infections. J. Antimicrob. Chemother. 59:246-253. [DOI] [PubMed] [Google Scholar]
- 6.Besier, S., A. Ludwig, K. Ohlsen, V. Brade, and T. A. Wichelhaus. 2007. Molecular analysis of the thymidine-auxotrophic small colony variant phenotype of Staphylococcus aureus. Int. J. Med. Microbiol. 297:217-225. [DOI] [PubMed] [Google Scholar]
- 7.Besier, S., et al. 2007. Prevalence and clinical significance of Staphylococcus aureus small-colony variants in cystic fibrosis lung disease. J. Clin. Microbiol. 45:168-172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Besier, S., et al. 2008. The thymidine-dependent small-colony-variant phenotype is associated with hypermutability and antibiotic resistance in clinical Staphylococcus aureus isolates. Antimicrob. Agents Chemother. 52:2183-2189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brouillette, E., G. Grondin, L. Shkreta, P. Lacasse, and B. G. Talbot. 2003. In vivo and in vitro demonstration that Staphylococcus aureus is an intracellular pathogen in the presence or absence of fibronectin-binding proteins. Microb. Pathog. 35:159-168. [DOI] [PubMed] [Google Scholar]
- 10.Brouillette, E., A. Martinez, B. J. Boyll, N. E. Allen, and F. Malouin. 2004. Persistence of a Staphylococcus aureus small-colony variant under antibiotic pressure in vivo. FEMS Immunol. Med. Microbiol. 41:35-41. [DOI] [PubMed] [Google Scholar]
- 11.Carryn, S., F. Van Bambeke, M. P. Mingeot-Leclercq, and P. M. Tulkens. 2002. Comparative intracellular (THP-1 macrophage) and extracellular activities of beta-lactams, azithromycin, gentamicin, and fluoroquinolones against Listeria monocytogenes at clinically relevant concentrations. Antimicrob. Agents Chemother. 46:2095-2103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chang, F. Y., et al. 2003. Staphylococcus aureus bacteremia: recurrence and the impact of antibiotic treatment in a prospective multicenter study. Medicine (Baltimore) 82:333-339. [DOI] [PubMed] [Google Scholar]
- 13.Chatterjee, I., et al. 2008. In vivo mutations of thymidylate synthase (encoded by thyA) are responsible for thymidine dependency in clinical small-colony variants of Staphylococcus aureus. J. Bacteriol. 190:834-842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ciampolini, J., and K. G. Harding. 2000. Pathophysiology of chronic bacterial osteomyelitis. Why do antibiotics fail so often? Postgrad. Med. J. 76:479-483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Clement, S., et al. 2005. Evidence of an intracellular reservoir in the nasal mucosa of patients with recurrent Staphylococcus aureus rhinosinusitis. J. Infect. Dis. 192:1023-1028. [DOI] [PubMed] [Google Scholar]
- 16.Clinical and Laboratory Standards Institute. 2008. Performance standards for antimicrobial susceptibility testing; fifteenth informational supplement (M100-S15). Clinical and Laboratory Standards Institute, Wayne, PA.
- 17.Frimodt-Moller, N. 1993. The mouse peritonitis model: present and future use. J. Antimicrob. Chemother. 31(Suppl. D):55-60. [DOI] [PubMed] [Google Scholar]
- 18.Grayson, M. L. 2006. The treatment triangle for staphylococcal infections. N. Engl. J. Med. 355:724-727. [DOI] [PubMed] [Google Scholar]
- 19.Gresham, H. D., et al. 2000. Survival of Staphylococcus aureus inside neutrophils contributes to infection. J. Immunol. 164:3713-3722. [DOI] [PubMed] [Google Scholar]
- 20.Hess, D. J., M. J. Henry-Stanley, E. A. Erickson, and C. L. Wells. 2003. Intracellular survival of Staphylococcus aureus within cultured enterocytes. J. Surg. Res. 114:42-49. [DOI] [PubMed] [Google Scholar]
- 21.Jensen, A. G., et al. 2002. Treatment and outcome of Staphylococcus aureus bacteremia: a prospective study of 278 cases. Arch. Intern. Med. 162:25-32. [DOI] [PubMed] [Google Scholar]
- 22.Johnson, L. B., M. O. Almoujahed, K. Ilg, L. Maolood, and R. Khatib. 2003. Staphylococcus aureus bacteremia: compliance with standard treatment, long-term outcome and predictors of relapse. Scand. J. Infect. Dis. 35:782-789. [DOI] [PubMed] [Google Scholar]
- 23.Jonsson, I. M., et al. 2003. Virulence of a hemB mutant displaying the phenotype of a Staphylococcus aureus small colony variant in a murine model of septic arthritis. Microb. Pathog. 34:73-79. [DOI] [PubMed] [Google Scholar]
- 24.Kahl, B., et al. 1998. Persistent infection with small colony variant strains of Staphylococcus aureus in patients with cystic fibrosis. J. Infect. Dis. 177:1023-1029. [DOI] [PubMed] [Google Scholar]
- 25.Lannergard, J., et al. 2008. Identification of the genetic basis for clinical menadione-auxotrophic small-colony variant isolates of Staphylococcus aureus. Antimicrob. Agents Chemother. 52:4017-4022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lemaire, S., F. Van Bambeke, M. P. Mingeot-Leclercq, and P. M. Tulkens. 2005. Activity of three beta-lactams (ertapenem, meropenem and ampicillin) against intraphagocytic Listeria monocytogenes and Staphylococcus aureus. J. Antimicrob. Chemother. 55:897-904. [DOI] [PubMed] [Google Scholar]
- 27.Lowry, O. H., N. J. Rosebrough, A. L. Farr, and R. J. Randall. 1951. Protein measurement with the Folin phenol reagent. J. Biol. Chem. 193:265-275. [PubMed] [Google Scholar]
- 28.Lowy, F. D. 1998. Staphylococcus aureus infections. N. Engl. J. Med. 339:520-532. [DOI] [PubMed] [Google Scholar]
- 29.Lowy, F. D. 2000. Is Staphylococcus aureus an intracellular pathogen? Trends Microbiol. 8:341-343. [DOI] [PubMed] [Google Scholar]
- 30.Mempel, M., et al. 2002. Invasion of human keratinocytes by Staphylococcus aureus and intracellular bacterial persistence represent haemolysin-independent virulence mechanisms that are followed by features of necrotic and apoptotic keratinocyte cell death. Br. J. Dermatol. 146:943-951. [DOI] [PubMed] [Google Scholar]
- 31.Miller, M. H., M. A. Wexler, and N. H. Steigbigel. 1978. Single and combination antibiotic therapy of Staphylococcus aureus experimental endocarditis: emergence of gentamicin-resistant mutants. Antimicrob. Agents Chemother. 14:336-343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Murchan, S., et al. 2003. Harmonization of pulsed-field gel electrophoresis protocols for epidemiological typing of strains of methicillin-resistant Staphylococcus aureus: a single approach developed by consensus in 10 European laboratories and its application for tracing the spread of related strains. J. Clin. Microbiol. 41:1574-1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nguyen, H. A., et al. 2009. Intracellular activity of antibiotics in a model of human THP-1 macrophages infected by a Staphylococcus aureus small-colony variant strain isolated from a cystic fibrosis patient: pharmacodynamic evaluation and comparison with isogenic normal-phenotype and revertant strains. Antimicrob. Agents Chemother. 53:1434-1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pelletier, L. L., Jr., M. Richardson, and M. Feist. 1979. Virulent gentamicin-induced small colony variants of Staphylococcus aureus. J. Lab. Clin. Med. 94:324-334. [PubMed] [Google Scholar]
- 35.Proctor, R. A., et al. 1998. Staphylococcal small colony variants have novel mechanisms for antibiotic resistance. Clin. Infect. Dis. 27(Suppl. 1):S68-S74. [DOI] [PubMed] [Google Scholar]
- 36.Proctor, R. A., P. van Langevelde, M. Kristjansson, J. N. Maslow, and R. D. Arbeit. 1995. Persistent and relapsing infections associated with small-colony variants of Staphylococcus aureus. Clin. Infect. Dis. 20:95-102. [DOI] [PubMed] [Google Scholar]
- 37.Proctor, R. A., et al. 2006. Small colony variants: a pathogenic form of bacteria that facilitates persistent and recurrent infections. Nat. Rev. Microbiol. 4:295-305. [DOI] [PubMed] [Google Scholar]
- 38.Quie, P. G. 1969. Microcolonies (G-variants) of Staphylococcus aureus. Yale J. Biol. Med. 41:394-403. [PMC free article] [PubMed] [Google Scholar]
- 39.Roder, B. L., et al. 1999. Clinical features of Staphylococcus aureus endocarditis: a 10-year experience in Denmark. Arch. Intern. Med. 159:462-469. [DOI] [PubMed] [Google Scholar]
- 40.Sadowska, B., et al. 2002. Characteristics of Staphylococcus aureus, isolated from airways of cystic fibrosis patients, and their small colony variants. FEMS Immunol. Med. Microbiol. 32:191-197. [DOI] [PubMed] [Google Scholar]
- 41.Sandberg, A., J. H. Hessler, R. L. Skov, J. Blom, and N. Frimodt-Moller. 2009. Intracellular activity of antibiotics against Staphylococcus aureus in the mouse peritonitis model. Antimicrob. Agents Chemother. 53:1874-1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schmidt-Ioanas, M., A. de Roux, and H. Lode. 2005. New antibiotics for the treatment of severe staphylococcal infection in the critically ill patient. Curr. Opin. Crit. Care 11:481-486. [DOI] [PubMed] [Google Scholar]
- 43.Schroder, A., R. Kland, A. Peschel, C. von Eiff, and M. Aepfelbacher. 2006. Live cell imaging of phagosome maturation in Staphylococcus aureus infected human endothelial cells: small colony variants are able to survive in lysosomes. Med. Microbiol. Immunol. 195:185-194. [DOI] [PubMed] [Google Scholar]
- 44.Seral, C., F. Van Bambeke, and P. M. Tulkens. 2003. Quantitative analysis of gentamicin, azithromycin, telithromycin, ciprofloxacin, moxifloxacin, and oritavancin (LY333328) activities against intracellular Staphylococcus aureus in mouse J774 macrophages. Antimicrob. Agents Chemother. 47:2283-2292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sifri, C. D., A. Baresch-Bernal, S. B. Calderwood, and C. von Eiff. 2006. Virulence of Staphylococcus aureus small colony variants in the Caenorhabditis elegans infection model. Infect. Immun. 74:1091-1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stalker, D. J., and G. L. Jungbluth. 2003. Clinical pharmacokinetics of linezolid, a novel oxazolidinone antibacterial. Clin. Pharmacokinet. 42:1129-1140. [DOI] [PubMed] [Google Scholar]
- 47.Tsuchiya, S., et al. 1980. Establishment and characterization of a human acute monocytic leukemia cell line (THP-1). Int. J. Cancer 26:171-176. [DOI] [PubMed] [Google Scholar]
- 48.Tulkens, P. M. 1991. Intracellular distribution and activity of antibiotics. Eur. J. Clin. Microbiol. Infect. Dis. 10:100-106. [DOI] [PubMed] [Google Scholar]
- 49.Van Bambeke, F., M. Barcia-Macay, S. Lemaire, and P. M. Tulkens. 2006. Cellular pharmacodynamics and pharmacokinetics of antibiotics: current views and perspectives. Curr. Opin. Drug Discov. Devel. 9:218-230. [PubMed] [Google Scholar]
- 50.Vaudaux, P., et al. 2002. Increased expression of clumping factor and fibronectin-binding proteins by hemB mutants of Staphylococcus aureus expressing small colony variant phenotypes. Infect. Immun. 70:5428-5437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Vesga, O., et al. 1996. Staphylococcus aureus small colony variants are induced by the endothelial cell intracellular milieu. J. Infect. Dis. 173:739-742. [DOI] [PubMed] [Google Scholar]
- 52.von Eiff, C. 2008. Staphylococcus aureus small colony variants: a challenge to microbiologists and clinicians. Int. J. Antimicrob. Agents 31:507-510. [DOI] [PubMed] [Google Scholar]
- 53.von Eiff, C., et al. 1997. Recovery of small colony variants of Staphylococcus aureus following gentamicin bead placement for osteomyelitis. Clin. Infect. Dis. 25:1250-1251. [DOI] [PubMed] [Google Scholar]
- 54.von Eiff, C., et al. 1997. A site-directed Staphylococcus aureus hemB mutant is a small-colony variant which persists intracellularly. J. Bacteriol. 179:4706-4712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.von Eiff, C., G. Peters, and K. Becker. 2006. The small colony variant (SCV) concept—the role of staphylococcal SCVs in persistent infections. Injury 37(Suppl. 2):S26-S33. [DOI] [PubMed] [Google Scholar]
- 56.von Eiff, C., R. A. Proctor, and G. Peters. 2000. Staphylococcus aureus small colony variants: formation and clinical impact. Int. J. Clin. Pract. Suppl. 2000:44-49. [PubMed] [Google Scholar]
- 57.Zander, J., et al. 2008. Influence of dTMP on the phenotypic appearance and intracellular persistence of Staphylococcus aureus. Infect. Immun. 76:1333-1339. [DOI] [PMC free article] [PubMed] [Google Scholar]